

Abstract
Intense light exposure causes photoreceptor apoptosis in dark-adapted adult albino zebrafish (Danio rerio). Subsequently, Müller glia increase expression of the Achaete-scute complex-like 1a (Ascl1a) and Signal transducer and activator of transcription 3 (Stat3) transcription factors and re-enter the cell cycle to yield undifferentiated neuronal progenitors that continue to proliferate, migrate to the outer nuclear layer, and differentiate into photoreceptors. A proteomic analysis of light-damaged retinal homogenates, which induced Müller glia proliferation when injected into an undamaged eye, revealed increased expression of tumor necrosis factor α (TNFα) signaling proteins relative to undamaged retinal homogenates. TNFα expression initially increased in apoptotic photoreceptors and later in Müller glia. Morpholino-mediated knockdown of TNFα expression before light damage diminished the expression of both Ascl1a and Stat3 in Müller glia and significantly reduced the number of proliferating Müller glia without affecting photoreceptor cell death. Knockdown of TNFα expression in the Müller glia resulted in fewer proliferating Müller glia, suggesting that Müller glial-derived TNFα recruited additional Müller glia to re-enter the cell cycle. While TNFα is required for increased Ascl1a and Stat3 expression, Ascl1a and Stat3 are both necessary for TNFα expression in Müller glia. Apoptotic inner retinal neurons, resulting from intravitreal injection of ouabain, also exhibited increased TNFα expression that was required for Müller glia proliferation. Thus, TNFα is the first molecule identified that is produced by dying retinal neurons and is necessary to induce Müller glia to proliferate in the zebrafish retinal regeneration response.
Introduction
The teleost retina is a model for studying cellular and molecular mechanisms underlying retinal regeneration (Brockerhoff and Fadool, 2011; Fleisch et al., 2011; Nelson and Hyde, 2012). Following neuronal death, Müller glia undergo cell division to yield neuronal progenitors that continue to proliferate, migrate, and differentiate into the lost retinal cell types (Yurco and Cameron, 2005; Fausett and Goldman, 2006; Bernardos et al., 2007; Fimbel et al., 2007; Kassen et al., 2007; Thummel et al., 2008a). While avian and mammalian Müller glia exhibit limited proliferation, they cannot regenerate significant numbers of neurons and restore vision (Rowan and Cepko, 2004; Bringmann et al., 2006; Fischer et al., 2009; Karl and Reh, 2010). Therefore, identifying the signaling events required for zebrafish retinal regeneration could yield approaches to induce mammalian retinal regeneration.
Several proteins have been identified that are necessary to regenerate the damaged zebrafish retina (Fausett et al., 2008; Thummel et al., 2008a; 2010; Calinescu et al., 2009; Qin et al., 2009; Ramachandran et al., 2010; Nelson et al., 2012; Powell et al., 2012; Wan et al., 2012). Heparin-binding epidermal-like growth factor (HB-EGF) is necessary and sufficient to induce Müller glia proliferation (Wan et al., 2012). However, HB-EGF is not expressed in damaged neurons, suggesting that unidentified molecules or processes induce Müller glia proliferation in the damaged zebrafish retina.
Tumor necrosis factor α (TNFα) is a secreted pro-inflammatory cytokine that signals several cellular processes, including apoptosis, cell survival, and proliferation (Tartaglia et al., 1993; Cheng, 1994; Chan and Leonard, 2000; Gupta, 2001). TNFα signaling activates pro-mitotic Stat proteins (Guo et al., 1998; Wang et al., 2000; Do et al., 2010), including Stat3 in a variety of cells and tissues (Guo et al., 1998; MacEwan, 2002; Miscia et al., 2002; Romanatto et al., 2007; Mori et al., 2011). TNFα activation of Stat3 promotes ex vivo human neuronal progenitor cell proliferation and gliogenesis (Peng et al., 2008, 2011). TNFα also stimulates Tumor necrosis factor associated protein-1 (TRAP1), which stabilizes the retinoblastoma protein during mitosis (Chen et al., 1996), regulates pro-mitotic gene expression (Liu et al., 2010), and mediates Stat3 phosphorylation and activation in the mouse brain (Kubota et al., 2009).
To determine whether dying photoreceptors induced Müller glia proliferation, we analyzed light-damaged retinal homogenates that stimulated Müller glial cell division when injected into undamaged eyes. This homogenate contained several proteins with increased expression relative to undamaged retinal homogenates, including TRAP1. We therefore characterized TNFα expression in the light-damaged retina and demonstrated that TNFα is expressed in apoptotic photoreceptors, followed by the Müller glia. TNFα was found to be necessary for both Stat3 and Asc1la expression, and Müller glia proliferation. Similarly, apoptotic inner retinal neurons and persistent Müller glia in the ouabain-damaged retina exhibit increased TNFα expression, which is required for maximal Müller glia proliferation. Thus, TNFα is a general signal from apoptotic neurons to initiate Müller glia proliferation through the Ascl1a and Stat3 proteins in the damaged zebrafish retina.
Materials and Methods
Fish maintenance.
Wild-type AB, albino, albino Tg[gfap:EGFP]nt11 (Kassen et al., 2007), and Tg[rho:Eco.NfsB-EGFP]nt19 (Montgomery et al., 2010) lines of zebrafish (Danio rerio) were maintained in the Center for Zebrafish Research at the University of Notre Dame Freimann Life Science Center. The adult zebrafish used in these studies were 6–18 months old and 4–5 cm in length. Fish were maintained under a light and dark cycle of 14 h light (250 lux) and 10 h of dark at 28.5°C. All experimental protocols in this study were approved by the University of Notre Dame Animal Care and Use Committee and are in compliance with the Association for Research in Vision and Ophthalmology statement for the use of animals in vision research.
Retinal damage paradigms.
Constant intense-light treatment (Vihtelic and Hyde, 2000; Vihtelic et al., 2006) was used to kill rod and cone photoreceptors in adult albino zebrafish. The albino fish were dark-adapted for 14 d and then placed in clear polycarbonate tanks between four 54 W mercury fluorescent bulbs producing a light intensity of 20,000 lux. The constant light was maintained for up to 4 d, after which they were returned to standard light conditions.
Inner retinal neurons (ganglion cells, amacrine cells, and bipolar cells) were killed by intravitreal injection of ouabain at a final vitreal concentration of 2 μm (Fimbel et al., 2007). A double-edged sapphire microknife (World Precision Instruments) was used to make a small incision in the posterior cornea adjacent to the lens and the appropriate volume (0.2–0.5 μl) of a freshly prepared ouabain solution (MP Biomedicals) was injected using a blunt-end 33 gauge Hamilton syringe.
Preparation and intravitreal injection of retinal lysates.
Total retinal lysates were generated by isolating 40–60 retinas either from dark-adapted controls or after 16 h of constant intense light and suspending them in 1.5 ml centrifuge tubes kept frozen on dry ice containing 0.4 μl of 1× PBS, pH 7.4, containing a protease inhibitor (Roche Diagnostics) per retina and then briefly sonicated. The recipient adult zebrafish were anesthetized in 2-phenoxyethanol (diluted 1:1000 in system water). A double-edged sapphire microknife was used to make a small incision in the cornea and 0.5 μl of homogenized retinal lysate was injected using a 33 gauge Hamilton syringe into the intravitreal space of the eye. The fish were revived and returned to standard light conditions.
Intravitreal injection of recombinant HB-EGF.
Recombinant HB-EGF (R&D Systems) was diluted in sterile 1× PBS, 0.1% bovine serum albumin (BSA; vehicle) to a concentration of either 200 ng/μl or 100 ng/μl as described previously (Wan et al., 2012). A small incision in the posterior cornea adjacent to the lens was created using a double-edged sapphire blade (World Precision Instruments). A blunt-end 33 gauge Hamilton syringe was used to inject either the PBS/0.1% BSA vehicle solution (control) or 2 μl of 100 ng/μl or 1 μl of 200 ng/μl HB-EGF solution into the vitreous of the left eye of each fish for 3 consecutive days. At 2 or 3 d after the third injection, the fish were killed and the eyes were enucleated and processed for immunohistochemistry.
Proteomic analysis.
Total dorsal retinal proteins were isolated from either 100 undamaged dorsal retinas or from 100 16 h light-damaged dorsal retinas, suspended in 1.5 ml centrifuge tubes, and kept frozen on dry ice containing 1 μl of 1× PBS containing a protease inhibitor (Roche Diagnostics) and 1% Triton X-100 per retina and then briefly sonicated. The homogenates were separately labeled with either Cy3 or Cy5, combined, and then separated by 2D gel electrophoresis (Applied Biomics). Changes in protein expression levels were quantified by comparing the fluorescence intensities of control and 16 h light damaged spots. Spots that increased in intensity by at least twofold were picked from the gels, trypsin digested, and analyzed by MALDI-TOF mass spectrometry (Applied Biomics). Combined use of peptide mass fingerprinting of the mass spectrometry spectra and peptide fragmentation mapping-derived sequences were analyzed by GPS Explorer software equipped with the MASCOT search engine to identify proteins from primary sequence databases. As noted in the text, the full list of identified proteins is found in Table 1.
Table 1.
Top ranked protein names and accession numbers
| Spot number | Top ranked protein name (Danio rerio) | Accession number |
|---|---|---|
| 1 | PREDICTED: similar to AT motif binding factor 1 | gi|189521821 |
| 2 | Heat shock protein 4, like | gi|47550833 |
| 3 | Novel protein | gi|126632590 |
| 4 | Heat shock protein 9 0kDa beta, member 1 | gi|38016165 |
| 5 | PREDICTED: similar to LOC567732 protein | gi|189528839 |
| 6 | PREDICTED: similar to tripartite motif-containing 39 | gi|125821174 |
| 7 | Complement component 9 | gi|220941693 |
| 8 | Heat shock protein 5 | gi|39645428 |
| 9 | Novel protein similar to heat shock cognate 70 kDa | gi|169158081 |
| 10 | Dynein, axonemal, heavy polypeptide 9 | gi|41055024 |
| 11 | Transferrin-a | gi|220672954 |
| 12 | Transferrin-a | gi|220672954 |
| 13 | Sb:cb26 protein | gi|33417203 |
| 14 | PREDICTED: similar to transketolase | gi|125829873 |
| 15 | Hypothetical protein LOC767787 | gi|115529393 |
| 16 | Hpx protein | gi|112180551 |
| 17 | Hpx protein | gi|112180551 |
| 18 | Hpx protein | gi|112180551 |
| 19 | Hemopexin | gi|162287365 |
| 20 | Serpin peptidase inhibitor, clade A (alpha-1 antiproteinase, antitrypsin), member 7 | gi|54400446 |
| 21 | Pkm2a protein | gi|45501385 |
| 22 | Hpx protein | gi|112180551 |
| 23 | Serpina1 protein | gi|38541769 |
| 24 | Novel protein similar to serine (or cysteine) proteinase inhibitor, clade A (alpha-1 antiproteinase) | gi|55962788 |
| 25 | Zgc:123103 | gi|116284149 |
| 26 | 26–29 kDa proteinase protein | gi|49901179 |
| 27 | Kinesin family member 14 | gi|113678899 |
| 28 | S-antigen; retina and pineal gland (arrestin) | gi|76253800 |
| 29 | Glyceraldehyde-3-phosphate dehydrogenase, spermatogenic | gi|47085833 |
| 30 | Novel protein similar to vertebrate phosphoglycerate mutase family member 4 | gi|220678446 |
| 31 | Hypothetical protein LOC560055 | gi|154426250 |
| 32 | Novel pentaxin family domain containing protein | gi|126540682 |
| 33 | Apolipoprotein A-I | gi|18858281 |
| 34 | PREDICTED: hypothetical protein | gi|189529246 |
| 35 | Recoverin | gi|41152377 |
| 36 | Hypothetical protein LOC322327 | gi|194578933 |
| 37 | Hypoxia upregulated 1 | gi|47086637 |
| 38 | Heat shock protein HSP 90 beta | gi|109835356 |
| 39 | Platelet-derived growth factor receptor alpha | gi|148726416 |
| 40 | Novel protein similar to gelsolin, like 1 | gi|148725532 |
| 41 | Aconitase 2, mitochondrial | gi|38707983 |
| 42 | Keratin 18 [D. rerio] | gi|29335504 |
| 43 | TNF receptor-associated protein 1 | gi|213627589 |
| 44 | Radixin isoform 1 | gi|51972166 |
| 45 | Guanine monphosphate synthetase | gi|220672808 |
| 46 | Tyrosyl-tRNA synthetase, cytoplasmic | gi|109895056 |
| 47 | Hypothetical protein LOC767787 | gi|115529393 |
| 48 | RuvB-like 2 | gi|27819634 |
| 49 | Zgc:123103 | gi|157422889 |
| 50 | Brain creatine kinase B | gi|27545193 |
| 51 | PREDICTED: similar to transketolase | gi|125829873 |
| 52 | Ceruloplasmin | gi|94732872 |
Western blotting.
Dorsal retinas were isolated, homogenized in 1× PBS/1% Triton X-100 and protease inhibitor (Roche Diagnostics), and incubated on ice for 1 h. Lysates were briefly centrifuged to remove Triton X-100-insoluble debris and the equivalent of two dorsal retinas was combined with 2× Laemmli sample buffer and 10× reducing agent (Invitrogen). Proteins were processed, electrophoresed, and transferred to Hybond PDVF membranes (GE Healthcare) as described previously (Nelson et al., 2012). The membranes were blocked in PBS/5% nonfat dry milk/0.1% Tween 20 for 1 h at room temperature and then incubated with either rabbit anti-TNFα (1:2000; AnaSpec) or mouse anti-actin (1:10,000; EMD Millipore) overnight at 4°C. The membranes were washed, incubated with horseradish peroxidase-conjugated secondary antibodies (1:10,000; GE Healthcare), and detected using the ECL-Prime detection system (GE Healthcare) as described previously (Nelson et al., 2012).
Immunohistochemistry.
Zebrafish were killed at designated times during treatment by an overdose of 2-phenoxyethanol anesthesia. Eyes were enucleated, fixed in either 9:1 ethanolic formaldehyde (100% ethanol:37% formaldehyde) or 4% PFA (4% paraformaldehyde, 5% sucrose, 1× PBS), processed, and sectioned as described previously (Thummel et al., 2010; Nelson et al., 2012).
Slides were incubated at 50°C for 20–30 min and then rehydrated in 1× PBS at room temperature for 20 min. Sections that underwent antigen retrieval (labeling with either anti-PCNA or anti-Ascl1a on 4% PFA fixed tissues) were boiled in 10 mm sodium citrate, pH 6.0, with 0.1% Tween 20 for 15 min and then cooled at room temperature (over ∼40 min). All retinal sections were blocked and incubated with primary antibodies overnight at 25°C as previously described (Thummel et al., 2010; Nelson et al., 2012). Primary antibodies used in this study were the mouse anti-PCNA monoclonal antibody (1:1000, clone PC10; Sigma Chemical), rabbit anti-PCNA polyclonal antiserum (1:750; Abcam), mouse anti-EGFP monoclonal antibody (1:1000; Invitrogen,), rabbit anti-EGFP polyclonal antiserum (1:1000; Invitrogen), rabbit anti-Stat3 polyclonal antiserum (1:100; Kassen et al., 2007), rabbit anti-Ascl1 polyclonal antiserum (1:50; Sigma-Aldrich), rabbit anti-TNFα polyclonal antiserum (1:30; AnaSpec), mouse monoclonal antibody 4C4 (1:250; generous gift from Dr. Jonathan Scholes, University College London), mouse anti-HuC/D monoclonal antibody (1:30; Invitrogen), rabbit anti-rhodopsin polyclonal antiserum (1:5000; Vihtelic et al., 1999), rabbit anti-UV opsin polyclonal antiserum (1:1000; Vihtelic et al., 1999), and rabbit anti-blue opsin polyclonal antiserum (1:250; Vihtelic et al., 1999). Sections were washed and incubated in secondary antibody diluted 1:500 in 1× PBS/0.05% Tween 20 for 1 h at room temperature. Secondary antibodies used in this study were Alexa Fluor goat anti-primary IgG 488, 568, 594, and 647 (1:500; Invitrogen). Nuclei were labeled with TO-PRO-3 (Invitrogen) diluted 1:1000 in 1× PBS/0.05% Tween 20. Slides were washed and mounted with glass coverslips and Vectashield mounting medium (Vector Laboratories).
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) was performed to identify apoptotic cells in frozen retinal sections using the ApoAlert DNA Fragmentation Assay kit (Clontech) in label buffer containing 2% biotinylated dUTP (New England BioLabs) as previously described (Bailey et al., 2010). Sections were processed and dUTP was detected as described previously (Bailey et al., 2010).
In situ hybridization.
Total RNA was isolated from zebrafish embryos at 48 h postfertilization (hpf) using Trizol (Invitrogen) and then reverse transcribed using random primers with the Superscript III Preamplification System (Invitrogen). The tnfα cDNA was amplified using Platinum Taq (Invitrogen) and two primers (Forward 5′ GCTGGTGATGGTGTCTAGGAGGA 3′ and Reverse 5′TGTTGGCGGCACATTGCCAAGA 3′) with an annealing temperature of 58°C. The ∼750 bp PCR product was gel purified (QIAquick Gel Extraction; Qiagen), cloned into pCRII-TOPO, and then sequenced to confirm the identity of the tnfα cDNA.
The tnfα cDNA was subcloned into the pCRII-TOPO (Invitrogen) vector and antisense and sense digoxigenin (DIG)-labeled RNA probes were in vitro transcribed (Roche DIG RNA Labeling Kit SP6/T7). The tnfα cDNA plasmid was linearized with either BamHI (antisense probe) or NotI (sense probe), precipitated, and in vitro transcribed with either T7 or SP6 RNA polymerase, respectively. The in vitro transcription reactions were terminated by adding 0.2 m EDTA and the riboprobes were precipitated using ammonium acetate and 100% ethanol. The quality of the in vitro transcribed RNA was confirmed by electrophoresis through a 1% agarose formaldehyde gel.
The hb-egfa cDNA was synthesized from total RNA isolated from undamaged retinas using Trizol and reverse transcribed as described above. The single-stranded DNA was PCR amplified using Platinum TaqPCR, the hb-egfa Forward primer TGAGTTCTCTGGGGATTATGGG and the Reverse primer ATTAGGGCAGGACGAAGTTGGT (Wan et al., 2012) at an annealing temperature of 55.5°. The 982 bp hb-egfa cDNA was gel purified and cloned into the pCRII-TOPO vector. The antisense and sense DIG-labeled RNA probes were synthesized in vitro using BamHI-linearized vector and T7 RNA polymerase (antisense probe) or NotI-linearized vector and SP6 RNA polymerase (sense probe).
Whole eyes were fixed overnight at 4°C in 4% PFA, cryopreserved, and sectioned at 14 μm. In situ hybridization was performed on retinal cryosections as previously described (Hitchcock and Kakuk-Atkins, 2004) with minor modifications. The procedure was performed at room temperature unless otherwise noted. Retinal sections were thawed from −80°C to room temperature for 1 h and rehydrated in an ethanol/DEPC water series of 100, 95, 70, and 50% for 1 min each. The slides were washed in 2× SSC for 1 min followed by a proteinase K (30 μl of 20 mg/ml stock into 50 ml of buffer [0.1 m Tris-HCl, pH 8.0, and 50 mm EDTA]) treatment at 37°C for 4.5 min. The proteinase K was washed off with DEPC water, the sections treated with 0.1 m triethanolamine (TEA) for 3 min, and then incubated in 0.1 m TEA with acetic anhydride for 10 min. The slides were dehydrated in an ethanol/DEPC series: 2× SSC, 50% ethanol, 70% ethanol, 95% ethanol, and twice with 100% ethanol. Sections were dried out at room temperature for 3 h. The probe was diluted into 2 ml of hybridization buffer (200 μl of 10× salt [NaCl, Tris-HCl, Tris-Base, sodium phosphate monobasic anhydrous, sodium phosphate dibasic monohydrous], 1 ml of deionized formamide, 400 μl 50% dextran sulfate, 200 μl tRNA (50 mg/ml), 40 μl 50× Denhardt's solution, and 160 μl DEPC water) and heated to 95°C for 5 min, snap cooled on ice for 2 min, placed over sections (100 μl at 1 ng/1 μl), and coverslipped with HybridSlips (Grace Bio-Labs). Sections were hybridized with riboprobe overnight at 55°C in a humidity chamber. After hybridization, the sections were washed three times with a 1:1 ratio of 2× SSC and deionized formamide plus 0.1% Tween 20 at 55°C for 30 min each, rinsed with MABT (100 mm maleic acid, pH 7.5, 150 mm NaCl, 0.1% Tween 20) for 30 min at 55°C, rinsed in MABT for 30 min at room temperature, and then blocked in MABT with 10% heat-inactivated sheep serum for 1 h. Sections were incubated overnight in a dark humidity chamber at 4°C with anti-DIG AP fragments (Roche)/MABT diluted 1:1500. After antibody incubation, the sections were washed three times for 20 min with MABT, washed twice for 10 min with stain buffer (100 mm Tris-HCl, 100 mm NaCl, 50 mm MgCl2, pH >9.0), and then processed with 22.5 μl NBT/17.5 μl BCIP (both 50 mg/ml stock solution) into 5 ml of stain buffer at room temperature. The reaction was stopped with 0.1 m Tris-HCl, coverslipped, and imaged on a Leica DM 5500B.
Confocal microscopy and statistical analysis.
Confocal microscopy was performed with a Leica TCS SP5 confocal microscope. Only retinal sections containing or immediately adjacent to the optic nerve were examined to maintain consistency across experimental and control groups. Labeled cell counts from retinal sections were obtained from either 5-μm-thick confocal z-stacks (Stat3 and Ascl1a immunolabeling) or 5- to 8-μm-thick confocal z-stacks (all other immunolabeling) that began at the surface of the 14 μm tissue sections closest to the coverslip using either the entire retina (retinal lysate injection experiments) or a 350 μm linear distance across the dorsal retina centered between the circumferential marginal zone and the optic nerve that consistently received the same degree of damage and reliably resulted in comparable subsequent high levels of Müller glia proliferation (Thomas et al., 2012). Between 10 and 15 experimental and control retinas were examined for each experimental time point. Low-intensity signals were enhanced and background signals were reduced in representative images using the levels function in Adobe Photoshop (Adobe Systems). Levels were adjusted identically to all layers within a panel and to all panels in a figure.
Statistical significance was calculated by using a Student's t test (two-tailed and two-sample unequal variance). The data met the assumptions of the test in all instances. Actual p values are found in the text, with p values < 0.05 and 0.01 being considered significant and highly significant, respectively.
Electroporation of morpholinos into the adult zebrafish retina.
Lissamine-tagged morpholinos (Gene Tools) used in this study included: anti-tnfα morpholino (5′-AGCTTCATAATTGCTGTATGTCTTA-3′) that is complementary to the 5′ UTR of tnfα (Matthews et al., 2009), anti-tnfα 5-base mismatch control morpholino (5′-ACCTTGATAATTCCTCTATCTCTTA-3′) that has the mismatched bases italicized, anti-stat3 morpholino that is complementary to the 5′ UTR of stat3 (Kassen et al., 2009), anti-ascl1a morpholino that is complementary to the 5′ UTR of ascl1a (Fausett et al., 2008), anti-lin28a that is complementary to the 5′ UTR of lin28a (Ramachandran et al., 2010), anti-hb-egfa morpholino that is complimentary to the 5′ UTR of hb-egfa (Wan et al., 2012), or a Standard Control morpholino (Gene Tools) that is not complementary to any known sequence in the zebrafish genome. Morpholinos were resuspended in water at a working concentration of 3 mm and electroporated into the adult zebrafish retina as previously described (Thummel et al., 2008a). The electroporated fish were revived before being placed into constant intense light or injected with ouabain.
Quantitative real-time PCR.
Total RNA was isolated from dorsal retinas collected before and at 16 h of constant light damage using the RNAqueous (Ambion) protocol and reagents. The cDNA was reverse transcribed by Superscript III first strand synthesis (Invitrogen) and quantified using a NANODROP spectrophotometer (Thermo Scientific). The gene-specific primers used for stat3 and ascl1a amplification were described previously (Nelson et al., 2012). The protocols for sample preparation and run conditions described in the ABI 7500 (Applied Biosystems) user manual were followed and described previously (Nelson et al., 2012). Agarose gel electrophoresis and dissociation curve analysis verified that single products were produced with each of the primer pairs used in these experiments. The ΔΔCT method was used for data analysis as described previously (Kassen et al., 2007). The 18S rRNA was used as the reference gene (ΔCT) and three biological replicates were performed in three technical replicates. The normalized ΔCT values for each gene from each time point were normalized against the control group to yield the ΔΔCT, which were used to calculate the log2-fold change in gene expression levels.
Results
Photoreceptor cell death produces a factor that induces Müller glia proliferation
Previous studies demonstrated that very low numbers of Müller glia actively divide in the undamaged adult zebrafish retina (Thummel et al., 2008a). Following light-induced photoreceptor cell death, there is a significant increase in the number of Müller glia that re-enter the cell cycle (Vihtelic and Hyde, 2000; Thummel et al., 2008a). One potential mechanism for this induction of Müller glia proliferation is that dying photoreceptors express a factor that induces Müller glia to re-enter the cell cycle. We hypothesized that this Müller glia proliferation inducing factor would be present at increased levels when photoreceptor apoptosis is maximal.
To determine the time of maximal photoreceptor cell death, we examined TUNEL during the first 31 h of light damage. While the dark-adapted control retinal sections contained 4.8 ± 2.5 TUNEL-positive nuclei in the outer nuclear layer (ONL), the retinas that had been light treated for 16 and 24 h possessed 88 ± 9 and 82 ± 13 TUNEL-positive nuclei in the ONL, respectively (data not shown). By 31 h of constant light, the number of TUNEL-positive nuclei decreased to 48 ± 16. Consistent with previous results (Vihtelic et al., 2006), maximal photoreceptor apoptosis was observed at 16 and 24 h of constant light exposure, which represents the time that the potential photoreceptor-derived factor that induces Müller glia proliferation was likely to be present. We selected the 16 h time point for analysis (over 24 h) because the regeneration process had already started by 16 h, with increased numbers of proliferating rod precursor cells and Stat3-expressing Müller glia relative to the undamaged retina (Vihtelic and Hyde, 2000; Kassen et al., 2007).
To determine whether the light-damaged retina expressed a factor that induced Müller glia proliferation, retinas that were either undamaged (control) or light damaged for 16 h were homogenized and separately injected into the vitreous of undamaged eyes (Fig. 1). Retinal sections containing the optic nerve from 10 individual eyes were labeled for rhodopsin expression to assess the morphology of rod photoreceptors at 1, 2, and 3 d post homogenate injection (dpi; Fig. 1A–F, red). The rhodopsin expression pattern did not change significantly from 1–3 dpi in either control or light-damaged retinal lysates indicating that neither the injection nor the lysates damaged the rods. The strong rhodopsin labeling in the intravitreal space at 1 and 2 dpi confirmed that the donor homogenate was successfully injected and persisted in the recipient eyes (Fig. 1A,B,D,E). PCNA-positive nuclei within the rhodopsin-positive homogenate in the vitreous colocalized with the 4C4 antibody, confirming that these are microglial cells (data not shown).
Figure 1.

Light-damaged retinal homogenate contains a factor that induces Müller glia proliferation. Retinal sections from wild-type eyes that were intravitreally injected with a homogenate from either undamaged retinas (A–C) or light-damaged retinas (D–L) were immunolabeled with anti-rhodopsin (red) to visualize rod outer segments and anti-PCNA (green) to detect proliferating cells in the ONL and INL (arrowheads and arrows, respectively) at 1, 2, and 3 dpi (days postinjection, A–F). Tg(gfap:EGFP)nt11 eyes were injected with light-damaged retinal homogenate and immunolabeled with anti-EGFP and anti-PCNA (G–I, green and red, respectively) at 1, 2, and 3 dpi. The number of PCNA-positive cells significantly increased (p < 0.05, n = 10) in the INL of light-damaged homogenate-injected eyes at 2 and 3 dpi relative to the controls (N), but not in the ONL (M). Injection of the light-damaged retinal homogenate did not cause any cell death as measured by the number of TUNEL-positive cells (J–L, O). Scale bars: (in A) A–F, J–L, 50 μm; (in G) G–I, 50 μm.
To confirm that neither the light-damaged homogenate nor the injections resulted in retinal cell death, which would lead to Müller glia proliferation, we examined TUNEL at the three time points. Low numbers of TUNEL-positive cells were found at all three time points in the light-damaged retinal homogenate-injected eyes (Fig. 1J–L, green). We quantified the number of TUNEL-positive nuclei in the uninjected control retina, the undamaged retinal homogenate-injected eyes, and the light-damaged retinal homogenate-injected eyes. There were no statistically significant differences in the number of TUNEL-positive nuclei in any of these retinas at the different time points (Fig. 1O). These data confirmed that neither the homogenate nor the injection resulted in any significant retinal cell death.
The retinal sections were colabeled for PCNA expression to identify PCNA-positive cells in both the ONL and inner nuclear layer (INL; Fig. 1A–F, green, arrowheads, and arrows, respectively). The number of proliferating ONL rod precursor cells was not statistically different between the uninjected control eyes and the undamaged retinal homogenate-injected eyes (Fig. 1M, red and blue bars, respectively). Similarly, there was no statistical difference in the number of PCNA-positive ONL cells in the undamaged retinal homogenate-injected eyes and the light-damaged retinal homogenate-injected eyes at any of the three time points (Fig. 1M; blue and green bars, respectively). In contrast, the light-damaged retinal homogenate-injected eyes had significantly more PCNA-positive INL cells than either the uninjected control or the undamaged retinal homogenate-injected eyes at 2 and 3 dpi (Fig. 1N; green, red, and blue bars, respectively). At 2 and 3 dpi, the light-damaged retinal homogenate-injected retinas possessed 8.2 ± 4.6 and 16.6 ± 4.1 PCNA-positive INL nuclei relative to 2.8 ± 1.8 and 4.1 ± 3.4 PCNA-positive INL nuclei in the undamaged retinal homogenate-injected eyes at 2 and 3 dpi, respectively (p < 0.005, 0.0001, respectively). The location and morphology of the PCNA-positive nuclei in the damaged retinal homogenate-injected retinas are consistent with that of Müller glia. Furthermore, the light-damaged retinal homogenate-injected retinas possessed small clusters of PCNA-positive nuclei at 3 dpi that were similar in morphology to the clusters of proliferating INL neuronal progenitors in the light-damaged retina (Vihtelic and Hyde, 2000; Kassen et al., 2007).
To confirm that the PCNA-positive INL cells in the light-damaged retinal homogenate-injected retinas were Müller glia, we injected the light-damaged homogenate into the Tg(gfap:EGFP)nt11 transgenic line (Fig. 1G–I, green), which expresses EGFP specifically in Müller glia (Kassen et al., 2007). At 2 dpi, when we first observed an increased number of PCNA-positive cells, the PCNA colocalized with the EGFP-positive Müller glia (Fig. 1H). Similarly at 3 dpi, the small clusters of PCNA-positive nuclei colocalized with diffuse EGFP-expression in the hypertrophied Müller glia (Fig. 1I). These observations demonstrated that the light-damaged retinal homogenate contained at least one factor that was sufficient to induce the Müller glia to re-enter the cell cycle in the absence of any significant retinal cell death.
TRAP1 is expressed at elevated levels in the light-damaged retinal homogenate
To identify proteins and candidate signaling pathways that were present in the light-damaged retinal homogenate that could induce Müller glia proliferation, we undertook a comparative proteomic analysis of the homogenates. Total dorsal retinal proteins were isolated from either undamaged retinas and labeled with Cy3 or from 16 h light-damaged retinas and labeled with Cy5. Both protein homogenates were combined and then separated by 2D gel electrophoresis. Greater than 50 protein spots, which were expressed at least twofold greater in the light-damaged retinal homogenate relative to the undamaged retinal homogenate, were isolated and analyzed via MALDI-TOF mass spectrometry to identify the proteins (Table 1). One of the proteins corresponded to TRAP1 (Table 1, Spot 43), which suggested that TNFα signaling was a candidate for inducing Müller glia proliferation.
TNFα expression is elevated in the light-damaged retina
While TNFα was not one of the proteins analyzed by MALDI-TOF, we examined the expression of TNFα using an immunoblot with total protein homogenates from retinas that were undamaged and exposed to constant light for 16 and 36 h. The control retinal lysates possessed a band of 28 kDa, which corresponded to the expected size of full-length pro-form zebrafish TNFα (Fig. 2). Following 16 h of constant light exposure, the expression of a 17 kDa protein, corresponding to the biologically relevant cleaved and secreted form of TNFα, increased relative to the control (Fig. 2). After 36 h of constant light, both the 28 and 17 kDa forms of TNFα were elevated (Fig. 2).
Figure 2.

TNFα expression increases during constant light treatment. Total protein was isolated from dark-adapted albino retinas after 0 (Control), 16, and 36 h of constant light treatment. Immunoblotting for TNFα expression reveals a 28 kDa band, likely corresponding to the uncleaved form of TNFα. After 16 h of constant light, a 17 kDa band is detected that corresponds to the mature, secreted form of TNFα. Total protein was also isolated from dark-adapted albino dorsal retinas that were injected and electroporated with either the tnfα 5-mis control morpholino (MM) or the tnfα morpholino (MO) and placed in constant intense light for 36 h. While the tnfα 5-mis control retina had approximately the same amount of TNFα as the uninjected light-damaged retina (36 h), the tnfα morphant possessed dramatically less TNFα protein. β-actin expression was detected as a loading control.
To determine the spatial expression of TNFα in the damaged retina, we immunolocalized the TNFα protein in the undamaged retina (Fig. 3A) and the light-damaged retina at 16 h of constant light (Fig. 3B,D,E) and at 36 h when the Müller glia begin proliferating (Fig. 3C,F,G). In control retinas, TNFα expression was observed within the outer plexiform layer (OPL) and the nerve fiber layer (NFL; Fig. 3A, arrowhead and double arrowhead, respectively). After 16 h of constant light, increased TNFα expression was associated with cells within the cone cell layer (Fig. 3B, arrows), ONL (open arrowhead), OPL (arrowhead), INL (double arrows), and NFL (double arrowhead). By 36 h of constant light, a similar TNFα expression pattern was observed, but the number and intensity of INL-labeled cells increased (Fig. 3C, double arrow) and the number and intensity of ONL and cone cell layer expression decreased (open arrowhead and arrow, respectively).
Figure 3.

TNFα is expressed in different cell types during the constant light treatment. Dark-adapted albino zebrafish were exposed to constant intense light for 0 h, 16 h, or 36 h. Cryosections were immunolabeled for TNFα (A–E, G, green; F, red) or in situ hybridized with tnfα antisense riboprobe (H–O). TNFα antisera weakly labeled the NFL and OPL in undamaged control retinas (A, double arrowhead and arrowhead, respectively). At 16 and 36 h of constant light (B and C, respectively), TNFα expression increased in the OPL (arrowhead), ONL (open arrowhead), photoreceptor outer segments (arrow), INL (double arrow), NFL (double arrowhead), and retinal pigmented epithelium (RPE; asterisk). Following 36 h of constant intense light (C), TNFα antisera labeling in the INL reveals cells with processes that span from the GCL to the ONL (double arrow). At 16 h of light, TNFα colocalized with some TUNEL-positive ONL cells (D, open arrowhead and inset) and ZPR1-positive double cones (E, arrows and inset). At 36 h, TNFα colocalized with EGFP-positive Müller glia (F, double arrow and inset) in the Tg(gfap:EGFP)nt11 transgenic line, but not with 4C4-positive microglia (G, red). In situ hybridization with a tnfα antisense probe (H–O, blue) demonstrates increased tnfα expression primarily in the ONL following 16 h of light (I) and then primarily in the INL at 36 h of treatment (J) relative to the undamaged control (H). Colocalization of the tnfα antisense riboprobe with TUNEL-positive rods and cones (K, arrows and arrowheads, respectively), blue opsin-expressing cones (L, arrow), and EGFP-expressing rod photoreceptors in the Tg[rho:Eco.NfsB-EGFP]nt19 transgenic line (M, arrows) occurred at 16 h of light damage. After 36 h of light damage, the EGFP-expressing Müller glia (N, green) in the Tg(gfap:EGFP)nt11 transgenic line expressed tnfα (N, arrows), while the 4C4-expressing microglia (O, green) did not express TNFα. The boxed insets (D–F) are magnified 2.9-fold to highlight the colocalization with TNFα. Scale bars: (in A) A–G, 50 μm; (in H) H–J, 50 μm; (in K) K–O, 50 μm.
Because TNFα is a secreted protein and many different cells are associated with the OPL, we performed in situ hybridization to identify the cells that expressed the tnfα transcript. In the undamaged retina, negligible amounts of tnfα were detected in both the INL and ONL (Fig. 3). After 16 h of constant light, the expression of tnfα greatly increased in the ONL and, to a lesser extent, cells in the INL (Fig. 3I). After 36 h of constant light, the predominant expression of tnfα shifted from the ONL to the INL (Fig. 3J). This suggests that the light damage initially results in increased tnfα expression in the ONL photoreceptors, followed by increased expression within a subset of the INL cells.
To determine the identity of the cells that express TNFα during the constant light treatment, retinal sections were either immunolabeled with anti-TNFα antibodies (Fig. 3D–G) or in situ hybridized with an in vitro transcribed tnfα riboprobe (Fig. 3K–O). After 16 h of constant light, the TUNEL signal in the ONL occasionally colocalized with anti-TNFα immunolabeling (Fig. 3D, open arrowhead). Similarly, strong colocalization was observed between the tnfα antisense probes (Fig. 3K, blue) and the TUNEL signal (green) within the ONL (arrows) and cone cell nuclei (arrowheads), which is consistent with the light-induced apoptotic rods and cones. Colocalization between TNFα antiserum (Fig. 3E, green) and the double-cone cell marker ZPR1 (red), as well as tnfα antisense probe (Fig. 3 L,M, blue) with either Blue opsin immunolabeling (Fig. 3L, green) or EGFP expression (Fig. 3M) in the rods of the Tg[rho:Eco.NfsB-EGFP]nt19 transgenic line (Montgomery et al., 2010), confirmed that TNFα was expressed in both rod and cone photoreceptors at 16 h of constant light. No colocalization was observed between the microglial 4C4 immunolabeling (Fig. 3G, red; O, green) and either TNFα immunolabeling (Fig. 3G, green) or tnfα antisense probes (Fig. 3O, blue). After 36 h of constant light, the Tg(gfap:EGFP)nt11 transgenic line, which expresses EGFP in all Müller glia (Fig. 3F,N, green), exhibited TNFα protein (Fig. 3F, red) and tnfα mRNA (Fig. 3N, blue) expression in the INL Müller glia. Together, these results demonstrate that TNFα expression increases initially in apoptotic rod and cone photoreceptors and then in Müller glia at a time when they are beginning to proliferate.
TNFα expression in the light-damaged retina is required for Müller glia proliferation, but not photoreceptor apoptosis
To determine whether TNFα plays a role in the light-damaged retina, we intravitreally injected and electroporated lissamine-tagged morpholinos into dark-adapted albino retinas immediately before placing the fish into constant intense light. One morpholino was designed to bind the 5′ untranslated region of tnfα (tnfα MO) to block translation of the TNFα protein, while a control morpholino (tnfα 5-mis) that contained five mismatched bases will not efficiently bind the tnfα 5′ untranslated region.
After 16 and 36 h of constant intense light, the retinas were immunolabeled for TNFα expression (Fig. 4A–D, green). At 16 h of constant light, control retinas that were electroporated with the 5-base mismatch morpholino (tnfα 5-mis) exhibited TNFα expression in photoreceptor outer segments (Fig. 4A, arrow), OPL (bracket), and the nerve fibers in the ganglion cell layer (GCL; asterisk). Following 36 h of constant light, the tnfα 5-mis control morphant retinas revealed increased TNFα expression in the INL Müller glia (Fig. 4C, arrowheads). At both time points, light-damaged retinas that were not electroporated with a morpholino were indistinguishable from the tnfα 5-mis control morphant retinas (data not shown). In contrast, the tnfα morphant retinas either lacked or possessed reduced TNFα expression in all of these retinal cell types or layers at 16 and 36 h of constant light (Fig. 4B,D, respectively). After 36 h of constant light, immunoblotting of protein lysates isolated from dorsal retinas showed similar expression of both the 28 and 17 kDa TNFα protein bands in both controls, while the tnfα morphant retinas exhibited a dramatic knockdown of both the 28 and 17 kDa TNFα protein expression (Fig. 2).
Figure 4.
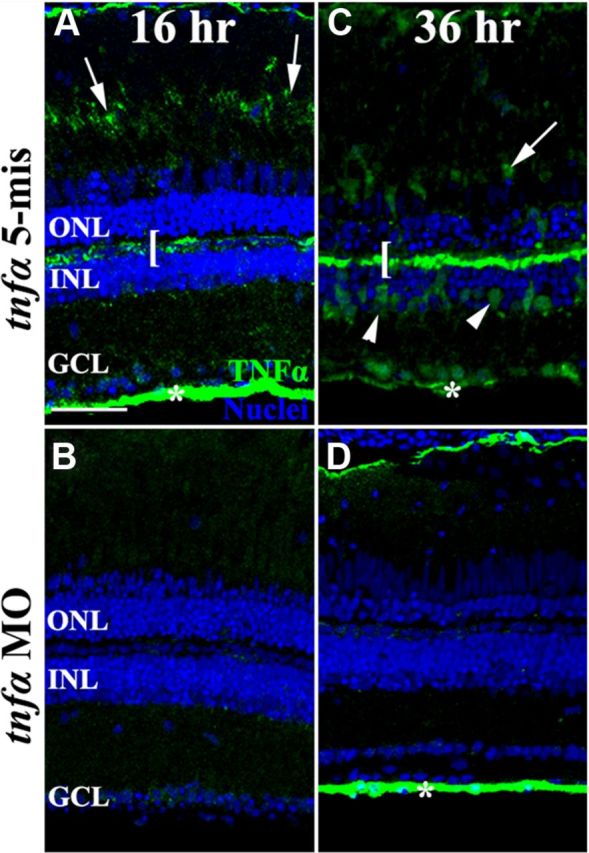
In vivo knockdown of TNFα during constant light treatment. Dark-adapted albino zebrafish were intravitreally injected and electroporated with either lissamine-tagged tnfα 5-mismatch morpholinos (tnfα 5-mis, A, C) or translation-inhibiting tnfα morpholinos (tnfα MO, B, D) and then exposed to constant intense light for either 16 h (A, B) or 36 h (C, D). The tnfα morphant retinas showed decreased intensity of TNFα immunolabeling relative to both the uninjected (data not shown) and tnfα 5-mis controls, in the outer segments (arrows), OPL (bracket), Müller glia (arrowheads), and the NFL (asterisk). Scale bar: (in A) A–D, 50 μm.
To determine whether loss of TNFα signaling affected light-induced photoreceptor cell death, uninjected, tnfα 5-mis control and tnfα morphant retinas were labeled for TUNEL after 16 h of constant light damage. Uninjected (data not shown) and tnfα 5-mis control retinal sections (Fig. 5A) contained 32.8 ± 4.4 and 28.9 ± 3.7 TUNEL-positive ONL nuclei respectively (Fig. 5C, blue and red bars, respectively). Similarly, the tnfα morphant retinas (Fig. 5B) possessed 30.5 ± 4.5 TUNEL-positive ONL nuclei (Fig. 5C). Thus, there was no significant difference in the number of TUNEL-positive cells in the tnfα morphant retina relative to either of the control retinas (p = 0.72 and 0.79, respectively), suggesting that TNFα is not required for light-induced photoreceptor cell death.
Figure 5.

Knockdown of TNFα does not affect photoreceptor apoptosis during constant light treatment. Dark-adapted albino fish were intravitreally injected and electroporated with either lissamine-tagged tnfα 5-mismatch morpholinos (tnfα 5-mis, A) or translation inhibiting tnfα morpholinos (tnfα MO, B) immediately before starting 16 h of constant light. Retinal cryosections were colabeled for TUNEL (A, B, green) and nuclei (To-Pro-3, blue) and the number of TUNEL-positive cells were quantified (C). No significant difference was observed in the number of TUNEL-positive cells between the controls (blue and red bars) and the tnfα morphant (green bar, p > 0.05, n = 10). Scale bar: (in A) A, B, 50 μm.
To examine if knockdown of TNFα affected cell proliferation in the light-damaged retina, dark-adapted albino zebrafish were either uninjected (data not shown) or injected and electroporated with either tnfα 5-mis control morpholino or the tnfα morpholino and placed in constant intense light. The eyes were collected at specific time points and retinal sections were immunolabeled for PCNA-positive cells (Fig. 6A–H, green).
Figure 6.

TNFα is required for Müller glial proliferation in the light-damaged retina. Dark-adapted albino zebrafish that were injected and electroporated with either the 5-base mismatch control morpholino (tnfα 5-mis, A–D) or the tnfα morpholino (tnfα MO, E–H) and exposed to constant intense light for either 16 h (A, E), 36 h (B, F), 51 h (C, G), or 96 h (D, H). The sections were immunolabeled with anti-PCNA to label the nuclei of dividing cells (green) and To-Pro-3 (blue) to label all nuclei. PCNA-positive cell counts were quantified in the INL (I) from the uninjected and tnfα 5-mis controls (blue and red bars, respectively) and the tnfα MO (green bars). The tnfα morphants contained significantly fewer PCNA-positive INL cells relative to either control (p < 0.0001, n = 10) at 36, 51, and 96 h of light treatment. Scale bar: (in A) A–H, 50 μm.
After 16 h of light damage, an average of <2.0 PCNA-positive cells were observed in the INL of the uninjected, tnfα 5-mis control, and tnfα morphant retinal sections (Fig. 6I). Following 36 h of constant light damage, the tnfα morphant retinal sections contained significantly fewer PCNA-positive INL cells (4.3 ± 1.2; Fig. 6F,I) compared with the uninjected (26.7 ± 3.7) and tnfα 5-mis control sections (21 ± 3.5; Fig. 6I, p < 0.001 for each). After 51 h of constant light, tnfα morphant retinas also contained significantly fewer PCNA-positive INL cells (9.1 ± 2.7; Fig. 6G,I) relative to the uninjected (38.2 ± 4.5) and tnfα 5-mis control sections (28.4 ± 4.4; Fig. 6I, p < 0.001 for each). After 96 h of constant light, tnfα morphant retinas still contained significantly fewer PCNA-positive INL cells (20.9 ± 4.8; Fig. 6H,I) relative to the uninjected (41.0 ± 4.3) and tnfα 5-mis control sections (40.6 ± 4.4; Fig. 6I, p < 0.006 for each). While there was no significant difference in the number of PCNA-positive ONL cells in the three treatments through 51 h of constant light, at 96 h of light the tnfα morphant retinas contained significantly fewer PCNA-positive ONL cells (11.5 ± 2.8) relative to the uninjected (67.1 ± 5.3) and tnfα 5-mis control sections (52.9 ± 7.4; p < 0. 001 for each). At 96 h of light, these PCNA-positive ONL cells likely correspond to both resident ONL rod precursor cells and INL-derived neuronal progenitors that migrated to the ONL. Thus, TNFα is required for the maximal proliferation of the INL Müller glia and neuronal progenitor cells beginning at 36 h of constant light.
The number of proliferating Müller glia is amplified through Müller glia expression of TNFα
TNFα is expressed at 16 h in the dying/damaged photoreceptors and then at 36 h in the Müller glia as they begin to proliferate (Fig. 3). Electroporation of the tnfα morpholino at the start of constant light treatment inhibited TNFα expression in both the photoreceptors and Müller glia, resulting in a significant reduction in the number of PCNA-positive Müller glia (Fig. 6). To assess the role of TNFα expression primarily in the Müller glia, dark-adapted albino zebrafish were either uninjected or injected and electroporated with either the tnfα 5-mis control morpholino or the tnfα morpholino into the retina following 16 h of light damage. The retinas were collected and labeled with anti-TNFα antisera after a total of 36 h of light treatment. The uninjected retinas (data not shown) were indistinguishable from the tnfα 5-mis control retinas, with robust anti-TNFα labeling in both the OPL and the INL Müller glia (Fig. 7A, bracket and double arrows, respectively). In contrast, the tnfα morphant retinas had dramatically reduced TNFα labeling relative to the controls (Fig. 7E). Injection and electroporation of the tnfα morpholino after 16 h of light treatment appears to have effectively knocked down TNFα expression in the Müller glia, the apoptotic photoreceptors, and the NFL at 36 h. Because the majority of TNFα expression in the apoptotic photoreceptors occurred at or before 16 h (Fig. 3B) and most of the TNFα expression in the Müller glia occurred after 16 h (Fig. 3C), the electroporation of the tnfα morpholino after 16 h of light treatment primarily reduced TNFα expression in the Müller glia.
Figure 7.

Müller glia-derived TNFα expression is required for inducing additional Müller glia to re-enter the cell cycle. Dark-adapted albino zebrafish were exposed to constant intense light for 16 h and then intravitreally injected and electroporated with either lissamine-tagged tnfα 5-mismatch morpholinos (tnfα 5-mis, A–D) or translation-inhibiting tnfα morpholinos (tnfα MO, E–H) before being placed back into intense light until 36 h (A, B, E, F), 51 h (C, G), or 68 h of treatment (D, H). Retinal sections were immunolabeled with either anti-TNFα (A, E, green) or anti-PCNA (B–D, F–H, green) and To-Pro-3 (A–H, blue) to label nuclei. Injection and electroporation of the tnfα MO after 16 h of constant light efficiently knocked down TNFα expression in the OPL, INL, and NFL relative to the control. INL PCNA-positive cell were quantified (I) from uninjected and tnfα 5-mismatch control eyes (blue and red bars, respectively) and the tnfα morphant (green bars) at 36, 51, and 68 h of light. The number of PCNA-labeled INL cells was significantly decreased in the tnfα morphant relative to both controls (p < 0.005, n = 10) at all three time points. PCNA-positive INL cell clusters were quantified (J) from uninjected and tnfα 5-mismatch control eyes (blue and red bars, respectively) and the tnfα morphant (green bars) at 68 h of light. The number of PCNA-labeled cell clusters was significantly decreased in the tnfα morphant relative to both controls (p < 0.05, n = 10). Scale bar: (in A) A–H, 50 μm.
We knocked down the expression of TNFα after 16 h of intense light and examined PCNA expression in retinal sections after 36, 51, or 68 h of total constant light (Fig. 7B–D,F–H, green). After 36 h of light, the uninjected and tnfα 5-mis control morphant eyes contained significantly greater numbers of PCNA-positive INL cells (28.8 ± 2.4 and 31.1 ± 3.4, respectively; Fig. 7I, blue and red bars, respectively), relative to the tnfα morphant sections (17.7 ± 3.2; Fig. 7I, green bars, p = 0.013 and 0.009, respectively). After 51 h of light damage, uninjected and tnfα 5-mis control retinas contained significantly more PCNA-positive INL cells (38.5 ± 4.0 and 41.9 ± 3.4, respectively; Fig. 7I, blue and red bars, respectively) compared with the tnfα morphant retinal sections (27.5 ± 3.3; Fig. 7I, green bars, p = 0.047 and 0.007, respectively). By 68 h of light damage, uninjected and tnfα 5-mis control retinas contained a highly significant increase in the number of PCNA-positive INL cells (74.3 ± 7.4 and 68.9 ± 5.3, respectively; Fig. 7I, blue and red bars, respectively) relative to the tnfα morphant retinal sections (36.5 ± 6.1; Fig. 7I, p < 0.001 for each).
To determine whether this significant reduction in the number of PCNA-positive cells was due to either fewer Müller glia re-entering the cell cycle or reduced numbers of proliferating neuronal progenitors in each cluster, we quantified the number of clusters of PCNA-positive cells, with the assumption that a cluster of proliferating neuronal progenitor cells was derived from a single Müller glial cell. At 68 h of constant light, uninjected and tnfα 5-mis control retinal sections contained 18.9 ± 1.3 and 18.1 ± 1.4 clusters of PCNA-positive cells relative to only 13.3 ± 1.2 clusters in the tnfα morphants (Fig. 7J, p < 0.02). This significantly reduced number of PCNA-positive cell clusters in the tnfα morphant relative to the controls demonstrated that TNFα expression in the Müller glia was necessary to increase the number of proliferating Müller glia in the light-damaged retina.
TNFα expression in the light-damaged retina is required for Stat3 and Ascl1a expression
To determine whether TNFα signaling is required for the increased expression of the transcription factors Stat3 or Ascl1a in the light-damaged retina (Fausett et al., 2008; Nelson et al., 2012), we immunolocalized Stat3 and Ascl1a in tnfα 5-mis albino control retinas (Fig. 8A,B, respectively), and tnfα morphant albino retinas (Fig. 8D,E) after 36 h of constant intense light. Uninjected retinas contained 62.7 ± 4.0 Stat3-positive cells and 31.8 ± 2.8 Ascl1a-positive cells (Fig. 8C, blue bars). The tnfα 5-mis control retinas contained similar, high numbers of Stat3-positive and Ascl1a-positive INL cells (Fig. 8A–C, red bars). In contrast, tnfα morphant retinas contained only 14.7 ± 4.6 Stat3-positive and 9.3 ± 3.0 Ascl1a-positive cells (Fig. 8C, green bars, D,E), which were both significantly reduced in number relative to the tnfα 5-mis control retinas (p = 0.003 and 0.02, respectively).
Figure 8.

TNFα expression is required for Stat3 and Ascl1a expression. Dark-adapted albino zebrafish retinas were intravitreally injected and electroporated with either the lissamine-tagged tnfα 5-mismatch morpholino (tnfα 5-mis, A, B) or translation-inhibiting tnfα morpholino (tnfα MO, D, E) and then exposed to constant intense light for 36 h. Retinal cryosections were immunolabeled for PCNA (A, B, D, E; red) and either Stat3 (A, D; green) or Ascl1a (B, E; green) and Stat3- and Ascl1a-labeled cells were quantified across a 350 μm region of the central dorsal retina (C). Some Stat3-labeled cells were PCNA positive and others were PCNA negative in the control (A, arrows and arrowheads, respectively). In contrast, all the Ascl1a-labeled cells coexpressed PCNA in the control (B). Relative to both controls (C; blue and red bars), the tnfα morphant (green bars) possessed significantly fewer Stat3-positive cells and Ascl1a-labeled cells than either the uninjected or tnfα 5-mis controls (p < 0.05, n = 10). Total RNA was isolated from dorsal retinas of dark-adapted albino fish that were not light damaged (0 h uninject), placed in constant light for 16 h (16 h Lt uninject), and fish that were intravitreally injected and electroporated with either the lissamine-tagged tnfα 5-mismatch morpholinos (tnfα 5-mis) or translation-inhibiting tnfα morpholinos (tnfα MO) and light damaged for 16 h. The log2 of the mean expression for stat3 and ascl1a determined by qRT-PCR (H; blue and red bars, respectively; n = 3) was plotted. The stat3 and ascl1a expression was significantly increased in both 16 h light-damaged control groups relative to the undamaged 0 h control (p < 0.05). Additionally, the expression of both stat3 and ascl1a was significantly reduced in the light-damaged tnfα morphants relative to either of the light-damaged controls (p < 0.04). Scale bar: (in A), A, B, D, E, 50 μm.
To confirm that TNFα is required for the expression of Stat3 and Ascl1a, we used quantitative real-time PCR (qRT-PCR) to examine the expression of the stat3 and ascl1a transcripts in dark-adapted albino zebrafish that were either uninjected or injected and electroporated with either the tnfα 5-mis control morpholino or the tnfα morpholino. As expected (Kassen et al., 2007; Bailey et al., 2010; Nelson et al., 2012), both stat3 and ascl1a RNA expression increased significantly within the first 16 h of constant light treatment in both the uninjected and tnfα 5-mis morpholino controls (Fig. 8F). However, both stat3 and ascl1a RNA expression was significantly reduced in the tnfα morphant after 16 h of light relative to either the uninjected (Fig. 8F, p = 0.01 and 0.04, respectively) or tnfα 5-mis control morphant (Fig. 8F, p = 0.02 and 0.04, respectively). While knockdown of TNFα expression could reduce the number of Müller glia (Thummel et al., 2008a), which in turn would reduce the overall expression of stat3 and ascl1a in the light-damaged retina, we observed equal numbers of Müller glia after 48 h of constant light in the tnfα morphant and uninjected control retinas (data not shown). Additionally, we did not detect TUNEL-positive INL cells in either the tnfα morphant or uninjected control retinas after 48 h of constant light (data not shown). Together, these data indicated that TNFα signaling resides upstream and is required for both Stat3 and Ascl1a expression during the earliest stages of retinal regeneration.
We previously described a model where the expression of Lin28a, Ascl1a, and Stat3 are all required in a subset of proliferating Müller glia to induce a different subset of Müller glia to re-enter the cell cycle (Nelson et al., 2012). Because Müller glial expression of TNFα appeared to be required to induce additional Müller glia to proliferate (Fig. 7), we examined if either Lin28a, Ascl1a, or Stat3 are required for TNFα expression in the Müller glia. Dark-adapted albino zebrafish were injected and electroporated with either the Standard Control morpholino (S.C. MO, which is not complementary to any known sequence in the zebrafish genome), the tnfα morpholino, stat3 morpholino, ascl1a morpholino, or the lin28a morpholino, and then exposed to constant intense light for 36 h. The Standard Control morphant retinas exhibited TNFα expression in the OPL and the Müller glia in the INL (Fig. 9A, bracket and arrows, respectively). The tnfα morphant retinas lacked the TNFα immunostaining in both the OPL and INL (Fig. 9B). In contrast, the stat3 (Fig. 9C), ascl1a (Fig. 9D), and lin28a (Fig. 9E) morphant retinas possessed a significant amount of TNFα immunostaining in the OPL, but showed dramatically less TNFα expression in the Müller glia relative to the Standard Control morphant. Thus, Stat3, Ascl1a, and Lin28a expression are required for TNFα expression in the Müller glia, but not in either the OPL or NFL.
Figure 9.

Müller glia, but not outer plexiform, expression of TNFα requires Stat3, Ascl1a, and Lin28a. Dark-adapted adult albino zebrafish retinas were intravitreally injected and electroporated with either standard control morpholino (S.C. MO, A), tnfα morpholino (tnfα MO, B), stat3 morpholino (stat3 MO, C), ascl1a morpholino (ascl1a MO, D), or lin28a morpholino (lin28a MO, E) and then exposed to constant intense light for 36 h. Retinal cryosections were immunolabeled for TNFα (green). Robust TNFα expression was observed in the OPL (bracket) and INL (arrows) of the S.C. morphant control retina (A). Both OPL and INL TNFα labeling was nearly abolished in the tnfα morphant retinas (B). The OPL immunolabeling was unaffected in the stat3, ascl1a, and lin28a morphants (C–E, brackets), but the INL expression of TNFα was nearly absent. Scale bar: (in A) A–E, 50 μm.
Morpholino-mediated knockdown of TNFα does not block rod and cone regeneration in the light-damaged retina
We examined the consequences of TNFα knockdown on Müller glia proliferation at 1 and 3 d following 96 h of constant light treatment. At 1 d after light treatment, the tnfα morphant retinas contained a significantly greater number of PCNA-positive INL cells (52.8 ± 4.3; Fig. 10C,E, green bars) relative to either the uninjected (33.1 ± 8.1) or tnfα 5-mismatch controls (26.0 ± 4.5; Fig. 10A,E, blue and red bars, respectively, p < 0.01). Similarly, at 3 d after light treatment, the tnfα morphant possessed significantly more PCNA-positive INL cells (95.4 ± 7.2; Fig. 10D,E, green bars) relative to either uninjected (42.3 ± 4.5) or tnfα 5-mismatch controls (41.4 ± 6.0; Fig. 10B,E, blue and red bars, respectively). Thus, the tnfα morphant exhibited delayed Müller glia proliferation relative to controls.
Figure 10.
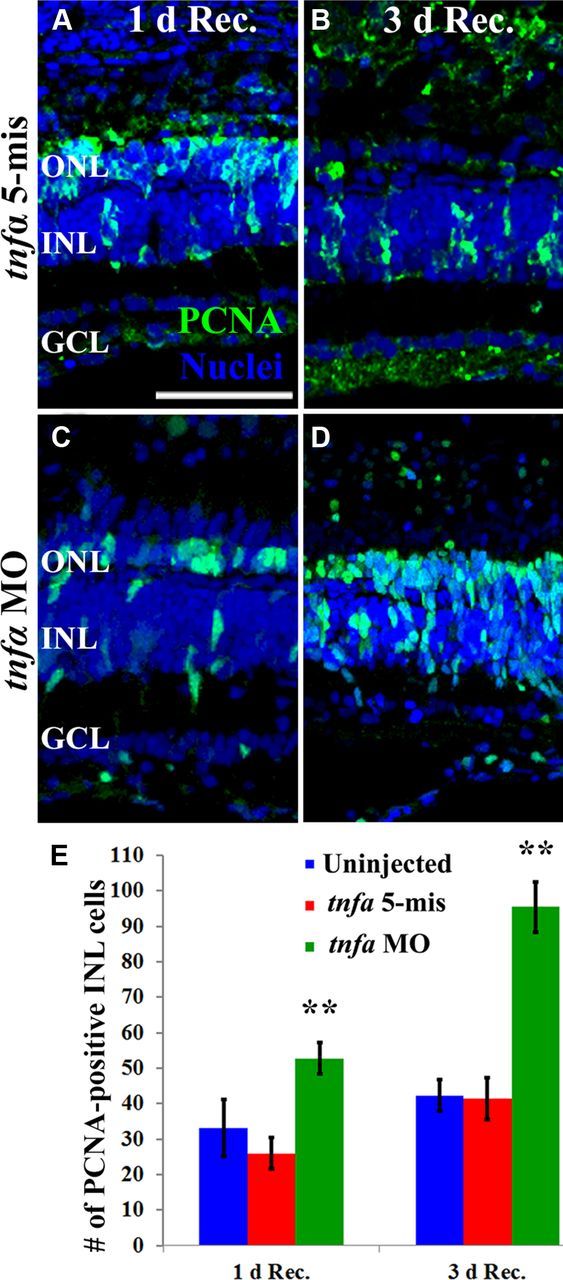
TNFα knockdown delays Müller glial proliferation in the light-damaged retina. Dark-adapted albino zebrafish that were injected and electroporated with either the 5-base mismatch control morpholino (tnfα 5-mis, A, B) or the tnfα morpholino (tnfα MO, C, D) and exposed to constant intense light for 96 h followed by either 1 (A, C) or 3 d of recovery (B, D). The sections were immunolabeled with anti-PCNA to label the nuclei of dividing cells (green) and To-Pro-3 (blue) to label all nuclei. PCNA-positive INL cell counts were quantified (E) from the uninjected and tnfα 5-mis controls (blue and red bars, respectively) and the tnfα MO (green bars). The tnfα morphants contained significantly more PCNA-positive INL cells relative to either control (p < 0.001, n = 10) at both 1 and 3 d recovery from 96 h of light treatment. Scale bar: (in A) A–D, 50 μm.
We examined if the delayed Müller glia proliferation in the light-damaged tnfα morphant still regenerated rod and cone photoreceptors. We confirmed the extent of rod and cone photoreceptor cell loss in the light-damaged tnfα morphants relative to the controls. After 96 h of constant light, uninjected (data not shown), tnfα 5-mismatch retinas (Fig. 11A,E,I) and tnfα morphant retinas (Fig. 11B,F,J) contained very low levels of Rhodopsin-, Blue opsin-, and UV opsin-expressing photoreceptors, suggesting a similar extent of photoreceptor cell loss in the tnfα morphant and the control retinas. However, the control retinas possessed a thicker and more densely packed ONL relative to the tnfα morphant ONL. This is likely due to the tnfα morphants failing to generate large numbers of progenitor cells that migrated to the ONL by 96 h of light treatment (Fig. 6).
Figure 11.

Aberrant rod photoreceptor regeneration following TNFα knockdown. Dark-adapted albino zebrafish that were injected and electroporated with either the 5-base mismatch control morpholino (tnfα 5-mis, A, C, E, G, I, K) or the tnfα morpholino (tnfα MO, B, D, F, H, J, L) and exposed to constant intense light for 96 h (A, B, E, F, I, J) followed by 14 d of recovery (C, D, G, H, K, L). Retinal cryosections were immunolabeled with either anti-Rhodopsin (A–D, green), anti-Blue opsin (E–H, green), or anti-UV opsin (I–L, green) to label rods, double cones, and UV cones, respectively, and To-Pro-3 (blue) to label all nuclei. At 96 h of treatment, tnfα morphants and controls contained low levels of opsin-positive photoreceptors (A, B, E, F, I, J, green). Following 14 d recovery, high levels of opsin expression were observed in tnfα 5-mis and tnfα morphants. However, the rod outer segment (ROS) length in tnfα morphants was decreased relative to the control. Scale bar: (in A) A–L, 50 μm.
We next examined the extent of photoreceptor cell regeneration 14 d after terminating 96 h of constant light. Uninjected (data not shown) and tnfα 5-mis retinas (Fig. 11C,G,K) contained high levels of Rhodopsin-, Blue opsin-, and UV opsin-labeled photoreceptor outer segments, indicating that rods, double cones, and UV cones had regenerated, respectively. The tnfα morphants possessed similar levels of double and UV cone outer segments relative to the controls (Fig. 11H,L, respectively), suggesting that cones had regenerated in the tnfα morphant. The tnfα morphants also possessed rhodopsin-positive rod outer segments, although they were shorter than the controls, suggesting that rod photoreceptor regeneration was delayed in the tnfα morphant relative to the controls.
TNFα is required for Müller glia proliferation in the ouabain-damaged retina
We were interested if TNFα is expressed in only dying photoreceptors or if it is expressed in other dying retinal neurons as a prerequisite to induce Müller glia to re-enter the cell cycle. This would reveal whether TNFα served as a specific signal to Müller glia to proliferate and regenerate only photoreceptor cells or if TNFα was a general signal to induce Müller glia to re-enter the cell cycle to regenerate retinal neuronal classes in addition to photoreceptors.
To distinguish between these two possibilities, we intravitreally injected a low concentration of ouabain to induce the death of ganglion, amacrine, and bipolar cells without causing any significant photoreceptor cell death (Fimbel et al., 2007). We immunolocalized TNFα expression at 0, 1, and 3 d post ouabain injection. In the undamaged retina, TNFα expression was weakly detected in the OPL and NFL (Fig. 12A,B). At 1 d post ouabain injection, TNFα expression increased dramatically in the OPL, the NFL, and cells within the GCL and INL that were morphologically different from Müller glia (Fig. 12C,D, bar, asterisks, and arrowheads, respectively). Some of the TNFα-positive cells in the INL and GCL coexpressed HuC/D (Fig. 12D, red), demonstrating that they were amacrine and ganglion cells, respectively. Colocalization of TNFα and TUNEL at 1 d post ouabain injection confirmed that TUNEL-positive ganglion cells and INL cells expressed TNFα (Fig. 12E). At 3 d postinjection, the TNFα expression persisted in the OPL, INL, and GCL, but was now also detected within INL cells that were morphologically similar to Müller glia (Fig. 12G,H, arrows). Immunolocalization of TNFα in the ouabain-damaged Tg(gfap:EGFP)nt11 transgenic retinas confirmed that many TNFα-positive INL cells were Müller glia at 3 d after ouabain injection (Fig. 12I,J, arrows). Thus, ouabain-induced inner retinal damage stimulated TNFα expression in apoptotic inner retinal neurons and subsequently the Müller glia.
Figure 12.

TNFα expression increased in apoptotic inner retinal neurons and Müller glia in the ouabain-damaged retina. Adult zebrafish were either uninjected (A, B) or intravitreally injected with ouabain to a final concentration of 2 μm (C–J). At 1 (C–F) and 3 d (G–J) after ouabain injection, TNFα (A–H, green; I, J, red) expression was immunolabeled with either HuC/D (D, red), TUNEL (E, red), EGFP in the Tg(gfap:EGFP)nt11 transgenic line (I, green) or To-Pro-3 to identify the nuclear layers (A, C, G, blue). TNFα was observed in undamaged control retinas in the OPL and NFL (bracket and asterisk, respectively). At 1 dpi, TNFα expression increased in the OPL (bracket), NFL (asterisk), INL, and GCL (arrowheads). Some of the TNFα-positive INL and GCL cells colabeled with HuC/D and TUNEL-positive nuclei (D, E, respectively). At 3 dpi, increased TNFα expression persisted in the OPL (bracket) and the INL (arrows). The TNFα-positive INL cells (red) colocalized with EGFP (green) in the Tg(gfap:EGFP)nt11 transgenic retinas (I, J). The inset in D corresponds to the dashed box region. Scale bar: (in A) A–J, 50 μm.
To determine whether TNFα expression was required to induce Müller glia to re-enter the cell cycle in the ouabain-damaged retina, in the absence of significant photoreceptor cell damage (Fimbel et al., 2007), we intravitreally injected a final concentration of 2 μm ouabain immediately following injecting nothing (Fig. 13A, Ouabain control) or injecting and electroporating either the tnfα 5-mismatch control morpholino or the tnfα morpholino (Fig. 13B,C). At 3 d postinjection, both the uninjected and the tnfα 5-mis control retinas contained similar numbers of PCNA-positive cells, 57.0 ± 4.9 and 52.8 ± 4.0, respectively (Fig. 13A,B,D; blue and red bars, respectively). In contrast, the tnfα morphants contained significantly fewer PCNA-positive cells, 29.9 ± 6.5 (Fig. 13C,D, green bar, p = 0.008 relative to the tnfα 5-mis control). These results demonstrated that both apoptotic photoreceptors and INL/GCL neurons expressed TNFα that was required for inducing maximal numbers of proliferating Müller glia. Furthermore, this demonstrates that TNFα plays a general role to initiate Müller glia proliferation in response to the loss of any neuronal cell-type within the zebrafish retina.
Figure 13.

TNFα is required for the maximal number of Müller glia to re-enter the cell cycle in the ouabain-damaged retina. Adult zebrafish were either uninjected (A) or intravitreally injected and electroporated with lissamine-tagged tnfα 5-mismatch morpholinos (tnfα 5-mis, B) or translation-inhibiting tnfα morpholinos (tnfα MO, C) and then intravitreally injected with ouabain to a final concentration of 2 μm. After 3 d, retinal cryosections were immunolabeled for PCNA to detect the nuclei of dividing cells (A–C, green) and stained with To-Pro-3 to identify the nuclear layers (blue). Both controls possessed significantly more PCNA-positive INL cells relative to the tnfα morphant at 3 dpi (D; blue bar, red bar, green bar, respectively; p < 0.01, n = 10). Scale bar: (in A) A–C, 50 μm
Human-recombinant HB-EGF is not sufficient to induce Müller glia proliferation
Because HB-EGF was previously shown to be both necessary and sufficient for Müller glia proliferation (Wan et al., 2012), we sought to examine if HB-EGF was sufficient to induce TNFα expression in either neurons or Müller glia. We intravitreally injected either human recombinant HB-EGF protein (Fig. 14C,D), which was sufficient to stimulate Müller glia proliferation (Wan et al., 2012), or vehicle (1× PBS, 0.1% BSA; Fig. 14A,B). The eyes were collected at either 1 (Fig. 14A,C) and 3 (Fig. 14B,D) dpi and immunolabeled for PCNA to identify proliferating Müller glia (Fig. 14A–D). PCNA-positive INL cell counts were acquired from optic nerve containing sections to ensure consistency between HB-EGF-injected eyes and vehicle controls. HB-EGF-injected retinas contained 1.3 ± 0.3 and 1.6 ± 0.5 PCNA-positive INL cells at 1 and 3 dpi, respectively, which was not significantly different relative to vehicle-injected retinas at 1 and 3 dpi (1.2 ± 0.3 and 1.5 ± 0.4 PCNA-positive cells, respectively; p = 0.84 and 0.87, respectively). The number of PCNA-expressing cells in the ONL of HB-EGF-injected eyes (15.0 ± 2.4 and 20.3 ± 1.7 at 1 and 3 dpi, respectively) was also not statistically different relative to vehicle-injected eyes at 1 and 3 dpi (17.1 ± 3.0 and 17.3 ± 2.8, respectively; p = 0.60 and 0.37, respectively).
Figure 14.
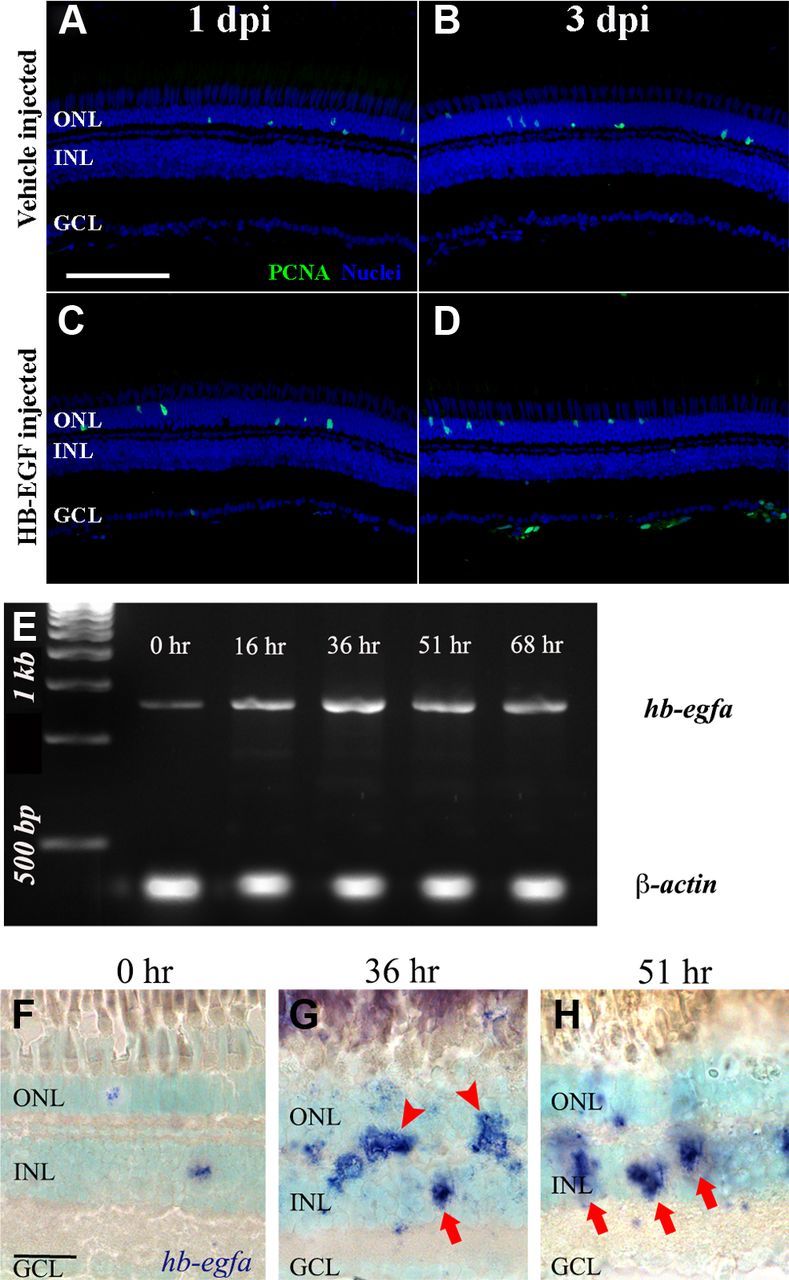
HB-EGF, which increases in expression in the Müller glia of the light-damaged retina, is not sufficient to induce Müller glia proliferation. Adult zebrafish were either injected with 1× PBS, 0.1% BSA vehicle solution (A, B), or human recombinant HB-EGF reconstituted in the vehicle solution (C, D). The eyes were collected at 1 (A, C) and 3 (B, D) dpi and processed for immunolabeling with anti-PCNA antibodies to detect the nuclei of dividing cells and To-Pro-3 nuclear stain (A–D, green and blue, respectively). Dark-adapted albino zebrafish were placed in constant intense light for 0, 16, 36, 51, and 68 h. Total RNA was isolated from the retinas and the hb-egfa and β-actin cDNAs were amplified by RT-PCR and analyzed by gel electrophoresis (E). Dark-adapted albino zebrafish were exposed to constant intense light for 0 h (F), 36 h (G), or 51 h (H) h and retinal cryosections were in situ hybridized with hb-egfa antisense riboprobe and counterstained with To-Pro-3. Low levels of hb-egfa were detected in the INL at 0 h. In the light-damaged retinas, hb-egfa was detected in both the INL and the OPL (G, arrows and arrowheads, respectively). Scale bars: (in A) A–D, 50 μm; (in F) F–H, 20 μm.
Because HB-EGF was not sufficient to induce Müller glia proliferation in the undamaged retina, we examined the expression pattern of hb-egfa mRNA in the light-damaged retina. Using RT-PCR to examine the temporal expression of hb-egfa, we found that the mRNA increased in expression from 0 to 16 h of light and then again at 36 h of light (Fig. 14E). This is in agreement with Wan et al. (2012), which described a rapid increase in hb-egfa expression after the retinal puncture wound. Using in situ hybridization, a low level of hb-egfa was detected in both the ONL and INL in the undamaged retina (Fig. 14F). At 36 h of constant light, there was a dramatic increase of hb-egfa expression in both the ONL and INL (Fig. 14G). Several of the hb-egfa-positive cells in the INL possessed a morphology that is consistent with Müller glia (Fig. 14G, arrows), while the remaining labeled cells likely represent proliferating Müller glia that are migrating to the ONL (Figure 14G, arrowheads; Qin et al., 2009; Wan et al., 2012). At 51 h, the hb-egfa expression localized primarily to the INL and possessed a Müller glial morphology (Fig. 14H, arrows). Surprisingly, morpholino-mediated knockdown of HB-EGFa expression in the light-damaged retina did not significantly reduce the number of proliferating Müller glia relative to either the uninjected or Standard Control morphant light-damaged retinas (data not shown). Together, hb-egfa expression increases in the Müller glia, but it does not appear to play a significant role in Müller glia proliferation in the light-damaged retina.
Discussion
TNFα represents the first and only signal identified that is produced by apoptotic retinal neurons to induce Müller glia proliferation at the initiation of zebrafish retinal regeneration. Photoreceptor-derived TNFα was also required for Ascl1a and Stat3 expression in the Müller glia. In contrast, Lin28a, Ascl1a, and Stat3 are all necessary for TNFα expression in the Müller glia and the maximal number of proliferating Müller glia in the light-damaged retina.
Zebrafish Müller glia respond to retinal damage by re-entering the cell cycle (Yurco and Cameron, 2005; Fausett and Goldman, 2006; Kassen et al, 2007; Fimbel et al., 2007; Thummel et al., 2008a,b; Bailey et al., 2010; Craig et al., 2010). We hypothesized that apoptotic neurons produced a signal that induced Müller glia proliferation. Intravitreal injection of light-damaged retinal homogenates collected after 16 h of constant light was sufficient to induce significantly more proliferating Müller glia, without causing neuronal cell death, relative to undamaged retinal homogenates. While the light-damaged homogenate induced far fewer proliferating Müller glia relative to the light-damaged retina, this could be due to diffusion of the homogenate's signaling molecules into the vitreous, reduced penetration of the signals through the retina, or degradation of the signaling molecules. Additionally, the reduced number of proliferating Müller glia may be due to the absence of any cell death and the retention of cell–cell contacts.
Immunolocalization and in situ hybridization confirmed that TNFα expression increased in apoptotic photoreceptors and later in Müller glia. Knockdown of TNFα expression before the start of the light treatment dramatically reduced TNFα expression throughout the retina and significantly reduced the number of PCNA-labeled INL cells by 82% relative to control retinas. In contrast, knockdown of TNFα expression after 16 h of light treatment inhibited TNFα expression in the Müller glia, but not in photoreceptor cells, the OPL, and NFL. This latter knockdown significantly reduced the number of PCNA-labeled INL cells by 40% relative to controls, similar to the 50% reduction of proliferating Müller glia in the stat3 morphant retina relative to controls (Nelson et al., 2012). This suggests that Müller glial-derived TNFα and Stat3 function at a similar point that induces maximal numbers of proliferating Müller glia. Additionally, knockdown of TNFα expression in only the Müller glia yielded fewer PCNA-labeled INL cell clusters, rather than fewer neuronal progenitor cells per cluster. Because each proliferating INL cell cluster likely represents the neuronal progenitor cells derived from a single proliferating Müller glial cell, the reduced number of proliferating clusters likely corresponds to fewer Müller glia re-entering the cell cycle. Thus, apoptotic photoreceptor-derived TNFα expression induced an initial population of Müller glia to re-enter the cell cycle, while the Müller glia-derived TNFα expression induced a secondary population of proliferating Müller glia.
These two populations of proliferating Müller glia are consistent with a model of dying photoreceptors activating a signaling cascade (Lin28a activating Stat3 through Ascl1a) within a subset of Müller glia (Primary Proliferating Müller glia, PPMg; Fig. 14; Ramachandran et al., 2010; Nelson et al., 2012). Lin28a and Ascl1a, but not Stat3, are required for PPMg proliferation; however, all three are necessary to stimulate a subsequent subset of Müller glia to proliferate (Secondary Proliferating Müller glia, SPMg). This study demonstrates that TNFα is a signal produced by dying neurons that is necessary for expressing Stat3 and Ascl1a in Müller glia, as well as Müller glia proliferation (Fig. 15). Additionally, knockdown of Lin28a, Ascl1a, or Stat3 dramatically reduced TNFα expression only in the Müller glia, suggesting that TNFα is a signal produced by the PPMg to induce the SPMg to proliferate in a Stat3-dependent manner. Consistent with that hypothesis, knockdown of TNFα expression after 16 h of light significantly reduced the number of proliferating Müller glia or clusters of proliferating INL cells. These data indicate that TNFα is required for Müller glia to become responding progenitor cells, which enter the cell cycle, but not for the subsequent proliferation and expansion of the retinal progenitors.
Figure 15.

Model of TNFα signaling in the initiation and amplification of Müller glia proliferation during retinal regeneration. The light-damaged zebrafish retina possesses three classes of Müller glia: PPMg, SPMg, and Quiescent Müller glia (QMg). Apoptotic retinal neurons upregulate TNFα expression, which activates Ascl1a expression in the PPMg. Ascl1a drives the PPMg to re-enter the cell cycle (express PCNA) and induces Stat3 expression. Stat3 activates TNFα expression in the PPMg, which in turn, activates the expression of Ascl1a in the SPMg. The Ascl1a in the SPMg stimulates their proliferation and induces Stat3 expression. Thus, the PPMg proliferate in a TNFα-dependent and Stat3-independent manner, while re-entry of the SPMg into the cell cycle requires Stat3- and Ascl1a-dependent TNFα expression in the PPMg. The QMg express Stat3, but do not express Ascl1a and do not proliferate. It remains unknown what activates Stat3 expression in the QMg.
While knockdown of TNFα expression significantly reduced the number of proliferating Müller glia during the light treatment, the number eventually recovered and the rods and cones regenerated (Fig. 11). This suggests that TNFα expression may increase after the transient morpholino-mediated knockdown ends. However, this TNFα expression would have to come from cells other than dying rods and cones, which are largely lost by 3 or 4 d of light damage. These TNFα-expressing cells could be the few remaining photoreceptors or some other retinal cell type that produces sufficient TNFα to induce the delayed Müller glia proliferation response. Alternatively, a delayed signal other than TNFα may induce Müller glia to re-enter the cell cycle, although the identity and source of this signal is unknown.
Morpholino-mediated knockdown of HB-EGF in puncture-damaged retinas and injecting human HB-EGF into undamaged eyes demonstrated that HB-EGF was both necessary and sufficient for Müller glia proliferation (Wan et al., 2012). Because hb-egf was expressed in Müller glia rather than damaged neurons (Wan et al., 2012; Fig. 14), it was unlikely to be required for TNFα expression in the dying neurons. To confirm this, we tested if HB-EGF-induced proliferating Müller glia in the undamaged retinas induced TNFα expression. However, we could not induce Müller glia proliferation by intravitreal injection of HB-EGF into undamaged retinas. There are several reasons why our results differed from those of Wan et al. (2012). We used a sapphire blade to make an incision in the eye and inserted the needle through the opening to avoid the blunt force trauma of forcing the needle to penetrate into the eye. Further, we used a blunt needle rather than a beveled needle (Wan et al., 2012). The latter requires the needle to be inserted beyond the beveled edge to successfully inject the solution into the vitreous, which could damage the retina. Finally, injecting 2.0 μl of 100 ng/μl HB-EGF (Wan et al., 2012), which is greater than the adult intravitreal volume (Fimbel et al., 2007), could result in high intraocular pressure and retinal damage. Because of this concern, we injected either 1.0 μl of 200 ng/μl or 2.0 μl of 100 ng/μl recombinant HB-EGF solution into the vitreous, which represents the same amount of recombinant HB-EGF described previously (200 ng; Wan et al., 2012). While injecting either HB-EGF volume resulted in some solution flowing out of the vitreous, neither volume induced significant numbers of proliferating Müller glia relative to controls. It is possible that injecting 2.0 μl without the prior incision minimized the outflow and significantly increased the intraocular pressure. Some combination of these differences likely resulted in retinal stress or damage and HB-EGF could be sufficient to induce Müller glia proliferation under these conditions (Wan et al., 2012).
Because HB-EGF was not sufficient to induce Müller glia proliferation in the undamaged retina, we tested if it was necessary for Müller glia proliferation in the light-damaged retina to examine the relationship of HB-EGF and TNFα. We injected and electroporated hb-egfa morpholinos into the retina immediately before starting the constant light treatment and found that it failed to significantly reduce the number of proliferating Müller glia relative to the Standard Control morphant retina (data not shown). While this difference between our result and Wan et al. (2012) could be due to the different damage models, HB-EGF is not required for inducing Müller glia proliferation in response to photoreceptor damage.
Ouabain damage to inner retinal neurons (Fimbel et al., 2007) increased TNFα expression in ganglion and INL cells at 1 dpi and in Müller glia by 3 dpi. Knockdown of TNFα before ouabain injection resulted in significantly fewer proliferating Müller glia relative to controls, suggesting that TNFα stimulates Müller glia to re-enter the cell cycle in response to retinal damage. However, numerous PCNA-positive Müller glia are present in the tnfα morphant retinas 3 d after ouabain injection (Fig. 13D). This suggests that at least one other unidentified TNFα-independent signal induces Müller glia proliferation.
The mechanism by which TNFα induces Stat3 expression in all Müller glia and Ascl1a in only proliferating Müller glia remains unclear (Kassen et al., 2007; Nelson et al., 2012). While TNFα expression may stimulate Stat3 expression in all Müller glia, it is unclear why Ascl1a, which lies downstream of TNFα but upstream of Stat3 (Nelson et al., 2012), is expressed in only the proliferating Müller glia. It is possible that a regulatory loop exists between Stat3 and Ascl1a. Alternatively, TNFα may function with another retinal damage signal to differentially induce Ascl1a expression in only proliferating Müller glia and Stat3 in all Müller glia. The identification of the TNFα receptor(s) and intracellular proteins regulated by the TNFα-bound receptor(s) will elucidate this mechanism. Ultimately, identifying the signaling pathways involved in Müller glia dedifferentiation and proliferation in the regenerating zebrafish model may reveal important mechanisms and therapeutic targets for enhancing mammalian retinal regeneration. Properly regulating these pathways may potentially induce Müller glia dedifferentiation and proliferation or promote exogenous pluripotent cells to actively and correctly regenerate lost neurons in the diseased or damaged human retina.
Footnotes
This study was supported by a grant from the National Eye Institute–National Institutes of Health to D.R.H. (R01-EY018417) and the Center for Zebrafish Research, University of Notre Dame. We appreciate the assistance of the Freimann Life Science technicians for their care of the zebrafish and members of the Hyde lab for thoughtful discussions.
The authors declare no competing financial interests.
References
- Bailey TJ, Fossum SL, Fimbel SM, Montgomery JE, Hyde DR. The inhibitor of phagocytosis, O-phospho-L-serine, suppresses Müller glia proliferation and cone cell regeneration in the light-damaged zebrafish retina. Exp Eye Res. 2010;91:601–612. doi: 10.1016/j.exer.2010.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardos RL, Barthel LK, Meyers JR, Raymond PA. Late-stage neuronal progenitors in the retina are radial Müller glia that function as retinal stem cells. J Neurosci. 2007;27:7028–7040. doi: 10.1523/JNEUROSCI.1624-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, Osborne NN, Reichenbach A. Müller cells in the healthy and diseased retina. Prog Retin Eye Res. 2006;25:397–424. doi: 10.1016/j.preteyeres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Brockerhoff SE, Fadool JM. Genetics of photoreceptor degeneration and regeneration in zebrafish. Cell Mol Life Sci. 2011;68:651–659. doi: 10.1007/s00018-010-0563-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calinescu AA, Vihtelic TS, Hyde DR, Hitchcock PF. Cellular expression of midkine-a and midkine-b during retinal development and photoreceptor regeneration in zebrafish. J Comp Neurol. 2009;514:1–10. doi: 10.1002/cne.21999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FK, Lenardo MJ. A crucial role for p80 TNF-R2 in amplifying p60 TNF-R1 apoptosis signals in T lymphocytes. Eur J Immunol. 2000;30:652–660. doi: 10.1002/1521-4141(200002)30:2<652::AID-IMMU652>3.3.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Chen CF, Chen Y, Dai K, Chen PL, Riley DJ, Lee WH. A new member of the hsp90 family of molecular chaperones interacts with the retinoblastoma protein during mitosis and after heat shock. Mol Cell Biol. 1996;16:4691–4699. doi: 10.1128/mcb.16.9.4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect neurons against metabolic-excitotoxic insults and promote maintenance of calcium homeostasis. Neuron. 1994;12:139–153. doi: 10.1016/0896-6273(94)90159-7. [DOI] [PubMed] [Google Scholar]
- Craig SE, Thummel R, Ahmed H, Vasta GR, Hyde DR, Hitchcock PF. The zebrafish galectin Drgal1–L2 is expressed by proliferating Müller glia and photoreceptor progenitors and regulates the regeneration of rod photoreceptors. Invest Ophthalmol Vis Sci. 2010;51:3244–3252. doi: 10.1167/iovs.09-4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do H, Pyo S, Sohn EH. Suppression of iNOS expression by fucoidan is mediated by regulation of p38 MAPK, JAK/STAT, AP-1 and IRF-1, and depends on up-regulation of scavenger receptor B1expression in TNF-α-and IFN-γ-stimulated C6 glioma cells. J Nutr Biochem. 2010;21:671–679. doi: 10.1016/j.jnutbio.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Fausett BV, Goldman D. A role for alpha1 tubulin-expressing Müller glia in regeneration of the injured zebrafish retina. J Neurosci. 2006;26:6303–6313. doi: 10.1523/JNEUROSCI.0332-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fausett BV, Gumerson JD, Goldman D. The proneural basic helix-loop-helix gene ascl1a is required for retina regeneration. J Neurosci. 2008;28:1109–1117. doi: 10.1523/JNEUROSCI.4853-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimbel SM, Montgomery JE, Burket CT, Hyde DR. Regeneration of inner retinal neurons after intravitreal injection of ouabain in zebrafish. J Neurosci. 2007;27:1712–1724. doi: 10.1523/JNEUROSCI.5317-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AJ, Scott MA, Tuten W. Mitogen-activated protein kinase-signaling stimulates Müller glia to proliferate in acutely damaged chicken retina. Glia. 2009;57:166–181. doi: 10.1002/glia.20743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisch VC, Fraser B, Allison WT. Investigating regeneration and functional integration of CNS neurons: lessons from zebrafish genetics and other fish species. Biochim Biophys Acta. 2011;1812:364–380. doi: 10.1016/j.bbadis.2010.10.012. [DOI] [PubMed] [Google Scholar]
- Guo D, Dunbar JD, Yang CH, Pfeffer LM, Donner DB. Induction of Jak/STAT signaling by activation of the type 1 TNF receptor. J Immunol. 1998;160:2742–2750. [PubMed] [Google Scholar]
- Gupta S. Molecular steps of tumor necrosis factor receptor-mediated apoptosis. Curr Mol Med. 2001;1:317–324. doi: 10.2174/1566524013363780. [DOI] [PubMed] [Google Scholar]
- Hitchcock P, Kakuk-Atkins L. The basic helix-loop-helix transcription factor neuroD is expressed in the rod lineage of the teleost retina. J Comp Neurol. 2004;477:108–117. doi: 10.1002/cne.20244. [DOI] [PubMed] [Google Scholar]
- Karl MO, Reh TA. Regenerative medicine for retinal diseases: activating endogenous repair mechanisms. Trends Mol Med. 2010;16:193–202. doi: 10.1016/j.molmed.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassen SC, Ramanan V, Montgomery JE, T Burket C, Liu CG, Vihtelic TS, Hyde DR. Time course analysis of gene expression during light-induced photoreceptor cell death and regeneration in albino zebrafish. Dev Neurobiol. 2007;67:1009–1031. doi: 10.1002/dneu.20362. [DOI] [PubMed] [Google Scholar]
- Kassen SC, Thummel R, Campochiaro LA, Harding MJ, Bennett NA, Hyde DR. CNTF induces photoreceptor neuroprotection and Müller glial cell proliferation through two different signaling pathways in the adult zebrafish retina. Exp Eye Res. 2009;88:1051–1064. doi: 10.1016/j.exer.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Kubota K, Inoue K, Hashimoto R, Kumamoto N, Kosuga A, Tatsumi M, Kamijima K, Kunugi H, Iwata N, Ozaki N, Takeda M, Tohyama M. Tumor necrosis factor receptor-associated protein 1 regulates cell adhesion and synaptic morphology via modulation of N-cadherin expression. J Neurochem. 2009;110:496–508. doi: 10.1111/j.1471-4159.2009.06099.x. [DOI] [PubMed] [Google Scholar]
- Liu D, Hu J, Agorreta J, Cesario A, Zhang Y, Harris AL, Gatter K, Pezzella F. Tumor necrosis factor receptor-associated protein 1 (TRAP1) regulates genes involved in cell cycle and metastases. Cancer Lett. 2010;296:194–205. doi: 10.1016/j.canlet.2010.04.017. [DOI] [PubMed] [Google Scholar]
- MacEwan DJ. TNF receptor subtype signalling: differences and cellular consequences. Cell Signal. 2002;14:477–492. doi: 10.1016/S0898-6568(01)00262-5. [DOI] [PubMed] [Google Scholar]
- Matthews RP, Lorent K, Mañoral-Mobias R, Huang Y, Gong W, Murray IV, Blair IA, Pack M. TNFalpha-dependent hepatic steatosis and liver degeneration caused by mutation of zebrafish S-adenosylhomocysteine hydrolase. Development. 2009;136:865–875. doi: 10.1242/dev.027565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miscia S, Marchisio M, Grilli A, Di Valerio V, Centurione L, Sabatino G, Garaci F, Zauli G, Bonvini E, Di Baldassarre A. Tumor necrosis factor alpha (TNF-alpha) activates Jak1/Stat3-Stat5B signaling through TNFR-1 in human B cells. Cell Growth Differ. 2002;13:13–18. [PubMed] [Google Scholar]
- Montgomery JE, Parsons MJ, Hyde DR. A novel model of retinal ablation demonstrates that the origin of regenerated rod photoreceptors is dependent on the extent of damage. J Comp Neurol. 2010;518:800–814. doi: 10.1002/cne.22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T, Miyamoto T, Yoshida H, Asakawa M, Kawasumi M, Kobayashi T, Morioka H, Chiba K, Toyama Y, Yoshimura A. IL-1β and TNFα-initiated IL-6–STAT3 pathway is critical in mediating inflammatory cytokines and RANKL expression in inflammatory arthritis. Int Immunol. 2011;23:701–712. doi: 10.1093/intimm/dxr077. [DOI] [PubMed] [Google Scholar]
- Nelson CM, Hyde DR. Müller glia as a source of neuronal progenitor cells to regenerate the damaged zebrafish retina. Adv Exp Med Biol. 2012;723:425–430. doi: 10.1007/978-1-4614-0631-0_54. [DOI] [PubMed] [Google Scholar]
- Nelson CM, Gorsuch RA, Bailey TJ, Ackerman KM, Kassen SC, Hyde DR. Stat3 defines three populations of Müller glia and is required for initiating maximal Müller glia proliferation in the regenerating zebrafish retina. J Comp Neurol. 2012;520:4294–4311. doi: 10.1002/cne.23213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, Whitney N, Wu Y, Tian C, Dou H, Zhou Y, Zheng J. HIV-1-infected and/or immune-activated macrophage-secreted TNF-alpha affects human fetal cortical neural progenitor cell proliferation and differentiation. Glia. 2008;56:903–916. doi: 10.1002/glia.20665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, Sun L, Jia B, Lan X, Zhu B, Wu Y, Zheng J. HIV-1-infected and immune-activated macrophages induce astrocytic differentiation of human cortical neural progenitor cells via the STAT3 pathway. PLoS One. 2011;6:e19439. doi: 10.1371/journal.pone.0019439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell C, Elsaeidi F, Goldman D. Injury-dependent Müller glia and ganglion cell reprogramming during tissue regeneration requires Apobec2a and Apobec2b. J Neurosci. 2012;32:1096–1109. doi: 10.1523/JNEUROSCI.5603-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z, Barthel LK, Raymond PA. Genetic evidence for shared mechanisms of epimorphic regeneration in zebrafish. Proc Natl Acad Sci U S A. 2009;106:9310–9315. doi: 10.1073/pnas.0811186106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Fausett BV, Goldman D. Ascl1a regulates Müller glia dedifferentiation and retinal regeneration through a Lin-28-dependent, let-7 microRNA signalling pathway. Nat Cell Biol. 2010;12:1101–1107. doi: 10.1038/ncb2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanatto T, Cesquini M, Amaral ME, Roman EA, Moraes JC, Torsoni MA, Cruz-Neto AP, Velloso LA. TNF-alpha acts in the hypothalamus inhibiting food intake and increasing the respiratory quotient–effects on leptin and insulin signaling pathways. Peptides. 2007;28:1050–1058. doi: 10.1016/j.peptides.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Rowan S, Cepko CL. Genetic analysis of the homeodomain transcription factor Chx10 in the retina using a novel multifunctional BAC transgenic mouse reporter. Dev Biol. 2004;271:388–402. doi: 10.1016/j.ydbio.2004.03.039. [DOI] [PubMed] [Google Scholar]
- Tartaglia LA, Pennica D, Goeddel DV. Ligand passing: the 75-kDa tumor necrosis factor (TNF) receptor recruits TNF for signaling by the 55-kDa TNF receptor. J Biol Chem. 1993;268:18542–18548. [PubMed] [Google Scholar]
- Thomas JL, Nelson CM, Luo X, Hyde DR, Thummel R. Characterization of multiple light damage paradigms reveals regional differences in photoreceptor loss. Exp Eye Res. 2012;97:105–116. doi: 10.1016/j.exer.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thummel R, Kassen SC, Montgomery JE, Enright JM, Hyde DR. Inhibition of Müller glial cell division blocks regeneration of the light-damaged zebrafish retina. Dev Neurobiol. 2008a;68:392–408. doi: 10.1002/dneu.20596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thummel R, Kassen SC, Enright JM, Nelson CM, Montgomery JE, Hyde DR. Characterization of Müller glia and neuronal progenitors in zebrafish adult retinal regeneration. Exp Eye Res. 2008b;87:433–444. doi: 10.1016/j.exer.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thummel R, Enright JM, Kassen SC, Montgomery JE, Bailey TJ, Hyde DR. Pax6a and Pax6b control novel regulatory points during neuronal progenitor cell proliferation in the regenerating zebrafish retina. Exp Eye Res. 2010;90:572–582. doi: 10.1016/j.exer.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihtelic TS, Hyde DR. Light-induced rod and cone cell death and regeneration in the adult albino zebrafish (Danio rerio) retina. J Neurobiol. 2000;44:289–307. doi: 10.1002/1097-4695(20000905)44:3<289::AID-NEU1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Vihtelic TS, Doro CJ, Hyde DR. Cloning and characterization of six zebrafish photoreceptor opsin cDNAs and immunolocalization of their corresponding proteins. Vis Neurosci. 1999;16:571–585. doi: 10.1017/s0952523899163168. [DOI] [PubMed] [Google Scholar]
- Vihtelic TS, Soverly JE, Kassen SC, Hyde DR. Retinal regional differences in photoreceptor cell death and regeneration in light-lesioned albino zebrafish. Exp Eye Res. 2006;82:558–575. doi: 10.1016/j.exer.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Wan J, Ramachandran R, Goldman D. HB-EGF is necessary and sufficient for Müller glia dedifferentiation and retina regeneration. Dev Cell. 2012;22:334–347. doi: 10.1016/j.devcel.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wu TR, Cai S, Welte T, Chin YE. Complex to inhibit NF-kB activation factor alpha receptor 1-TRADD signaling Stat1 as a component of tumor necrosis. Mol Cell Biol. 2000;20:4505–4512. doi: 10.1128/MCB.20.13.4505-4512.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurco P, Cameron DA. Responses of Müller glia to retinal injury in adult zebrafish. Vision Res. 2005;45:991–1002. doi: 10.1016/j.visres.2004.10.022. [DOI] [PubMed] [Google Scholar]