

Background and synopsis of history
Idiopathic pulmonary arterial hypertension (IPAH), formerly known as primary pulmonary hypertension (PPH), was first described by Dresdale and colleagues [1] in 1951. This report was soon followed by a second publication in 1954, in which the same author described the occurrence of pulmonary hypertension among several members of the same family, comprising the first report of familial PPH, now known as familial pulmonary arterial hypertension (FPAH) [2].
After this initial description, slow progress in understanding the genetics of FPAH was made during the next 30 years, with individual reports describing 13 different families reported in the United States. In 1984, a follow-up analysis of these 13 families, including interval description of 8 new cases in 9 of the families and an additional 14th family, were reviewed. The additional family contained the largest number of affected family members described to that time, with six deaths caused by PPH during two generations [3]. That publication described inheritance patterns, including vertical transmission and father-to-son transmission, which suggested autosomal dominant pattern of inheritance indicative of a single gene defect. This report also caused experts to speculate that cases of PPH that seemed to occur in isolation may, in fact, have a familial basis, although this was difficult to recognize partly because of skip generations that resulted from incomplete penetrance [4].
The National Institutes of Health (NIH) PPH prospective registry [5] in the mid-1980s provided the benchmark for the clinical definition of IPAH, and facilitated interaction of participating investigators to collect and organize sufficient numbers of FPAH families to provide robust statistical power for a genome-wide search for FPAH loci. Two separate teams of investigators independently established linkage of a locus for the gene, named PPH1, for familial PPH to chromosome 2q31–32 in 1997 [6,7]. A few years later, both teams also showed that mutations in the gene encoding bone morphogenetic protein receptor type 2 (BMPR2), a receptor that is a member of the transforming growth factor beta (TGF-β) superfamily [8,9], was responsible for FPAH.
Epidemiology of pulmonary arterial hypertension
The frequency of PPH, which encompasses IPAH and FPAH, is estimated to be one to two cases per year per 1 million in the general population, with a female-to-male prevalence of 2:1 but with similar disease severity and outcome [10,11]. Even after 2 decades, the best available clinical information on the natural history of IPAH derives from the prospective NIH study of patients who had PPH and were followed up in the United States from 1981 to 1985. This report remains the benchmark because it meticulously described and followed up 187 patients who had PPH at 32 centers in the United States, and was conducted before effective therapies were developed. The ethnic background of patients was comparable to that of the U.S. population. The registry confirmed that PPH can develop at any age, with a mean age at diagnosis of 36 years. In general, women tended to present in the third decade of life and men in the fourth. The median survival after diagnosis was 2.8 years. The mean time from symptom onset to diagnosis was 2 years for the entire cohort, but this interval was shorter for patients who had a family history of PPH. In terms of familial prevalence, the NIH registry reported prevalence of 6% based on the finding that 12 of the 187 patients reported one or more immediate family member affected with PAH. These 12 FPAH patients did not differ from the 175 other patients in hemodynamic data or symptoms [5,12].
Similar findings were confirmed and extended in a national prospective registry of patients who have PAH that was established in France in 2000 and which recently reported 674 enrolled PAH cases. Investigators reported a PAH prevalence of 15 cases per 1 million adult French inhabitants. They identified a female-to-male ratio of 1.9:1, consistent with the NIH registry data. The distribution of PAH causes in the 674 cases showed 39.2% from IPAH, 3.9% from FPAH, 15.3% associated with connective tissue diseases (systemic sclerosis was leading cause), 11.3% related to congenital heart diseases, 10.4% related to portal hypertension, 9.5% associated with anorexigen use, 6.2% related to HIV, and 4.3% related to other causes. The design of this registry did not include children [13].
Genetics and the diagnosis of familial pulmonary arterial hypertension
IPAH and FPAH share the same clinical features, histopathology, and clinical course. The true incidence of FPAH is unknown and is difficult to completely elucidate [14]. Some investigators speculate that as knowledge of the complex genetics of PAH increases and mutation detection improves, the percentage of patients whose disease is identified as familial will continue to climb. Establishing the role of familial transmission is often difficult for several reasons. First, despite increasing awareness of the disease entity, misdiagnosis of pulmonary hypertension continues to occur. Second, despite the autosomal dominant nature of the genetic defects in the BMPR2 gene, the penetrance of disease expression is low and varies among families. As a result, generations of individuals carrying mutation may not develop clinical expression of FPAH. Estimates indicate that only approximately 20% of individuals with a known genetic mutation in BMPR2 will develop PAH during their entire life, and that phenotypic expression may occur at any age. Finally, the high mobility of our society may hinder complete knowledge of family genealogic history, which can impair recognition of families not otherwise known to be related [15].
Only gender has been shown to influence clinical expression of the BMPR2 mutation, demonstrated by the 2:1 female predominance. This finding implicates the role of gender in facilitating a predisposition to disease, suggesting some hormonal influence or X chromosome association for clinical expression [16]. Neither suggestion has been confirmed, but an abnormal gender ratio among progeny of obligate gene carriers has been described. Increased ratio of female live births suggests possible selective wastage of male fetuses or an abnormal primary sex ratio [17]. The fact that more women are affected may be caused by the loss of male fetuses from an unrecognized defect in embryologic development.
Data from national prospective registries in the United States and France described a 6% and 3.9% prevalence, respectively, of FPAH among patients who have PAH [5,13]. Discovery of the molecular basis of FPAH and IPAH has advanced investigations into the true impact of genetics on the diagnosis of PAH. The family reported to be most heavily affected with FPAH was described in an investigation that connected five previously independent Tennessee families, including 18 affected members, 12 of whom were erroneously believed to have IPAH. These five family branches, not formerly known to the investigators to be related, shared the same BMPR2 missense mutation and a common ancestry [18]. Currently, 23 patients (20 women, and 3 men) who have FPAH have been identified in the extended kindred (Fig. 1), including a new family branch discovered in 2006.
Fig. 1.
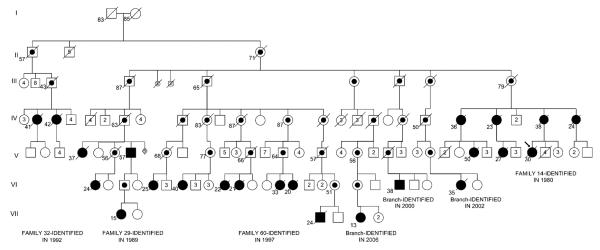
Pedigree of an extended kindred with familial pulmonary arterial hypertension (FPAH). This pedigree now links six families that were previously labeled as independent, and 23 total patients who had FPAH. Solid symbols represent individuals who have disease; circles represent women, squares represent men. Line through symbol represents death. Dot inside symbol represents obligate carrier of the BMPR2 mutation. Numbers below symbols represent age at death or current living age. Numbers inside symbols represent numbers of unaffected siblings of each gender. S inside symbol represents stillbirth. Diamond symbol represents sex unknown.
FPAH seems to show no predilection for geographic location; it has been reported around the globe. FPAH predominantly affects Caucasians in the United States, but ethnic or racial distribution has not been investigated in depth [15]. The registry at Vanderbilt University currently contains 100 families that have two or more affected members. Of the 3750 total individuals followed up, 352 subjects meet criteria for PAH.
Reduced penetrance
Initial pedigree studies of families with PAH showed phenotypic expression in fewer individuals than expected. In fact, individuals known to be at risk for disease through pedigree analysis did not express PAH consistently [3]. From genetic analysis, mutant BMPR2 alleles are known to display reduced penetrance among PAH kindreds, indicating that heterozygous mutation of the gene is required, but not sufficient, to precipitate clinical expression of disease [10]. The mechanisms of reduced penetrance are unknown, and are the subject of intense investigation because of the possible role of genetic or environmental modifiers of disease expression [19].
The registry at Vanderbilt continues to display reduced penetrance. In fact, of the 3750 total subjects followed up, approximately 2250 are within family bloodlines, but only 15.6% of subjects were diagnosed with PAH. Furthermore, pedigree analysis of the six most heavily affected families shows that 24.2% of first-degree relatives of patients who have PAH were also diagnosed with this disease (Eric D. Austin, MD; Lisa Wheeler; James E. Loyd, MD, unpublished data, 2006). These heavily affected families provide a valuable opportunity for genetic and environmental evaluation of disease modifiers, which is underway.
Genetic anticipation
Several reports have shown earlier age of FPAH onset in subsequent generations. This early onset is one manifestation of genetic anticipation that was observed in some of the earliest reports of individual families in the 1960s, and was again substantiated in a summary analysis of multiple families in 1984 [3]. The age at death in each successive generation decreased serially by approximately a decade. For example, age of death in successive generations statistically decreased from a mean of 45.6 years to 36.3 years to 24.2 years. Historically, genetic anticipation was attributed to ascertainment and other possible bias. In a method designed to overcome artefactual bias, 60 pairs, including an affected parent and child, were analyzed with the finding that age at death was significantly less (15 years younger) for the affected child than for the parent, confirming a biologic basis for genetic anticipation as best possible [20].
The molecular mechanism of genetic anticipation in FPAH remains unknown, despite the recent advances in knowledge about central pathogenesis. Many other disease examples of genetic anticipation exist and may lend clues. Several neurologic diseases, including fragile X syndrome, myotonic dystrophy, and Huntington's disease, exhibit genetic anticipation in association with trinucleotide repeat expansion (TRE). In these diseases, TRE has been confirmed as the molecular basis for not only genetic anticipation but also incomplete penetrance and variable age of onset [21,22]. All three phenomena are also similarly observed in FPAH. TRE has not been identified in the primary gene defects in BMPR2, but efforts continue to identify relevant regions containing expanded repeats. Meanwhile, speculation continues that TRE might be present in a modifier gene (not yet identified) that facilitates disease expression. Alternatively, another molecular mechanism, not yet recognized, may cause genetic anticipation.
Mutations in BMPR2 cause familial pulmonary arterial hypertension
The discovery of the genetic basis of FPAH was made possible by the collaborative development of family cohorts with banked DNA, and facilitated by advances in molecular genetics and availability of information from the human genome project [23]. As families were identified with PPH, DNA was collected and stored, so that by the mid-1990s the statistical power was sufficient to locate the genetic cause of PAH. Two separate groups used microsatellite markers and linkage analysis to focus the search to chromosome 2q33. Using DNA collected from 19 affected and 58 unaffected family members of six families with PPH that had pedigrees suggestive of an autosomal dominance pattern of gene transmission, Nichols and colleagues [6] used a microsatellite marker search to establish linkage to a 30 million base pair region on chromosome 2q33. Similarly, Morse and colleagues [24] independently identified the locus of a familial PPH gene to chromosome 2q31–32.
Because the segment on chromosome 2 was longer than could readily be sequenced in that era, the investigators used a positional candidate gene approach, targeting and testing several genes in this region with known gene products of potential relevance [15]. Because it is a member of the TGF-β superfamily of receptors, BMPR2 was selected for evaluation. In 2000, two separate teams of investigators characterized multiple heterozygous mutations in this large gene, which resides on chromosome 2q33 and has 13 exons, as the cause of disease in several affected kindreds [8,9].
Prevalence of BMPR2 mutations in familial pulmonary arterial hypertension
Since the discovery of BMPR2 mutations in several families with FPAH in 2000, investigators have worked to identify mutations in other families and in those with apparently “sporadic” PAH (IPAH). Molecular testing for BMPR2 abnormalities has shown mutations among many different ethnic groups [25–27]. By 2005, approximately 65% of known FPAH families had BMPR2 mutations identifiable through gene sequencing. A summary report described 144 distinct mutations among 210 subjects, and most of these coded frameshift, nonsense, or splice site donor/acceptance site mutations [28]. By expanding the mutation screening methods in 2005, Cogan and colleagues [29] showed that 48% of the 21 mutations discovered in 30 unrelated affected families were deletions or duplications of exons. These investigators used Multiplex Ligation-dependent Probe Amplification (MLPA) analysis of genomic DNA, confirmed with real-time polymerase chain reaction. They concluded that these techniques improved the identification of genetic abnormalities in BMPR2 in FPAH, showing that 70% or more of families have identifiable BMPR2 mutations. A different group also recently reported using MLPA analysis of genomic DNA, with mutations discovered in 28% of families previously reported to lack BMPR2 mutations [30]. These reports further support the primary role of BMPR2 in the central pathogenesis of disease in FPAH. In the Vanderbilt cohort, DNA specimens are available from at least one affected patient in 55 families, and using current methods BMPR2 mutations are found to be present in 46 families, but not in 9. Fig. 2 displays the distribution of most reported recurrent mutations in the BMPR2 gene, including their domain location.
Fig. 2.

Distribution of mutations across the BMPR2 gene. (A) Line graph illustrating the distribution and frequency of recurrent mutations. The x-axis represents the position of mutations relative to the BMPR2 cDNA below, and the y-axis defines the number of unrelated probands observed to harbor each mutation. The contiguous black and dotted line identifies the two amino acid substitutions recurrent at position 491. (B) Proportional representation of the BMPR2 cDNA, with exons 1–13 indicated. The boxed regions delineate the extracellular (ECD), transmembrane (TM), kinase (KD), and cytoplasmic (CD) domains of the receptor depicted below. Large gene rearrangements (deletions and duplications) identified in PAH cases are arrayed below the cDNA. (C) Table summarizing the total number of PAH-causing mutations observed in each functional domain and nondomain encoding sequence. (From Machado RD, Aldred MA, James V, et al. Mutations of the TGF-β type II receptor BMPR2 in pulmonary arterial hypertension. Human Mutation 2006; 27(2):121–32; with permission).
BMPR2 mutations in sporadic or idiopathic PAH
Although further analysis of larger IPAH cohorts is needed, it is already clear that a significant proportion of patients who have IPAH harbor mutations in the BMPR2 gene. These mutations may occur in the setting of low-penetrance germline mutations, in which other family members transmit the mutation without disease expression, or the mutations may develop de novo [31]. This fact is not surprising, given that low penetrance and genetic anticipation are hallmarks of FPAH. Although the rate of BMPR2 mutation in the general population is not known, it seems to be extremely low given that no mutations have been detected among a few hundred control samples [15].
Reports of mutation prevalence in patients who have IPAH are limited, and these depend partly on which detection techniques were used, because these techniques continue to improve. The true prevalence is currently unknown, with reports ranging from 11% to 40% [14,26,27]. As with FPAH, more mutations were detected when newer molecular techniques were applied to IPAH cohorts. Aldred and colleagues [30] noted that 6 of 106 (5.7%) patients who had IPAH previously labeled BMPR2 mutation–negative had detectable gene rearrangements. Parental evaluation in three of these patients showed that two of the mutations were de novo, whereas one was inherited from an unaffected mother.
BMPR2 and the expression of disease
Haploinsufficiency describes the condition in which heterozygosity for a gene mutation leads to insufficient protein product, with decreased protein function leading to phenotypic expression of disease. In 2001, Machado and colleagues [25] examined the role of BMPR2 mutation in terms of genotype–phenotype relationships, proposing that a model of haploinsufficiency explains the development of pulmonary hypertension from a molecular perspective. They suggested that 60% of the reported pathogenic mutations should result in premature truncation codons of the BMPR2 transcript, which would lead to nonsense-mediated decay. Recent studies have supported this by showing deletions within exons 2–13, which would confer nonfunctional peptide [29,30]. These studies suggest that haploinsufficiency contributes to the molecular mechanisms underlying PAH through reduction in the available level of BMPR2 protein, at least in some cases.
Investigation of downstream molecular effects of the BMPR2 signaling pathway have also shown that specific disease-causing mutations in the ligand-binding and kinase domains of BMPR2 seem to exert dominant negative effects on BMP signaling. Therefore, disruption of BMP signaling may lead to absence of critical mechanisms in the antiproliferative and differentiation mechanisms in the pulmonary vasculature [28]. In fact, functional studies have shown dominant negative effects on BMP signaling in vitro in the setting of point mutations or truncations in the kinase domain of BMPR2 [32].
Multiple “hits”: Is one abnormal BMPR2 allele sufficient to cause pulmonary arterial hypertension?
The observation of reduced penetrance implies that mutation of the BMPR2 gene is required but is not sufficient alone for phenotypic expression. This fact suggests that other genetic or environmental modifiers likely mediate the clinical expression of disease [10]. Given this information, individuals who carry the BMPR2 mutation and others may express disease in the setting of a “multiple hit” model, analogous to models of sequential mutations in the genesis of neoplasia.
Plexiform lesions are a characteristic histopathologic microvascular abnormality found in the lungs of many patients who have PAH [33]. Some investigators have suggested that they represent a neoplastic-like proliferative process, with resultant endothelial and smooth muscle cell proliferation and diminished apoptosis. In support of this, evidence of monoclonal proliferation of local endothelial cells and the migration and local proliferation of smooth muscle cells exists [34]. Furthermore, investigations suggest a role for BMPR2 signaling in these events [35]. In contrast, the plexiform lesions in patients who have secondary pulmonary hypertension contain endothelial cells that are polyclonal [36]. In addition, experts have suggested that circulating endothelial precursor cells that migrate to the pulmonary vasculature may participate in the pathogenesis of PAH [37].
Immunohistochemical staining of lung tissue from patients who have FPAH shows nearly complete absence of BMPR2 protein in those who demonstrate heterozygosity through genetic testing [38]. This finding is somewhat unexpected, because at least some protein production is anticipated in the setting of one wild-type and one germline mutated allele. To examine this, Machado and colleagues [19] investigated whether inactivation of the remaining wild-type BMPR2 allele through somatic mutation prompts disease expression, similar to the loss of a second tumor suppressor gene in cancer development. However, results suggested that microsatellite instability with somatic loss of function in the remaining wild-type BMPR2 allele did not contribute to disease development. This area needs more investigation to evaluate whether de novo mutations or common polymorphisms in other genes function as multiple genetic “hits” that interact with the heterozygous BMPR2 mutation to create a molecular environment suitable to trigger phenotypic expression of disease [39].
BMPR2 signaling pathway
Expressed ubiquitously, BMPR2 is a receptor for a family of cytokines known as bone morphogenetic proteins (BMPs). BMPs are members of the TGF-β superfamily of proteins. The TGF-β family of proteins and their receptors are highly conserved throughout nature. Originally identified for their role in ectopic bone and cartilage formation, BMPs play a crucial role in regulating mammalian development, such as embryonic lung morphogenesis [40]. Although genetic studies strongly implicate the TGF-β superfamily in the regulation of pulmonary vascular cell growth and differentiation, investigations have not shown the precise molecular mechanisms involved. Nonetheless, progress has been made in evaluating their contribution to lung vascular development.
The BMP signaling pathway involves heterodimerization of the serine/threonine transmembrane kinases, BMPR1 and BMPR2. Four functional domains comprise BMPR2: ligand binding, kinase, transmembrane, and cytoplasmic domains. Activation of the BMPR2/BMPR1 receptor complex leads to phosphorylation of a series of cytoplasmic mediators, which include the Smad family. Specifically, Smad proteins 1, 5, and 8 are phosphorylated, and subsequently complex with Smad 4 for translocation into the nucleus to regulate target gene transcription. The Smad signaling pathway seems to participate in the inhibition of cell growth and induction of apoptosis [41]. Perhaps mutations in BMPR2 eliminate a critical growth regulatory function in pulmonary vascular cells. Why other types of vasculature in the body are spared remains a mystery.
In addition to Smad pathway activation, other substrates are affected by BMPR2/BMPR1 receptor activation, including mitogen-activated protein kinases (MAPKs), NH2-terminal kinase, and others [42]. Recent studies have implicated not only Smad signaling but also unopposed MAPK function in association with BMPR2 mutations. Yang and colleagues [32] showed that kinase domain mutations of BMPR2 found in PAH patients result in down-regulation of Smad signaling in pulmonary artery smooth muscle cells (PASMCs), with resultant loss of antiproliferative effect. The resulting imbalance between Smad and MAPK function may lead to proproliferative and antiapoptotic effects that promote the development of PAH.
BMPR2 mutations in other disease states
The discovery of mutations in BMPR2 in patients who have FPAH and IPAH has prompted searches for this abnormality in other patients who have pulmonary hypertension. Mutations in this gene generally have not been found consistently, further supporting the relationship between these two disease entities, which may in fact exist together along a continuum.
Patients have been evaluated who have pulmonary veno-occlusive disease (PVOD), a rare form of pulmonary hypertension in which the vascular changes originate in the small pulmonary veins and venules. Runo and colleagues [43] documented a novel mutation in exon 1 of the BMPR2 gene, resulting in a truncated protein predicted to have no function, in a 36-year-old woman who had biopsy-confirmed PVOD. Further investigation showed that the proband's mother died of pulmonary hypertension, whereas laboratory evaluation confirmed an identical BMPR2 mutation in the proband's healthy sister. Similarly, Aldred and colleagues [30] discovered a genetic mutation in exon 2 of BMPR2 in a patient who had PVOD. This patient had three first-degree relatives who died with the diagnosis of pulmonary hypertension. This mutation of BMPR2 had previously been described in a family that had FPAH with no evidence of PVOD. These results suggest that the molecular alterations associated with BMPR2 mutations may exhibit phenotypic heterogeneity (ie, the ability of an allelic mutation at a single locus to produce more than one expression of disease). Furthermore, PVOD and FPAH may represent different spectra of the same disease, with phenotype influenced by genetic or environmental modifiers [43].
Another cause of PAH is associated with drug exposure. Specifically, PAH has been reported in conjunction with exposure to appetite suppressants, such as fenfluramine and dexfenfluramine [44,45]. The exact mechanism of PAH promotion has not been elucidated, but these drugs may serve as an environmental mediator to facilitate disease expression, possibly in genetically susceptible individuals. Individual factors of susceptibility are particularly plausible given the low numbers of PAH in those exposed to fenfluramine (approximately 1 case per 10,000 people exposed) [46]. Humbert and colleagues [47] found distinct BMPR2 mutations, each predicted to result in diminished receptor function, in 9% of unrelated patients who had PAH associated with fenfluramine exposure. They found no BMPR2 mutations in 130 normal control subjects. When compared with other patients who had PAH associated with fenfluramine, patients who had BMPR2 mutation expressed disease after a shorter interval of exposure to fenfluramine, which was also statistically significant. This information, although in requiring further study, again emphasizes the importance of additional genetic and environmental factors required to produce disease expression.
Insufficient data exist for an exhaustive discussion of congenital heart disease and BMPR2 mutations. A report by Roberts and colleagues [48] noted that 6% of 106 children and adults who had congenital heart disease possessed mutations of the BMPR2 gene. They noted that this finding agreed with reports in mouse models of vasculo-genesis and cardiac anomalies, which implicate abnormalities of the TGF-β pathway. These findings clearly warrant further investigation.
The serotonin pathway and pulmonary arterial hypertension mediation
Although the modifier genes of PAH pathogenesis are currently unknown, investigations are underway to elucidate these pathways. The neurotransmitter serotonin (5-hydroxytryptamine [5-HT]) has been implicated. Common genetic variations (polymorphisms) in the serotonin pathway or polymorphisms in the serotonin transporter (SERT) gene may contribute, either independently or related to the presence of a BMPR2 mutation. Serotonin has been shown to be a cellular mitogen, and stimulates PASMC proliferation through a signaling pathway mediated by SERT [49].
In 1995, Herve and colleagues [50] described increased plasma serotonin levels caused by abnormal platelet serotonin storage in 16 patients who had PPH. Subsequently, Eddahibi and colleagues [51,52] found increased growth of PASMCs in culture from patients who had PPH compared with controls when stimulated by serotonin or serum, and this proliferative response was augmented in the setting of hypoxia. The investigators attributed these mitogenic effects to increased expression of SERT. Furthermore, they proposed a clinical link to the molecular cause for SERT overexpression in the form of variations in the SERT gene promoter by comparing 89 people who had severe PPH and 84 control subjects. Homozygosity for a long promoter variant (L-allelic variant) was present in approximately 65% of patients (LL genotype individuals) who had PPH but only 27% of controls [52].
Continued work on the role of serotonin as a cellular mitogen has shown that internalization of 5-HT by SERT leads to downstream activation of the MAPK cascade, partly through the production of reactive oxygen species, which ultimately stimulates transcription of genes to drive cellular proliferation. PASMCs respond strongly to these stimulatory effects in vitro [53,54]. Therefore, up-regulation of the MAPK cascade as an effect of abnormal serotonin signaling has garnered attention as a potential antagonist to the growth inhibitory effects of BMP signaling pathways, and possibly confers susceptibility to PAH expression.
Genetic abnormalities may lead to an imbalance of these cellular processes that facilitates abnormal PASMC proliferation and the development of PAH. Seeking to confirm disease susceptibility alleles using a larger cohort, Machado and colleagues [55] recently examined the role of polymorphic variation within the SERT gene in the predisposition to and modification of PAH in 528 affected patients. This study evaluated 133 patients who had FPAH or IPAH and known BMPR2 mutation, 259 patients who had IPAH, 136 patients who had PAH associated with other diseases or exposures, and 353 control subjects. Results showed no difference in frequency of SERT gene alleles among any groups, suggesting that the SERT mutations evaluated are not likely to contribute to phenotypic expression of PAH in patients carrying a BMPR2 mutation and those who are not. Likewise, genetic variation of the SERT promoter gene locus did not correlate with differences in age of onset of disease, nor did this variation differ by gender, which is the only known risk factor for PAH development.
The authors' group also investigated the role of polymorphisms in the SERT gene, specifically the L allele, in a cohort of patients who had IPAH and FPAH. This cohort included 83 patients who had IPAH, 99 patients who had FPAH, 67 unaffected carriers of a known BMPR2 mutation, and 125 controls [54]. Overall, results comparable to those found in the study by Machado and colleagues [55] were found, including no difference in SERT genotype distribution among groups. Again, a lack of difference between diseased patients who had FPAH and unaffected patients carrying a BMPR2 mutation suggested that the polymorphisms studied do not affect disease penetrance. However, other polymorphisms within the SERT gene have not been investigated and could function as modifier genes. Lastly, although patients who had FPAH were diagnosed earlier than those who had IPAH, patients who had FPAH and were homozygous for the L allele (LL genotype) showed an even earlier age at diagnosis than patients who had FPAH and did not have this polymorphism. This finding may reflect unappreciated interactions between BMPR2 mutations and polymorphisms in the SERT gene that affect disease expression [56].
Hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu syndrome) and pulmonary arterial hypertension
Hereditary hemorrhagic telangiectasia (HHT), or Osler-Weber-Rendu syndrome, is a vascular dysplasia characterized by mucocutaneous telangiectasias that cause recurrent epistaxis, gastro-intestinal bleeding, and arteriovenous malformations of the pulmonary, hepatic, and cerebral circulations. Defects in additional components of the TGF-β signaling superfamily receptor complex—activin receptor-like kinase 1 (ALK1) located on chromosome 12 and endoglin on chromosome 9—are pivotal in the pathogenesis of HHT [57,58]. Pulmonary hypertension is also known to occur in HHT, typically a secondary form related to a high cardiac output state. However, pulmonary hypertension that is clinically and histologically identical to FPAH and IPAH has recently been described in multiple unique kindreds with HTT associated with detectable mutations in ALK1 [59,60]. Furthermore, a recent case report described an endoglin germline mutation in a patient who had HHT and PAH related to dexfenfluramine exposure [61]. Investigators also recently reported a child who had a mutation that resulted in abnormal splicing of the endoglin transcript that caused markedly reduced levels, suggesting haploinsufficiency leading to disease expression. This child initially presented at the age of 3 months with PAH and was later diagnosed with HHT at 8 years of age. The endoglin mutation was also detected in her asymptomatic father [62].
The finding of PAH in patients who have HHT and identifiable abnormalities of the TGF-β signaling pathway suggests that a common molecular pathway precipitates pulmonary vascular disease. A direct interaction between the gene products of the BMPR2 gene and ALK1 or endoglin genes has not been elucidated, and these receptors do not seem to share activating ligands [42,62]. However, each receptor mediates intracellular signaling through the Smad family of co-activators, suggesting interaction among these molecular receptors at some level [63]. TGF-β signaling pathway defects are likely to be diverse in type, interaction, and phenotypic expression, and require further study.
Additional mediators and mechanisms involved in pathogenesis
Many other mediators and mechanisms have been proposed to contribute to vasoconstriction and thrombosis and to pulmonary arterial obstruction caused by vascular proliferation and remodeling. Many of these topics are the focus of important ongoing studies but cannot be discussed in depth in this article. However, evaluation of these pathways at the molecular level may lead to important advances in earlier diagnosis, therapy, and outcomes [64].
Exuberant pulmonary vasoconstriction and inappropriate cell proliferation are strongly linked to endothelial cell dysfunction [65]. Endothelial cell dysfunction impairs local production of protective vasoactive compounds, such as prostacyclin and nitric oxide. Endothelial cell release of prostacyclin (prostaglandin-I2), an endogenous vasodilator (through cyclic adenosine monophosphate [cAMP]-dependent pathways) and an inhibitor of platelet aggregation and suppressor of vascular smooth muscle cell proliferation, is diminished in patients who have PAH [66]. The pulmonary endothelial cells of these patients express far less prostacyclin synthase, leading to an imbalance in the local prostacyclin-to-thromboxane A2 ratio. Thromboxane A2 is a vasoconstrictor and a potent stimulant of platelet aggregation [67], which is particularly relevant considering the belief that platelet dysfunction and the development of thrombotic lesions are important processes in PAH, and the potential role of the serotonin pathway [65,68].
Reduced nitric oxide synthase expression in pulmonary endothelial cells results in diminished local production of nitric oxide, a potent pulmonary vasodilator and an inhibitor of pulmonary vascular smooth muscle proliferation [69,70]. Nitric oxide and atrial natriuretic peptide modulate pulmonary vascular resistance, and possibly remodeling, at least partly through manipulating intracellular cyclic guanosine monophosphate (cGMP)–mediated pulmonary vasodilatation [66,71]. Investigators continue to examine other mediators that affect the cGMP pathway, including vasoactive intestinal peptide. Vasoactive intestinal peptide is a neuropeptide that has potent systemic and pulmonary vasodilator effects and contributes to the inhibition of vascular smooth muscle cell proliferation and platelet aggregation. Vasoactive intestinal peptide, the effects of which are mediated through cGMP- and cAMP-dependent pathways, is deficient in the serum and lung tissue of patients who have IPAH compared with controls. Investigations to evaluate the response to vasoactive intestinal peptide as a therapeutic agent are underway [72].
Under pathologic conditions, endothelial cells increase production of compounds with vasoconstrictive, proliferative, and inflammatory properties, such as the peptides of the endothelin system [66]. Convincing evidence also exists for the role of endothelial cell–derived endothelin-1 in the pathogenesis of PAH, including the finding of increased endothelin-1 plasma levels and expression in patients who have pulmonary hyper-tension [69,73,74]. Endothelin-1 effects are mediated through two distinct endothelin receptor isoforms: endothelin receptors type A (ETA) and B (ETB). Activation of these receptors by endothelins results in variable, often competing, signals, including vasodilatory versus vasoconstrictive, proliferative versus antiproliferative, and inflammatory versus anti-inflammatory [65,75]. Investigations continue to examine the role of ET receptor blockade in the treatment of PAH.
Pathways that modulate vascular smooth muscle tone and proliferation often do so through PASMC membrane potential (Em) regulation [76]. Models of hypoxia show the critical role of various types of potassium channels, which are the primary regulators of Em. Hypoxic inhibition of potassium channels, and depressed potassium channel function for other reasons, ultimately trigger PASMC contraction and proliferation [77]. Pathways counteractive to hypoxic responses include cGMP signaling, which activates large-conductance calcium-activated potassium channels, causing vasodilatation [65]. In addition, potassium channel activity is important in activating apoptosis, and therefore defective channel function may facilitate cell proliferation [78]. Several studies have shown diminished potassium channel expression and function in patients who have PAH, implicating loss of function in the pathogenesis of this disease [65,77,79,80]. Most anorectic drugs, including dexfenfluramine, directly inhibit potassium channels [81,82].
The TGF-β superfamily, of which BMPs are a member, clearly has a critical role in a diverse array of cellular processes. Additional growth factors have been implicated in the pathogenesis of PAH, including vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF). The exact function of VEGF in the lung is unknown, but it is expressed at high levels and required for both lung development and structural maintenance, including endothelial cell survival. Studies of plexiform lesions in PAH showed elevated levels of VEGF expression, but current understanding of the complex interactions of this compound has not clarified whether its actions promote or inhibit apoptosis and remodeling in a beneficial manner in PAH [65,66]. A potent mitogen and pulmonary vascular smooth muscle cell chemoattractant, PDGF is postulated to contribute to pulmonary vasculature remodeling in various forms of pulmonary hypertension. It is also a stimulant for other mitogens, including endothelin-1 [83]. Elevated levels of PDGF have been seen in the lungs of individuals who have PAH, implicating this compound in disease pathogenesis [84,85]. Additional growth factors warrant continued examination as modulators of cellular proliferation in PAH, such as angiopietin-1, basic fibroblast growth factor, epidermal growth factor, and insulin-like growth factor-1, but are beyond the scope of this article [65].
The role of inflammation and autoimmunity in the pathogenesis of PAH is also a subject of ongoing investigation. It is well known that PAH develops more often in diseases known to be associated with immune disturbance, such as scleroderma, systemic lupus erythematosus, autoimmune thyroiditis, and HIV infection [86]. Histologic evaluation of plexiform lesions in various types of PAH shows perivascular inflammatory cell infiltrates, typically lymphocytic in nature [87,88]. Furthermore, elevated levels of autoantibodies such an antinuclear antibodies, and abnormally high levels of circulating proinflammatory cytokines such as IL-1 and IL-6, have been detected in some patients who have PAH [89]. Recently, Tamby and colleagues [90] identified the presence of antiendothelial cell antibodies in patients who had PAH, implicating a loss of tolerance with subsequent pulmonary vasculature attack. Meanwhile, other investigators have suggested that immune dysregulation related to dysfunctional regulatory T-cell activity underlies all of these inflammatory processes and facilitates the expression of PAH, but continued work is needed to elucidate these potential mechanisms [91].
Genetic implications for clinical evaluation and therapy
Although the potential is growing, scant information is available for evaluating the role of genotype in predicting therapeutic response in patients who have PAH. A small proportion of these have good long-term response to calcium channel blockers, but models for predicting who will respond on an acute and chronic basis are lacking [92,93]. Elliott and colleagues [94] retrospectively compared the results of vasoreactivity testing in patients who had PAH with the presence of known BMPR2 gene changes that code for different amino acid products (nonsynonymous BMPR2 sequence variations). They found that patients who had FPAH and IPAH with nonsynonymous BMPR2 sequence variations were less likely to show vasoreactivity using standard testing. Their report highlights the potential to use information about known genetic variations in PAH to guide decisions about prognosis and treatment for individuals.
Screening and counseling
Siblings or children of patients who have FPAH, or of obligate heterozygotes for a BMPR2 mutation, have an overall risk of 50% for inheriting the abnormal gene. Because the gene penetrance is approximately 20%, this yields an estimated risk of 10% for expressing disease [95]. Therefore, most relatives will be asymptomatic.
Current recommendations for asymptomatic family members of individuals who have FPAH advise echocardiographic screening at 3- to 5-year intervals [95]. Investigators in Germany proposed a screening process that uses estimation of systolic pulmonary artery pressure at rest and during exercise using echocardiography, based on studies of families of patients who have IPAH and FPAH. Using a systematic clinical assessment of pulmonary artery pressure at rest and during exercise in members of families with PAH, Grunig and colleagues [96] attempted to detect individuals who had abnormal pulmonary vascular response to supine bicycle exercise, and subsequently linked this response to inheritance of an abnormal genetic locus ultimately discovered to include the gene for BMPR2. In 2005, this group also reported an abnormal pulmonary artery pressure response to hypoxia during echocardiographic evaluation in individuals deemed at risk for developing PAH based on a known BMPR2 mutation or a possible risk haplotype [97]. Although these efforts should be commended considering the profound need for earlier diagnosis of PAH, the use of stress echocardiography as a screening tool for PAH has not been widely accepted, and identifying a risk haplotype is challenging. Because both require more extensive evaluation and independent confirmation, they are not yet recommended for general use.
Genetic testing for mutations in the BMPR2 gene are now available clinically, and therefore patients who have IPAH and FPAH and their families should be instructed about the availability of genetic testing and the potential risk for family members to develop PAH.
Genetic testing should only be provided after professional genetic counseling [15,95]. A negative test result can be helpful to members of a family with a known BMPR2 mutation. If the test is negative, an individual FPAH family member's risk for disease declines from 1 in 10 to that of the population, which is approximately 1 in 1 million (100,000-fold risk reduction). Conversely, if an asymptomatic individual at risk is found to carry the known familial BMPR2 mutation, risk increases only modestly to 1 in 5 (twofold risk increase) for expressing clinical disease throughout their lifetime.
Given the vast number of potential mutations in the large BMPR2 gene, screening for a mutation is most efficient in an affected patient who has IPAH or FPAH, so that the specific mutation in their family can be identified if it is present. Clinical genetic testing of relatives of a patient who has PAH has no rationale unless a mutation is identified in the patient sample. The detection of a BMPR2 mutation in a patient who has IPAH is often surprising and alarming, specifically because the familial basis was concealed by absence of knowledge about other cases in the family, and therefore it converts the concept of disease from one of a sporadic finding to that of a potentially familial disease. These family members are thus at increased risk and should be offered counseling about their risk and about testing for the known mutation.
Summary
Great strides in the evaluation and management of PAH have been made in relation to the genetics and pathogenic mechanisms of this devastating disease. The discovery that mutations in the BMPR2 gene underlie most cases of FPAH and an important subset of IPAH cases has energized the field and prompted significant progress in understanding the molecular basis of PAH. Efforts continue to show the full genetic basis of FPAH and IPAH. Work is still needed to elucidate the causes of the variable penetrance of FPAH, including the identity and role of various modifiers of disease expression. These modifiers may also be the basis of genetic anticipation in FPAH. In addition, studies of genotype–phenotype interactions should include an exploration of the role of gender, which is the only known risk modifier.
Investigations should include evaluation of potential interactions between the SERT gene and members of the TGF-β signaling pathway, but also novel genes and proteins yet to be linked to PAH. The molecular mechanisms that converge to develop a milieu of cellular proliferation and vascular remodeling require aggressive investigation. Ultimately, a greater understanding of the genetic and molecular mechanisms of PAH should lead to earlier diagnosis, advanced pharmacogenetics, and, perhaps someday, disease prevention.
Acknowledgments
The authors thank the many patients and families who graciously contributed to this work, and Ms. Lisa Wheeler, whose service is invaluable as coordinator of the Vanderbilt Familial Primary Pulmonary Hypertension study.
This work was supported by NIH NHLBI PO1 HL72058 and The Vanderbilt GCRC grant M01 RR 00095 NCRR/NIH.
References
- [1].Dresdale DT, Schultz M, Michtom RJ. Primary pulmonary hypertension. I. Clinical and hemodynamic study. Am J Med. 1951;11(6):686–705. doi: 10.1016/0002-9343(51)90020-4. [DOI] [PubMed] [Google Scholar]
- [2].Dresdale DT, Michtom RJ, Schultz M. Recent studies in primary pulmonary hypertension, including pharmacodynamic observations on pulmonary vascular resistance. Bull N Y Acad Med. 1954;30(3):195–207. [PMC free article] [PubMed] [Google Scholar]
- [3].Loyd JE, Primm RK, Newman JH. Familial primary pulmonary hypertension: clinical patterns. Am Rev Respir Dis. 1984;129(1):194–7. doi: 10.1164/arrd.1984.129.1.194. [DOI] [PubMed] [Google Scholar]
- [4].Thomas AQ, Gaddipati R, Newman JH, et al. Genetics of primary pulmonary hypertension. Clin Chest Med. 2001;22(3):477–91. ix. doi: 10.1016/s0272-5231(05)70285-9. [DOI] [PubMed] [Google Scholar]
- [5].Rich S, Dantzker DR, Ayres SM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107(2):216–23. doi: 10.7326/0003-4819-107-2-216. [DOI] [PubMed] [Google Scholar]
- [6].Nichols WC, Koller DL, Slovis B, et al. Localization of the gene for familial primary pulmonary hypertension to chromosome 2q31–32. Nat Genet. 1997;15(3):277–80. doi: 10.1038/ng0397-277. [DOI] [PubMed] [Google Scholar]
- [7].Morse JH, Barst RJ. Detection of familial primary pulmonary hypertension by genetic testing. N Engl J Med. 1997;337(3):202–3. doi: 10.1056/NEJM199707173370315. [DOI] [PubMed] [Google Scholar]
- [8].Lane KB, Machado RD, Pauciulo MW, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nat Genet. 2000;26(1):81–4. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- [9].Deng Z, Morse JH, Slager SL, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67(3):737–44. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gaine SP, Rubin LJ. Primary pulmonary hypertension. Lancet. 1998;352(9129):719–25. doi: 10.1016/S0140-6736(98)02111-4. [DOI] [PubMed] [Google Scholar]
- [11].Runo JR, Loyd JE. Primary pulmonary hypertension. Lancet. 2003;361(9368):1533–44. doi: 10.1016/S0140-6736(03)13167-4. [DOI] [PubMed] [Google Scholar]
- [12].D'Alonzo GE, Barst RJ, Ayres AM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115(5):343–9. doi: 10.7326/0003-4819-115-5-343. [DOI] [PubMed] [Google Scholar]
- [13].Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. 2006;173(9):1023–30. doi: 10.1164/rccm.200510-1668OC. [DOI] [PubMed] [Google Scholar]
- [14].Thomson JR, Machado RD, Pauciulo MW, et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J Med Genet. 2000;37(10):741–5. doi: 10.1136/jmg.37.10.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Newman JH, Trembath RC, Morse JA, et al. Genetic basis of pulmonary arterial hypertension: current understanding and future directions. J Am Coll Cardiol. 2004;43(12 Suppl S):33S–9S. doi: 10.1016/j.jacc.2004.02.028. [DOI] [PubMed] [Google Scholar]
- [16].Humbert M, Nunes H, Sitbon O, et al. Risk factors for pulmonary arterial hypertension. Clin Chest Med. 2001;22(3):459–75. doi: 10.1016/s0272-5231(05)70284-7. [DOI] [PubMed] [Google Scholar]
- [17].Kuhn KP, Byrne DW, Arbogast PG, et al. Outcome in 91 consecutive patients with pulmonary arterial hypertension receiving epoprostenol. Am J Respir Crit Care Med. 2003;167(4):580–6. doi: 10.1164/rccm.200204-333OC. [DOI] [PubMed] [Google Scholar]
- [18].Newman JH, Wheeler L, Lane KB, et al. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med. 2001;345(5):319–24. doi: 10.1056/NEJM200108023450502. [DOI] [PubMed] [Google Scholar]
- [19].Machado RD, James V, Southwood M, et al. Investigation of second genetic hits at the BMPR2 locus as a modulator of disease progression in familial pulmonary arterial hypertension. Circulation. 2005;111(5):607–13. doi: 10.1161/01.CIR.0000154543.07679.08. [DOI] [PubMed] [Google Scholar]
- [20].Loyd JE, Butler MG, Foroud TM, et al. Genetic anticipation and abnormal gender ratio at birth in familial primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;152(1):93–7. doi: 10.1164/ajrccm.152.1.7599869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Warren ST, Ashley CT., Jr Triplet repeat expansion mutations: the example of fragile X syndrome. Annu Rev Neurosci. 1995;18:77–99. doi: 10.1146/annurev.ne.18.030195.000453. [DOI] [PubMed] [Google Scholar]
- [22].Ashley CT, Jr, Warren ST. Trinucleotide repeat expansion and human disease. Annu Rev Genet. 1995;29:703–28. doi: 10.1146/annurev.ge.29.120195.003415. [DOI] [PubMed] [Google Scholar]
- [23].Newman JH. Pulmonary hypertension. Am J Respir Crit Care Med. 2005;172(9):1072–7. doi: 10.1164/rccm.200505-684OE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Morse JH, Jones AC, Barst RJ, et al. Mapping of familial primary pulmonary hypertension locus (PPH1) to chromosome 2q31–q32. Circulation. 1997;95(12):2603–6. doi: 10.1161/01.cir.95.12.2603. [DOI] [PubMed] [Google Scholar]
- [25].Machado RD, Pauciulo MW, Thomson JR, et al. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet. 2001;68(1):92–102. doi: 10.1086/316947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Koehler R, Grunig E, Pauciulo MW, et al. Low frequency of BMPR2 mutations in a German cohort of patients with sporadic idiopathic pulmonary arterial hypertension. J Med Genet. 2004;41(12):e127. doi: 10.1136/jmg.2004.023101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Morisaki H, Nakanishi N, Kyotani S, et al. BMPR2 mutations found in Japanese patients with familial and sporadic primary pulmonary hypertension. Hum Mutat. 2004;23(6):632. doi: 10.1002/humu.9251. [DOI] [PubMed] [Google Scholar]
- [28].Elliott CG. Genetics of pulmonary arterial hypertension: current and future implications. Semin Respir Crit Care Med. 2005;26(4):365–71. doi: 10.1055/s-2005-916150. [DOI] [PubMed] [Google Scholar]
- [29].Cogan JD, Pauciulo MW, Batchman AP, et al. High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;174:590–8. doi: 10.1164/rccm.200602-165OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Aldred MA, Vijayakrishnan J, James V, et al. BMPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension. Hum Mutat. 2006;27(2):212–3. doi: 10.1002/humu.9398. [DOI] [PubMed] [Google Scholar]
- [31].Machado RD, Aldred MA, James V, et al. Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum Mutat. 2006;27(2):121–32. doi: 10.1002/humu.20285. [DOI] [PubMed] [Google Scholar]
- [32].Yang X, Long L, Southwood M, et al. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ Res. 2005;96(10):1053–63. doi: 10.1161/01.RES.0000166926.54293.68. [DOI] [PubMed] [Google Scholar]
- [33].Loyd JE, Atkinson JB, Pietra GG, et al. Heterogeneity of pathologic lesions in familial primary pulmonary hypertension. Am Rev Respir Dis. 1988;138(4):952–7. doi: 10.1164/ajrccm/138.4.952. [DOI] [PubMed] [Google Scholar]
- [34].Humbert M, Morrell NW, Archer SL, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- [35].Teichert-Kuliszewska K, Kutryk MJ, Kuliszewski MA, et al. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ Res. 2006;98(2):209–17. doi: 10.1161/01.RES.0000200180.01710.e6. [DOI] [PubMed] [Google Scholar]
- [36].Lee SD, Shroyer KR, Markham NE, et al. Monoclonal endothelial cell proliferation is present in primary but not secondary pulmonary hypertension. J Clin Invest. 1998;101(5):927–34. doi: 10.1172/JCI1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Stewart DJ. Bone morphogenetic protein receptor-2 and pulmonary arterial hypertension: unraveling a riddle inside an enigma? Circ Res. 2005;96(10):1033–5. doi: 10.1161/01.RES.0000168922.54339.47. [DOI] [PubMed] [Google Scholar]
- [38].Atkinson C, Stewart S, Upton PD, et al. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105(14):1672–8. doi: 10.1161/01.cir.0000012754.72951.3d. [DOI] [PubMed] [Google Scholar]
- [39].Yuan JX, Rubin LJ. Pathogenesis of pulmonary arterial hypertension: the need for multiple hits. Circulation. 2005;111(5):534–8. doi: 10.1161/01.CIR.0000156326.48823.55. [DOI] [PubMed] [Google Scholar]
- [40].De Caestecker M, Meyrick B. Bone morphogenetic proteins, genetics and the pathophysiology of primary pulmonary hypertension. Respir Res. 2001;2(4):193–7. doi: 10.1186/rr57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signaling. Nature. 2003;425(6958):577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- [42].Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113(6):685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- [43].Runo JR, Vnencak-Jones CL, Prince M, et al. Pulmonary veno-occlusive disease caused by an inherited mutation in bone morphogenetic protein receptor II. Am J Respir Crit Care Med. 2003;167(6):889–94. doi: 10.1164/rccm.200208-861OC. [DOI] [PubMed] [Google Scholar]
- [44].Simonneau G, Galie N, Rubin LJ, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):5S–12S. doi: 10.1016/j.jacc.2004.02.037. [DOI] [PubMed] [Google Scholar]
- [45].Abenhaim L, Moride Y, Brenot F, et al. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N Engl J Med. 1996;335(9):609–16. doi: 10.1056/NEJM199608293350901. [DOI] [PubMed] [Google Scholar]
- [46].Sztrymf B, Yaici A, Jais X, et al. Idiopathic pulmonary hypertension: what did we learn from genes? Sarcoidosis Vasc Diffuse Lung Dis. 2005;22(Suppl 1):S91–S100. [PubMed] [Google Scholar]
- [47].Humbert M, Deng Z, Simonneau G, et al. BMPR2 germline mutations in pulmonary hypertension associated with fenfluramine derivatives. Eur Respir J. 2002;20(3):518–23. doi: 10.1183/09031936.02.01762002. [DOI] [PubMed] [Google Scholar]
- [48].Roberts KE, McElroy JJ, Wong WP, et al. BMPR2 mutations in pulmonary arterial hypertension with congenital heart disease. Eur Respir J. 2004;24(3):371–4. doi: 10.1183/09031936.04.00018604. [DOI] [PubMed] [Google Scholar]
- [49].Lee SL, Wang WW, Moore BJ, et al. Dual effect of serotonin on growth of bovine pulmonary artery smooth muscle cells in culture. Circ Res. 1991;68(5):1362–8. doi: 10.1161/01.res.68.5.1362. [DOI] [PubMed] [Google Scholar]
- [50].Herve P, Launay JM, Scrobohaci ML, et al. Increased plasma serotonin in primary pulmonary hypertension. Am J Med. 1995;99(3):249–54. doi: 10.1016/s0002-9343(99)80156-9. [DOI] [PubMed] [Google Scholar]
- [51].Eddahibi S, Hanoun N, Lanfumey L, et al. Attenuated hypoxic pulmonary hypertension in mice lacking the 5-hydroxytryptamine transporter gene. J Clin Invest. 2000;105(11):1555–62. doi: 10.1172/JCI8678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Eddahibi S, Humbert M, Fadel E, et al. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J Clin Invest. 2001;108(8):1141–50. doi: 10.1172/JCI12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Liu Y, Suzuki YJ, Day RM, et al. Rho kinase-induced nuclear translocation of ERK1/ERK2 in smooth muscle cell mitogenesis caused by serotonin. Circ Res. 2004;95(6):579–86. doi: 10.1161/01.RES.0000141428.53262.a4. [DOI] [PubMed] [Google Scholar]
- [54].Lee SL, Wang WW, Finlay GA, et al. Serotonin stimulates mitogen-activated protein kinase activity through the formation of superoxide anion. Am J Physiol. 1999;277(2 Pt 1):L282–91. doi: 10.1152/ajplung.1999.277.2.L282. [DOI] [PubMed] [Google Scholar]
- [55].Machado RD, Koehler R, Glissmeyer E, et al. Genetic association of the serotonin transporter in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;173(7):793–7. doi: 10.1164/rccm.200509-1365OC. [DOI] [PubMed] [Google Scholar]
- [56].Willers ED, Newman John H, Loyd James E, et al. Serotonin transporter polymorphisms in familial and idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;173(7):798–802. doi: 10.1164/rccm.200509-1361OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8(4):345–51. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- [58].Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13(2):189–95. doi: 10.1038/ng0696-189. [DOI] [PubMed] [Google Scholar]
- [59].Trembath RC, Thomson JR, Machado RD, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2001;345(5):325–34. doi: 10.1056/NEJM200108023450503. [DOI] [PubMed] [Google Scholar]
- [60].Harrison RE, Flanagan JA, Sankelo M, et al. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J Med Genet. 2003;40(12):865–71. doi: 10.1136/jmg.40.12.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Chaouat A, Coulet F, Favre C, et al. Endoglin germline mutation in a patient with hereditary haemorrhagic telangiectasia and dexfenfluramine associated pulmonary arterial hypertension. Thorax. 2004;59(5):446–8. doi: 10.1136/thx.2003.11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Harrison RE, Berger R, Haworth SG, et al. Transforming growth factor-beta receptor mutations and pulmonary arterial hypertension in childhood. Circulation. 2005;111(4):435–41. doi: 10.1161/01.CIR.0000153798.78540.87. [DOI] [PubMed] [Google Scholar]
- [63].Fernandez LA, Sanz-Rodriguez F, Blanco FJ, et al. Hereditary hemorrhagic telangiectasia, a vascular dysplasia affecting the TGF-beta signaling pathway. Clin Med Res. 2006;4(1):66–78. doi: 10.3121/cmr.4.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation. 2006;114(13):1417–31. doi: 10.1161/CIRCULATIONAHA.104.503540. [DOI] [PubMed] [Google Scholar]
- [65].Perros F, Dorfmuller P, Humbert M. Current insights on the pathogenesis of pulmonary arterial hypertension. Semin Respir Crit Care Med. 2005;26(4):355–64. doi: 10.1055/s-2005-916149. [DOI] [PubMed] [Google Scholar]
- [66].Said SI. Mediators and modulators of pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2006;291:547–58. doi: 10.1152/ajplung.00546.2005. [DOI] [PubMed] [Google Scholar]
- [67].Christman BW, McPherson CD, Newman JH, et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327(2):70–5. doi: 10.1056/NEJM199207093270202. [DOI] [PubMed] [Google Scholar]
- [68].Herve P, Humbert M, Sitbon O, et al. Pathobiology of pulmonary hypertension. The role of platelets and thrombosis. Clin Chest Med. 2001;22(3):451–8. doi: 10.1016/s0272-5231(05)70283-5. [DOI] [PubMed] [Google Scholar]
- [69].Giaid A. Nitric oxide and endothelin-1 in pulmonary hypertension. Chest. 1998;114(3 Suppl):208S–12S. doi: 10.1378/chest.114.3_supplement.208s. [DOI] [PubMed] [Google Scholar]
- [70].Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995;333(4):214–21. doi: 10.1056/NEJM199507273330403. [DOI] [PubMed] [Google Scholar]
- [71].Steiner MK, Preston IR, Klinger JR, et al. Pulmonary hypertension: inhaled nitric oxide, sildenafil and natriuretic peptides. Curr Opin Pharmacol. 2005;5(3):245–50. doi: 10.1016/j.coph.2004.12.008. [DOI] [PubMed] [Google Scholar]
- [72].Petkov V, Mosgoeller W, Ziesche R, et al. Vasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertension. J Clin Invest. 2003;111(9):1339–46. doi: 10.1172/JCI17500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Stewart DJ, Levy RD, Cernacek P, et al. Increased plasma endothelin-1 in pulmonary hypertension: marker or mediator of disease? Ann Intern Med. 1991;114(6):464–9. doi: 10.7326/0003-4819-114-6-464. [DOI] [PubMed] [Google Scholar]
- [74].Giaid A, Yanagisawa M, Langleben D, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328(24):1732–9. doi: 10.1056/NEJM199306173282402. [DOI] [PubMed] [Google Scholar]
- [75].Barst RJ, Langleben D, Badesch D, et al. Treatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentan. J Am Coll Cardiol. 2006;47(10):2049–56. doi: 10.1016/j.jacc.2006.01.057. [DOI] [PubMed] [Google Scholar]
- [76].Martin KB, Klinger JR, Rounds SIS. Pulmonary arterial hypertension: new insights and new hope. Respirology. 2006;11(1):6–17. doi: 10.1111/j.1440-1843.2006.00778.x. [DOI] [PubMed] [Google Scholar]
- [77].Mauban JR, Remillard CV, Yuan JX. Hypoxic pulmonary vasoconstriction: role of ion channels. J Appl Physiol. 2005;98(1):415–20. doi: 10.1152/japplphysiol.00732.2004. [DOI] [PubMed] [Google Scholar]
- [78].Burg ED, Remillard CV, Yuan JX. K+ channels in apoptosis. J Membr Biol. 2006;209(1):3–20. doi: 10.1007/s00232-005-0838-4. [DOI] [PubMed] [Google Scholar]
- [79].Geraci MW, Moore M, Gesell T, et al. Gene expression patterns in the lungs of patients with primary pulmonary hypertension: a gene microarray analysis. Circ Res. 2001;88(6):555–62. doi: 10.1161/01.res.88.6.555. [DOI] [PubMed] [Google Scholar]
- [80].Yuan XJ, Wang J, Juhaszova M, et al. Attenuated K+ channel gene transcription in primary pulmonary hypertension. Lancet. 1998;351(9104):726–7. doi: 10.1016/S0140-6736(05)78495-6. [DOI] [PubMed] [Google Scholar]
- [81].Michelakis E. Anorectic drugs and vascular disease: the role of voltage-gated K+ channels. Vascul Pharmacol. 2002;38(1):51–9. doi: 10.1016/s1537-1891(02)00126-x. [DOI] [PubMed] [Google Scholar]
- [82].Michelakis ED, Weir EK. Anorectic drugs and pulmonary hypertension from the bedside to the bench. Am J Med Sci. 2001;321(4):292–9. doi: 10.1097/00000441-200104000-00009. [DOI] [PubMed] [Google Scholar]
- [83].Yu Y, Sweeney M, Zhang S, et al. PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am J Physiol Cell Physiol. 2003;284(2):C316–30. doi: 10.1152/ajpcell.00125.2002. [DOI] [PubMed] [Google Scholar]
- [84].Eddahibi S, Humbert M, Sediame S, et al. Imbalance between platelet vascular endothelial growth factor and platelet-derived growth factor in pulmonary hypertension. Effect of prostacyclin therapy. Am J Respir Crit Care Med. 2000;162(4 Pt 1):1493–9. doi: 10.1164/ajrccm.162.4.2003124. [DOI] [PubMed] [Google Scholar]
- [85].Humbert M, Monti G, Fartoukh M, et al. Platelet-derived growth factor expression in primary pulmonary hypertension: comparison of HIV seropositive and HIV seronegative patients. Eur Respir J. 1998;11(3):554–9. [PubMed] [Google Scholar]
- [86].Barst RJ, McGoon M, Torbicki A, et al. Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):40S–7S. doi: 10.1016/j.jacc.2004.02.032. [DOI] [PubMed] [Google Scholar]
- [87].Tuder RM, Groves B, Badesch DB, et al. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hyper-tension. Am J Pathol. 1994;144(2):275–85. [PMC free article] [PubMed] [Google Scholar]
- [88].Achcar RO, Yung GL, Saffer H, et al. Morphologic changes in explanted lungs after prostacyclin therapy for pulmonary hypertension. Eur J Med Res. 2006;11(5):203–7. [PubMed] [Google Scholar]
- [89].Dorfmuller P, Perros F, Balabanian K, et al. Inflammation in pulmonary arterial hypertension. Eur Respir J. 2003;22(2):358–63. doi: 10.1183/09031936.03.00038903. [DOI] [PubMed] [Google Scholar]
- [90].Tamby MC, Chanseaud Y, Humbert M, et al. Anti-endothelial cell antibodies in idiopathic and systemic sclerosis associated pulmonary arterial hypertension. Thorax. 2005;60(9):765–72. doi: 10.1136/thx.2004.029082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Nicolls MR, Taraseviciene-Stewart L, Rai PR, et al. Autoimmunity and pulmonary hypertension: a perspective. Eur Respir J. 2005;26(6):1110–8. doi: 10.1183/09031936.05.00045705. [DOI] [PubMed] [Google Scholar]
- [92].Montani D, Marcelin AG, Sitbon O, et al. Human herpes virus 8 in HIV and non-HIV infected patients with pulmonary arterial hypertension in France. AIDS. 2005;19(11):1239–40. doi: 10.1097/01.aids.0000176230.94226.06. [DOI] [PubMed] [Google Scholar]
- [93].Archer SL, Michelakis ED. An evidence-based approach to the management of pulmonary arterial hypertension. Curr Opin Cardiol. 2006;21(4):385–92. doi: 10.1097/01.hco.0000231410.07426.9b. [DOI] [PubMed] [Google Scholar]
- [94].Elliott CG, Glissmeyer EW, Havlena GT, et al. Relationship of BMPR2 mutations to vasoreactivity in pulmonary arterial hypertension. Circulation. 2006;113(21):2509–15. doi: 10.1161/CIRCULATIONAHA.105.601930. [DOI] [PubMed] [Google Scholar]
- [95].McGoon M, Gutterman D, Steen V, et al. Screening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126(1 Suppl):14S–34S. doi: 10.1378/chest.126.1_suppl.14S. [DOI] [PubMed] [Google Scholar]
- [96].Grunig E, Janssen B, Mereles D, et al. Abnormal pulmonary artery pressure response in asymptomatic carriers of primary pulmonary hypertension gene. Circulation. 2000;102(10):1145–50. doi: 10.1161/01.cir.102.10.1145. [DOI] [PubMed] [Google Scholar]
- [97].Grunig E, Dehnert C, Mereles D, et al. Enhanced hypoxic pulmonary vasoconstriction in families of adults or children with idiopathic pulmonary arterial hypertension. Chest. 2005;128(6 Suppl):630S–3S. doi: 10.1378/chest.128.6_suppl.630S-a. [DOI] [PubMed] [Google Scholar]