

Abstract
Caveolin proteins physically interact with and compartmentalize membrane-localized signaling proteins to facilitate high-fidelity intracellular signaling. Though primarily studied outside the nervous system, recent investigations have revealed that caveolin proteins are key modulators of a variety of neuronal intracellular signaling pathways. Through both protein aggregation and segregation, caveolin proteins can exert positive and negative influences on intracellular signaling. This review will detail recent findings regarding caveolin function in the brain.
Keywords: Caveolin, Caveolae, Intracellular signaling, Neuron, Membrane receptor, G-protein
Caveolins: Background
Originally the cellular membrane was thought of as a homogenous medium through which membrane receptor proteins freely circulated [1, 2]. This model posited that neurotransmitter and neuromodulator receptors relied solely on random diffusion for both ligand binding as well as activation of the appropriate downstream signaling molecules. Yet, this diffusion model does not account for the speed and fidelity with which intracellular signaling occurs. It is now clear that the cellular membrane is a highly organized structure in which discrete structural and functional membrane microdomains facilitate rapid and reliable signal transduction. To fully appreciate cellular function, we require a thorough understanding of the mechanisms by which membrane microdomains influence intracellular signaling.
Composed principally of caveolin proteins (CAVs), caveolae were among the first described membrane microdomains in epithelial cells [3]. Caveolae, literally meaning “little caves”, appear as flask-like invaginations in the plasma membrane of these cells [4]. There are three CAV proteins: caveolin 1 (CAV1), caveolin 2 (CAV2), and caveolin 3 (CAV3) [5–7]. CAV1 and CAV2 have overlapping patterns of expression throughout numerous tissues [8–11], while CAV3 is widely expressed in muscle [7, 12–14] as well the nervous system [9]. Disruption of CAV1 or CAV3 expression results in a loss of caveolae formation [15, 16]. Conversely, disruption of CAV2 expression does not affect caveolae formation in vivo, and thus it has been hypothesized that CAV2 only forms caveolae as hetero-oligomers with CAV1 (and possibly CAV3) [17].
It is thought that all CAVs share a similar tertiary structure. CAVs are hairpin structures with both the C- and N-termini residing in the cytoplasm. Post-translational lipid-modification proximate to the carboxy terminus, along with the lipophilic hairpin motif, likely facilitate CAV interaction with and localization to the plasma membrane. All three CAVs contain a caveolin scaffolding domain (CSD) that serves as the docking site for many intracellular signaling proteins, including but not limited to, Gα, Gβγ, adenylyl cyclase (AC), protein kinase A, protein kinase C, mitogen-activated protein kinase, c-src, PI3-kinase, and nitric oxide synthase (NOS) [4]. Given the plethora of signaling molecules with which the CSD interacts, CAVs are ideally suited to coordinate and regulate rapid, membrane-initiated signaling pathways.
Despite widespread expression in most other tissues, it was initially believed that CAV expression in the nervous system was limited to glial cells, and thus did not play a prominent role in neuronal intracellular signaling [18]. More recent studies have reported expression of all three CAV isoforms in neurons [8, 19, 20]. CAV action in neurons diverges from known mechanisms in non-neuronal cells in two significant ways. First, since immature neuronal-like cells have been demonstrated to lose CAV expression upon differentiation, investigators must carefully consider developmental influences when assaying for CAV expression and function in neurons [21]. Second, CAV expression in neurons has been detected independently of any conclusive evidence for caveolae [22]. CAV interaction with cytoskeletal components [23] may be sufficient to compartmentalize neuronal signaling molecules.
While little is known about CAV interaction with the cytoskeleton in neurons, post-synaptic density proteins (PSDs) are well-studied neuronal structural proteins that facilitate intracellular signaling at the post-synaptic site. Thus, one of the most interesting questions within the nervous system relates to the relationship between CAVs and PSDs. Proteins in this class (such as PSD-95) contain PDZ domains, which function similarly to the CSD in CAVs, allowing the protein to interact with various signaling and structural moieties [24]. As their name implies, PSD proteins are enriched at postsynaptic sites where they facilitate neurotransmitter signaling. It has yet to be determined whether CAVs also display a similar enrichment to post-synaptic sites. Despite similar functions as structural proteins that regulate intracellular signaling moieties, little is known about the potential interactions between PSDs and CAVs. One exception is a 1998 study that reported PSD co-localization to CAV1-enriched fractions [25]. These data suggest a hypothesis whereby CAVs and PSDs act cooperatively at the synapse to influence rapid intracellular signaling. Since little is known about the sub-cellular localization of membrane CAVs in neurons, future experiments should determine whether CAVs localize to the post-synaptic site, as well as whether CAVs and PSDs interact and co-localize in intact neurons. Data from these investigations will contribute to building a comprehensive framework for integrating the mechanisms by which these structural proteins interact to modulate intracellular signaling.
Independent of PSD interactions, two lines of evidence suggest that CAVs may play a role in synaptic maintenance and stabilization. First, CAVs bind cholesterol and require cholesterol for protein oligomerization [22, 26]. Second, cholesterol has been shown to play critical roles in synapse development in retinal ganglion cells [27], as well as synapse maintenance at the neuromuscular junction [28]. These lines of evidence suggest a hypothesis whereby localization of cholesterol-bound CAV to the synapse is a critical step for post-synaptic development and stabilization. Interactions with membrane constituents like cholesterol may represent a means of “secondary modulation” whereby CAVs influence the structure and composition of the synapse to facilitate the presence and function of intracellular signaling moieties.
The presence of all three CAV isoforms in the brain implies that they may exert profound regulatory effects on neurons. Indeed, CAVs have been demonstrated to play critical roles in regulating neuronal signaling pathways and overall nervous system function. Although initially described as negative regulators of intracellular signaling, a seminal study in 2004 was the first to demonstrate that CAVs can also facilitate intracellular signaling [29]. The remainder of this discussion will describe some specific examples of CAV-mediated regulation of neuronal intracellular signaling pathways in the context of their ability to exert both positive and negative influences on intracellular signaling. Since many studies investigating CAV regulation of neuronal signaling also utilize non-neuronal preparations, we will include several relevant examples in our discussion (see Table 1). Based on these initial studies, it is clear that we are only beginning to understand the full scope of CAV function. Future experiments will require a more comprehensive study of CAV isoforms, examine potential differences between cell types, and examine both short- and long-term effects of CAVs on receptor signaling events.
Table 1.
Summary of CAV regulation of neuronal-relevant intracellular signaling discussed in this review
| Signaler | Preparation | CAV isoform | Direction of regulation | Reference |
|---|---|---|---|---|
| 5HT2AR (Gαq) | HEK293/C6 glioma | CAV1 | Positive | [40] |
| ER/mGluR (G αq /G αi/o ) | Hippocampal and striatal neurons | CAV1/CAV3 | Positive | [ 20 ] |
| Group I mGluR (G αq ) | Hippocampal neurons/HEK293 | CAV1 | Both | [ 46 ] |
| L-type Ca2+ Channel | Smooth, skeletal, cardiac muscle | CAV1 | Positive | [47–50] |
| NMDAR | Cortical neurons | CAV1 | Positive | [ 51 ] |
| EEAC1 | C6 glioma cells | CAV1/CAV2? | Both | [56] |
| GLUT4 | Skeletal muscle cells | CAV3 | Positive | [57] |
| Cellular prion protein | Neuronal cell lines (N2A, 1C11, PC12, GN11) | CAV1 | Positive | [61–65] |
| Adenylate cyclase | C6 Glioma | CAV1 | Negative | [68] |
| Dopamine I receptor (Gαs) | COS7/HEK293 | CAV1 | Negative | [30] |
| bFGF | N2A neuroblastoma | CAV1 | Negative | [69] |
| iNOS | SK-N-MC neuroblastoma | CAV1 | Negative | [73] |
| AMPAR | Hippocampal neurons | CAV1 | Negative | [ 74 ] |
Studies conducted in neurons are highlighted in bold
Mechanisms of intracellular signaling regulation
CAVs influence membrane-initiated intracellular signaling via the clustering of proteins, the segregation of proteins, and the trafficking of proteins to and from the membrane (Fig. 1). Clustering refers to the ability of CAVs to facilitate rapid and specific intracellular signaling by assembling the appropriate signaling molecules into close physical proximity. Since there is no evidence to date supporting the existence of caveolae-like invaginations in neurons, this physical organization may occur in the context of membrane microdomains. Given that CAVs have been shown to interact with a wide variety of signaling proteins, this modulation is thought to occur via direct protein–protein interactions between CAVs and the target signaling molecules. Notably, it has also been demonstrated that in addition to facilitating the activation of signaling pathways, CAVs may isolate certain proteins from obligatory signaling partners to dampen signaling [30]. This capacity for bidirectional regulation is evidence of the profound influence that CAVs may have on intracellular signaling.
Fig. 1.
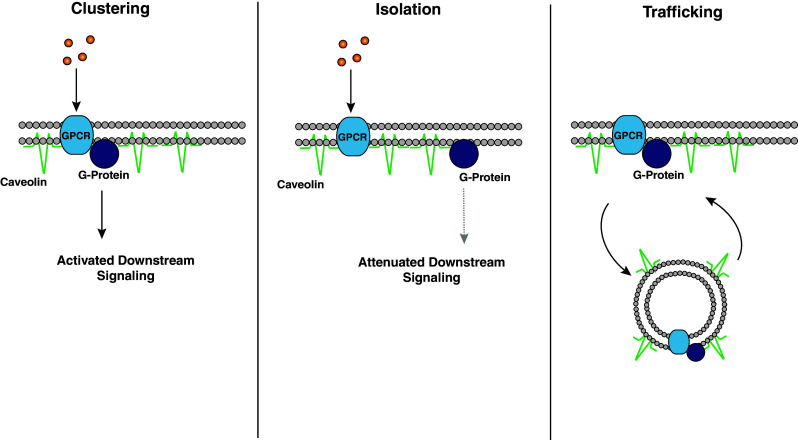
Examples of CAV-dependent modulation of intracellular signaling. a Clustering: CAVs can pair a GPCR and its obligate G-protein to facilitate intracellular signaling. b Isolation: CAVs can segregate a GPCR from a G-protein to attenuate intracellular signaling. c Trafficking: CAVs can transport membrane-localized signaling moieties to and from the plasma membrane to exert both positive and negative regulation of intracellular signaling
Trafficking, an additional mechanism by which CAVs regulate membrane-initiated signaling, refers to the ability of CAVs to move membrane receptors and intracellular signaling molecules to and from the plasma membrane [30–38]. Notably, this mechanism of action also allows for CAV-mediated bidirectional regulation of signal transduction. This form of trafficking provides multifaceted signaling dynamics in addition to the immediate presence or absence of a signaling unit in the membrane. For example, receptors may undergo endocytosis, a process of initial negative regulation. However, receptor endocytosis may provide protection from receptor desensitization and/or degradation. This would allow for receptor reinsertion into the membrane, ultimately providing a positive influence upon intracellular signaling [39]. That said, we will use positive/negative classification in reference to the immediate effect on the signaling molecule in question.
Positive regulation by CAVs
5-HT2AR signaling
CAVs facilitate intracellular signaling by clustering relevant signaling proteins into discrete functional microdomains, presumably through the CSD. Important neuron-relevant signaling molecules positively regulated by CAVs (specifically CAV1) appear to be several GPCRs that interact with Gαq G-proteins. For instance, CAV1 has been shown to facilitate both GPCR–Gαq interactions, as well as Gαq-mediated activation of downstream signalers such as extracellular signal-regulated kinase (ERK).
Using both transfected HEK293 cells and C6 glioma cells, Bhatnagar et al. [40] demonstrated that CAV1 positively regulates signaling of the serotonin type 2A receptor (5-HT2AR). Specifically, the authors found that while CAV1 is not required for the membrane targeting of 5-HT2ARs, it does interact with 5-HT2AR in transfected cell lines and whole-brain homogenate, supporting a role for CAV regulation of 5-HT2ARs in nervous system function. It should be noted that a limitation of using whole-brain homogenate is an inability to discriminate contributions from glia and neurons. The authors also found that this interaction likely facilitates downstream signaling. Specifically, CAV1 knock-down in C6 glioma cells results in abrogation of 5-HT2AR- and P2Y purinergic receptor-mediated intracellular Ca2+ mobilization (both Gαq-mediated). Furthermore, CAV1 knock-down in these cells results in a dysregulation of 5-HT2AR-mediated ERK signaling.
The exact nature by which CAV1 regulates the above signaling pathways is not entirely clear. Given that CAV1 expression is not necessary for targeting the receptor to the membrane surface, CAV1 must regulate 5-HT2ARs independent of receptor trafficking. The authors reported that CAV1 knock-down modestly attenuates signaling by a constitutively active Gαq subunit, and that CAV1 potentiates the interaction between Gαq and the 5-HT2AR. These data suggest a model whereby CAV1 positively regulates Gαq signaling by spatially congregating the receptor and the G-protein into a functional microdomain to facilitate efficacious activation of the signaling pathway upon neurotransmitter binding to the receptor. Importantly, these data from non-neuronal cells have generated testable hypotheses regarding CAV facilitation of 5-HT2AR and Gαq signaling which can now be examined in intact neuronal systems.
Steroid hormone/mGluR signaling
Though CAVs were not initially thought to be expressed in neurons [18], as early as 2004 it was hypothesized that in neurons, caveolar-like microdomains (CLMs) may localize steroid hormone receptors to the membrane as a novel way for steroids to facilitate activation of intracellular signaling pathways [41]. While caveolae have yet to be identified in neurons, CAVs have since been shown to be expressed in neurons [8, 19, 20]. Furthermore, a report from our lab that CAVs modulate rapid estrogen signaling in hippocampal neurons is consistent with these initial models of membrane-localized steroid hormone signaling [20].
Estrogen bidirectionally regulates the phosphorylation state of the activity-dependent transcription factor CREB in cultured hippocampal neurons [42]. Estrogen enhancement of CREB phosphorylation is mediated via estrogen binding to membrane-localized estrogen receptor alpha (ERα), and subsequent activation of mGluR1a (Gαq) signaling. In contrast, when a hippocampal neuron is depolarized in the presence of estrogen, estrogen binding to membrane-localized ERα or estrogen receptor β (ERβ) leads to mGluR2-(Gαi/o) dependent inhibition of AC, resulting in a subsequent decrease in L-type Ca2+ channel-mediated CREB phosphorylation.
In a follow-up study, CAVs were found responsible for functionally (and probably physically) segregating these two discrete neuronal signaling pathways [20]. Specifically, inhibition of CAV1 function or expression resulted in an abolishment of estrogen-induced CREB phosphorylation. Importantly, inhibition of CAV1 did not inhibit CREB phosphorylation that is induced by the mGluR1 agonist DHPG, suggesting that CAV1 functions by promoting or facilitating an interaction between ERα and mGluR1. Analogously, inhibition of CAV3 function resulted in a loss of the estrogen-mediated inhibition of L-type Ca2+ channel-dependent CREB phosphorylation. Since inhibition of L-type Ca2+ channel-mediated CREB phosphorylation by the mGluR2 agonist APDC is only minimally affected by CAV3 knock-down, CAV3 again likely facilitates signaling by promoting an interaction between mGluR2 and ERα/β. A similar mechanism occurs in striatal neurons as disruption of CAV1 eliminates ERα/mGluR5-mediated CREB phosphorylation, while disruption of CAV3 blocks ERα/β/mGluR3-mediated inhibition of L-type Ca2+ channel-mediated CREB phosphorylation (unpublished observations). Notably, the mechanism by which CAVs promote 5-HT2AR signaling (coupling GPCR to G-protein) is distinct from the mechanism by which they promote ER/mGluR signaling (coupling receptor to receptor). To determine whether CAVs facilitate distinct signaling pathways via diverse mechanisms, future studies will need to examine several Gαq signaling pathways in an intact neuronal preparation.
In support of a model where different CAVs facilitate ER membrane localization and interaction with distinct mGluRs, our co-immunoprecipitation experiments indicate that in neurons CAV1 interacts with ERα, while CAV3 interacts with both ERα and ERβ (unpublished observations). While these data suggest that CAVs function by facilitating the interactions between ERs and mGluRs at the membrane surface, they do not preclude the possibility that CAV1/3 also regulate these signaling pathways by trafficking ERα and ERβ to the membrane surface. This possibility is intriguing given the fact that ERs were initially thought of as solely ligand-activated transcription factors that do not localize to or function at the membrane surface [43, 44]. Thus, CAVs may function to traffic and localize non-canonical membrane-signaling proteins, thereby greatly amplifying the potential intracellular signaling opportunities in neurons.
Importantly, CAV1 regulation is limited to the ERα-mGluR1/5 pathway, while CAV3 regulation is limited to the ERα/β-mGluR2/3 pathway [20]. Together, these data suggest a model whereby CAV1 and CAV3 physically segregate these signaling proteins into either CAV1- or CAV3-containing microdomains. Consistent with this model, CAV1 was found not to co-localize with ERβ in a hippocampal-derived cell line [45]. This leads to the intriguing possibility that in the same neuron, distinct signaling pathways can be physically segregated into discrete microdomains that are comprised of different CAV isoforms. Thus, expression of signaling molecules may not be sufficient for a neuron to exhibit a given intracellular signaling pathway. Rather, function of CAVs or other structural proteins may be necessary for functional expression of that signaling pathway.
Francesconi et al. [46] recently demonstrated that CAVs can also directly interact with and regulate the function of Group I mGluRs. Using whole-brain homogenate, they reported that both mGluR1 and mGluR5 interact with CAV1 in the hippocampus and cerebellum (with the caveat that glial contributions cannot be ruled out here). In cell lines, they demonstrated that CAV1 facilitates surface expression of both mGluR1 and mGluR5. Further, mutation of the consensus CAV-binding-domain in mGluR1 attenuated its association with CAV1. Within hippocampal neurons, CAV1 was found to be involved in mGluR surface trafficking. While these data do not definitely establish a role for CAV1 in mGluR regulation in neurons, they generate specific hypotheses that can now be tested in neuronal systems.
CAV regulation of mGluR1 in cell lines reveals additional distinctions between membrane localization and facilitation of downstream signaling. Intriguingly, expression of CAV1 in HEK cells decreased mGluR-dependent activation of ERK [46]. Furthermore, basal levels of ERK activation are elevated in neurons of CAV1 knockout-mice, suggesting a role for CAV1 in the constitutive inhibition of downstream mGluR1 signaling. Thus, CAV1 may positively regulate transport of mGluR1 to the membrane surface, but then act to attenuate mGluR1 signaling possibly via physical segregation. Clearly further study is required. However, it is evident that CAV regulation of a given signaling protein may involve both positive and negative regulation depending on the particular physiological state of the cell. Relative to non-excitable cell lines, the complexity of CAV regulation may be amplified in neurons, which rapidly and repeatedly modulate their physiological state by modifying the electrical potential across their membrane. While CAVs may not be directly regulated by voltage, neuronal signaling pathways are, and thus the influence of CAVs on those signaling pathways may depend on these dynamic electrical conditions.
L-type Ca2+ channel/glutamate/VDAC signaling
In addition to G-protein signaling, CAVs have been shown to modulate ion channel signaling. For instance, in skeletal muscle [47], smooth muscle [48, 49] and cardiac cells [50] CAV function has been shown to be necessary for L-type Ca2+ function. Although not directly addressed in the ER/mGluR study [20], this is consistent with the hypothesis that CAV3 may facilitate estrogen-induced attenuation of depolarization-induced CREB phosphorylation by clustering ERα/β, mGluR2/3, and L-type Ca2+ channels into functional microdomains via direct protein–protein interactions. These data also suggest that distinct CAV isoforms may regulate specific signaling players across multiple cell types (i.e., CAV3 and L-type Ca2+ channels).
CAVs influence ionotropic glutamate receptor signaling in neuronal systems. CAV1 function was found to be necessary for N-methyl-d-aspartate receptor (NMDAR)-mediated activation of ERK and Src in cultured cortical neurons. CAV1 and the NMDAR subunit NMDAR2B co-localize in rat cortical neurons [51]. Although the authors did not mutate the CAV-binding motif to test its necessity in this interaction, NMDAR2B most likely interacts with CAV1 via a specific protein motif interaction, since the NMDAR2B contains two putative CAV-binding motifs [52]. This interaction also likely facilitates NMDA-mediated Src and ERK1/2 activation, as siRNA knock-down of CAV1 attenuates the ability of NMDARs to mediate this effect [51]. Finally, over-expression of CAV1 in CAV1 knock-out neurons rescues NMDA-mediated Src and ERK1/2 activation. Together, these data strongly suggest that CAV1 facilitates NMDAR signaling in cortical neurons via a specific protein–protein interaction with the NMDAR2B subunit. These results are especially exciting since they were obtained from primary neurons. Given the ubiquity of NMDAR expression in brain neurons, future investigations should determine whether CAV1 similarly facilitates NMDAR signaling in other neuronal types.
CAV regulation of ion channel signaling is also relevant to organism physiology and pathology. Head et al. [51] demonstrated that CAV1 facilitation of NMDAR-mediated ERK and Src activation is necessary for neuronal protection from ischemic cell death. CAV dysregulation may play a role in human pathologies as well. In post-mortem samples of human cortex and hippocampus, voltage-dependent anion channel (VDAC) and ERα form a complex with CAV1. This interaction is modified in pathological states, as samples from patients with Alzheimer’s showed excessive VDAC accumulation in caveolae [53]. Since VDAC is thought to be involved in mediating some aspects amyloid-β neurotoxicity in Alzheimer’s disease [54], CAV dysregulation may play a prominent role in its symptomology. It remains to be determined, however, whether these VDAC aggregations precede Alzheimer’s pathology and play a causal role in degeneration, or whether the aggregations are symptomatic of some more fundamental dysregulation.
In contrast to CAVs regulating glutamatergic signaling by affecting glutamate receptors themselves, CAVs have also been shown to modulate the localization of EEAC1, a sodium-sensitive glutamate transporter critical for the re-uptake and recycling of glutamate in neurons [55]. Gonzalez et al. found that CAV1 facilitates cell surface localization of EAAC1 [56]. Specifically, expression of EAAC1 on the cell surface of glioma cells was attenuated by either over-expression of a dominant negative CAV1, or siRNA knock-down of CAV1. Consistent with this result, cultured cortical neurons from CAV1 knock-out mice displayed attenuated membrane delivery kinetics of EAAC1 relative to wild-type controls. Not surprisingly, immuno-precipitation experiments revealed that CAV1 and CAV2 form complexes with EAAC1. This is consistent with the hypothesis that a direct interaction between CAVs and EAAC1 may be responsible for mediating the trafficking of the transporter to the membrane surface.
Similar to bidirectional mGluR regulation [46], it is interesting to note that CAVs may also negatively regulate EAAC1 [56]. For instance, CAV1 facilitates endocytosis of EAAC1, since endocytosis of EAAC1 is delayed when CAV expression or function is disturbed by use of siRNA knock-down of CAV1 or dominant negative CAV1 over-expression. Since CAV1 also facilitates EAAC expression at the membrane surface (see above), CAVs can participate in the full membrane-protein trafficking cycle and bidirectionally regulate the same protein depending on specific physiological conditions.
CAVs also affect the trafficking and membrane distribution of several other transporters. For instance, insulin-induced membrane trafficking of glucose transporter 4 (GLUT4) is impaired when CAV3 expression is eliminated in skeletal muscle cells [57]. Additionally, the disruption of lipid rafts has also been shown to attenuate glutamate [58], anandamide [59], and serotonin uptake [60]. Since these effects were shown by general disruption of lipid rafts, it still remains to be determined if CAVs are specifically involved in regulating the activity of these transporters. These studies were carried out in non-neuronal systems, and therefore it will also be necessary to determine whether CAV regulation of these membrane transporters occurs in neuronal systems.
Cellular prion protein signaling
Cellular prion protein (PrPC) is a glycosyl-phosphatidyl inositol linked cell surface proteins, whose aberrant form (PrPSC) is thought to underlie transmissible spongiform encephalopathies. In this disease state, it is thought that PrPC is internalized before its conversion to PrPSC. A seminal report using mouse neuroblastoma cells (N2A), found that PrPC forms a physical association with CAV1, which may explain the basis for its localization to membrane rafts [61].
While PrPSC was initially studied because of its pathological role, more recent attention has been focused on the physiological role of PrPC, which is highly enriched in neurons and membrane lipid rafts. These investigations have revealed a role for CAV1 in PrPC-mediated intracellular signaling. In fact, several reports detailed below have used neuronal cell lines as a model to investigate these issues. In one of the first reports investigating CAV1 influence of PrPC-mediated intracellular signaling in neuronal cell lines, Mouillet-Richard and colleagues [62] utilized the neuronal differentiation model 1C11, a neuroectodermal progenitor that acquires either a serotonergic or noradrenergic fate following specific induction paradigms. Although 1C11 cells express PrPC in both the differentiated an undifferentiated states, antibody cross-linking activation of PrPC results in activation of the tyrosine kinase Fyn only in differentiated cells [62]. In PC12 cells CAV1 association with PrPC may both depend on tyrosine phosphorylation of CAV1, and contribute to cell differentiation [63]. CAV1 function is necessary for this effect in 1C11 cells since immuno-sequestration of CAV1 prevented PrPC-mediated Fyn activation [62]. Moreover, PrPC co-immunoprecipitates with CAV1 only in lysates from differentiated cells. Thus, while there does not appear to be a dramatic developmental regulation of CAV1 expression in this paradigm (as in GnRH neurons [21]), there is a dynamic regulation of the localization and coupling of CAV1 to intracellular signaling pathways. These data reinforce the notion that CAV expression and/or activity may be closely linked to the developmental state of the neuron in question, and thus these variables must be carefully considered when designing future experiments.
CAV1 may also contribute to PrPSC signaling in the disease state by facilitating PrPC-CAV1-Fyn signaling. Application of the peptide fragment PrP 106–126, which imitates the neurotoxicity of PrPSC, resulted in overflow of reactive oxygen species, as well as activation of ERK1/2, SAPK, and apoptotic signaling in differentiated 1C11 cells [64]. Importantly, these effects depend on the PrPC-CAV1-Fyn signaling pathway, and only occur in differentiated neuronal cells, suggesting that CAV1 regulation of the signaling pathway is required for these effects. Thus, attenuating the association of CAV1 with PrP signaling in pathological states represents a putative target for interventional therapy.
The physical association between PrPC and CAV1 also occurs in GN11 cells, which are a neuronal cell line derived from migrating GnRH neurons. CAV1 interaction with PrPC in GN11 cells depends on an octapeptide repeat in the N-terminus of PrPC [65]. Density-gradient centrifugation confirmed co-localization of CAV1 and PrPC. In this system, this association is activity-dependent as antibody-mediated activation of PrPC resulted in its clustering with both CAV1 and Fyn. Since PrPC activation resulted in increases in downstream ERK1/2 phosphorylation, the authors attempted to demonstrate the necessity of CAV1. However, in agreement with results from a study discussed above [46] as well as investigations in non-neuronal cells [66, 67], knock-down of CAV1 expression resulted in elevated levels of phopho-ERK1/2. Given the consistency of this phenomenon across many cell types and preparations, CAV1-inhibition of ERK1/2 may be a fundamental regulatory feature. Whether CAV1 directly inhibits ERK1/2, or alternatively inhibits upstream modulators of ERK1/2 has yet to be determined, and warrants future study.
Negative regulation by CAVs
Gαs/AC signaling
CAVs can negatively regulate the initiation of intracellular signaling by sequestering membrane signaling proteins and preventing their interaction with the appropriate downstream signaling machinery. Furthermore, CAVs may remove membrane-signaling machinery from the membrane surface via endocytosis. Below we discuss several examples in which CAVs mediate negative regulation of intracellular signaling that is relevant to the nervous system.
CAVs have been shown to negatively regulate AC signaling. For instance, in C6 glioma cells, RNAi knock-down of CAV1 potentiates isoproterenol (β-adrenergic receptor), thyroid stimulating hormone (thyroid stimulating hormone receptor), and forskolin-(AC) induced cAMP accumulation [68]. The authors determined that while CAV1 knockdown actually resulted in decreased Gαs localization and reduced AC activity in caveolae, it attenuated agonist-induced Gαs internalization. These data are consistent with a model in which CAVs support agonist-induced internalization of the receptor complex, in this case leading to the observed reduction of cAMP production following various neurotransmitter agonists.
A second example of CAV-mediated attenuation of membrane-initiated signaling is CAV1-regulation of Gαs-coupled D1-dopamine receptors (D1R). In a study using COS7 and HEK293 cells, D1R was shown to both interact with CAV1 as well as co-localize in CAV-enriched fractions [30]. In contrast to other results [68], Gαs did not co-localize in CAV-enriched fraction. This contradiction may be due to differences in the cell lines used in each study.
This study also reported that agonist application causes D1R translocation to CAV fractions, and subsequent internalization via a CAV-dependent mechanism, both of which depend on the D1R CAV-binding motif [30]. However, CAV1-regulation of D1R signaling appears to be considerably more complicated. Disruption of the D1R CAV1 binding motifs result in constitutive desensitization of D1R signaling, while disruption of caveolae formation enhances D1R-mediated cAMP accumulation. Thus, blocking CAV1 expression is not equivalent to disrupting caveolae in this system, perhaps because other CAVs may be involved. As noted, this issue will require further study to be fully understood. Nevertheless, in non-neuronal cells, CAV1 negatively regulates D1R signaling by internalizing the receptor upon agonist binding. It is unknown whether the receptor is then targeted for proteosomal degradation or re-inserted into the membrane at a later time point. While a weakness of this study for addressing neuronal signaling is its use of non-neuronal cell lines, if this CAV regulation of D1R signaling occurs in the nervous system, it will have implications for a wide variety of human pathologies characterized by aberrant dopamine signaling.
Growth factor and nitric oxide signaling in neuroblastomas
CAVs have been shown to play a role in growth factor signaling in neuroblastoma cells. Specifically, over-expression of CAV1 can inhibit basic fibroblast growth factor (bFGF)-mediated neurite growth in N2a mouse neuroblastoma cells [69]. In particular, CAV1 attenuates the interaction between Rac/Cdc and Pak1 leading to an attenuation of downstream ERK activation, necessary for neurite outgrowth. The study reported that CAV1 interacts with these signaling molecules, preventing their interaction. It is not yet clear whether this occurs via sequestering or trafficking the molecules from the membrane. It also remains to be determined whether this regulation is relevant to embryonic development (CAV1 as a negative regulator of neurite outgrowth) and human disease such as Parkinson’s [70], or if it is exclusive to neuroblastoma cells. These are important considerations when extrapolating to neurons since cancer cells are often characterized by aberrant intracellular signaling. Nevertheless, CAV1-inhibition of neurite outgrowth in neuroblastoma cells may have implications for cancer treatment.
Using purified protein, CAVs have been shown to interact with and inhibit both neuronal NOS (nNOS) and inducible NOS (iNOS) function via the CSD [71, 72]. Thus, a study reporting the effects of CAV inhibition of iNOS in neuroblastomas may have direct relevance to CAV regulation of neuronal survival [73]. The authors reported that neuroblastoma cells exposed to hypoxic conditions for 15 h showed an increase in iNOS expression and downstream NO production. This led to a specific increase in CAV1 expression that caused feedback inhibition of iNOS expression. The authors note that since hypoxic neuroblastoma cells can become progenitor cells and lead to cancerous progression, this is a putative mechanism by which these cells adapt to these hypoxic conditions. Thus, an understanding of the exact mechanism by which CAV1 facilitates this adaptation will be important for developing effective clinical treatments for cancer therapy.
Glutamatergic signaling
Finally, the glutamate receptor, 2-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPAR) also seems to be negatively regulated by CAV1 through an indirect mechanism. Ligand binding to AMPARs is almost completely abolished by over-expression of the CAV1 CSD [74]. However, this modulation is not due to CAV1 binding the APMAR, but rather CAV1 modulation of the enzyme phospholipase (PLA2), which itself has been shown to promote APMAR ligand binding [75, 76]. PLA2 contains a CAV-binding domain and has been identified in CAV-enriched membrane fractions from hippocampal neurons (which may include glial contributions). The authors propose that CAV1 inhibits the enzymatic activity of PLA2, subsequently leading to modulation of AMPAR binding in neurons. CAV1 seems to divergently regulate glutamate signaling (facilitation of NMDAR signaling [51] versus attenuation of AMPAR signaling). This dichotomy is exciting given that these studies were both carried out in primary neurons. However, it remains to be determined whether this difference can be attributed to difference in neuronal type (hippocampal vs. cortical).
Conclusions
It is now clear that CAVs play a crucial role in achieving the temporal and spatial specificity that is required for effective execution of intracellular signaling in neurons. CAVs achieve this exquisite regulation by physically interacting with and sequestering the appropriate signaling molecules at the neuronal membrane and/or trafficking the molecules to and from the membrane surface. They may also influence signaling molecules via indirect mechanisms such as interacting with modulators of those pathways (as in the case of AMPARs). Importantly, these mechanisms allow for bidirectional regulation of particular signaling pathways, and in some cases bidirectional regulation of a single signaling molecule.
We have only begun to understand the pivotal role of CAVs in regulating neuronal intracellular signaling. Many of the studies here have utilized non-neuronal preparations to generate testable hypotheses about neuronal intracellular signaling pathways. Thus future investigators are challenged to investigate these possibilities in primary neuronal systems that allow a dissociation of neuronal and glia contributions. These preparations will also provide insight into the seemingly paradoxical notion that CAVs exert complex regulation in neurons in the absence of evidence for caveolae.
While these limitations prevent us from offering a comprehensive theory of CAV regulation of neuronal signaling, some themes have begun to emerge from the initial studies described here: CAV1 appears to positively regulate certain classes of signaling (Gαq signaling), and attenuate others (ERK1/2). Evidence from investigations of rapid ER effects in hippocampal neurons suggests that CAVs may facilitate membrane-initiated signaling by non-canonical proteins. This notion that CAVs may not only organize previously identified signaling cascades, but also facilitate previously unrecognized signaling pathways greatly amplifies the signaling possibilities in neurons. Furthermore, different CAV isoforms may be responsible for physically segregating distinct signaling pathways in neurons. Nonetheless, the overall impact of CAV on nervous system function will be an integration of CAV influence on specific signaling molecules and specific pathways throughout the duration of signaling events. This capacity for extensive but precise regulation of intracellular signaling dictates that CAVs are uniquely positioned to impact many aspects of neuronal signaling, and play a critical role in nervous system function and disease.
References
- 1.Singer SJ, Nicolson GL. The fluid mosaic model of the structure of cell membranes. Science. 1972;175:720–731. doi: 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- 2.Allen JA, Halverson-Tamboli RA, Rasenick MM. Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci. 2007;8:128–140. doi: 10.1038/nrn2059. [DOI] [PubMed] [Google Scholar]
- 3.Yamada E. The fine structure of the gall bladder epithelium of the mouse. J Biophys Biochem Cytol. 1955;1:445–458. doi: 10.1083/jcb.1.5.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel HH, Murray F, Insel PA. Caveolae as organizers of pharmacologically relevant signal transduction molecules. Annu Rev Pharmacol Toxicol. 2008;48:359–391. doi: 10.1146/annurev.pharmtox.48.121506.124841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glenney JR., Jr The sequence of human caveolin reveals identity with VIP21, a component of transport vesicles. FEBS Lett. 1992;314:45–48. doi: 10.1016/0014-5793(92)81458-X. [DOI] [PubMed] [Google Scholar]
- 6.Scherer PE, Okamoto T, Chun M, Nishimoto I, Lodish HF, Lisanti MP. Identification, sequence, and expression of caveolin-2 defines a caveolin gene family. Proc Natl Acad Sci USA. 1996;93:131–135. doi: 10.1073/pnas.93.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang Z, Scherer PE, Okamoto T, Song K, Chu C, Kohtz DS, Nishimoto I, Lodish HF, Lisanti MP. Molecular cloning of caveolin-3, a novel member of the caveolin gene family expressed predominantly in muscle. J Biol Chem. 1996;271:2255–2261. doi: 10.1074/jbc.271.4.2255. [DOI] [PubMed] [Google Scholar]
- 8.Galbiati F, Volonte D, Gil O, Zanazzi G, Salzer JL, Sargiacomo M, Scherer PE, Engelman JA, Schlegel A, Parenti M, Okamoto T, Lisanti MP. Expression of caveolin-1 and -2 in differentiating PC12 cells and dorsal root ganglion neurons: caveolin-2 is up-regulated in response to cell injury. Proc Natl Acad Sci USA. 1998;95:10257–10262. doi: 10.1073/pnas.95.17.10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ikezu T, Ueda H, Trapp BD, Nishiyama K, Sha JF, Volonte D, Galbiati F, Byrd AL, Bassell G, Serizawa H, Lane WS, Lisanti MP, Okamoto T. Affinity-purification and characterization of caveolins from the brain: differential expression of caveolin-1, -2, and -3 in brain endothelial and astroglial cell types. Brain Res. 1998;804:177–192. doi: 10.1016/S0006-8993(98)00498-3. [DOI] [PubMed] [Google Scholar]
- 10.Lisanti MP, Scherer PE, Vidugiriene J, Tang Z, Hermanowski-Vosatka A, Tu YH, Cook RF, Sargiacomo M. Characterization of caveolin-rich membrane domains isolated from an endothelial-rich source: implications for human disease. J Cell Biol. 1994;126:111–126. doi: 10.1083/jcb.126.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vogel U, Sandvig K, van Deurs B. Expression of caveolin-1 and polarized formation of invaginated caveolae in Caco-2 and MDCK II cells. J Cell Sci. 1998;111(Pt 6):825–832. doi: 10.1242/jcs.111.6.825. [DOI] [PubMed] [Google Scholar]
- 12.Song KS, Scherer PE, Tang Z, Okamoto T, Li S, Chafel M, Chu C, Kohtz DS, Lisanti MP. Expression of caveolin-3 in skeletal, cardiac, and smooth muscle cells. Caveolin-3 is a component of the sarcolemma and co-fractionates with dystrophin and dystrophin-associated glycoproteins. J Biol Chem. 1996;271:15160–15165. doi: 10.1074/jbc.271.25.15160. [DOI] [PubMed] [Google Scholar]
- 13.Way M, Parton RG. M-caveolin, a muscle-specific caveolin-related protein. FEBS Lett. 1995;376:108–112. doi: 10.1016/0014-5793(95)01256-7. [DOI] [PubMed] [Google Scholar]
- 14.Chang WJ, Ying YS, Rothberg KG, Hooper NM, Turner AJ, Gambliel HA, De Gunzburg J, Mumby SM, Gilman AG, Anderson RG. Purification and characterization of smooth muscle cell caveolae. J Cell Biol. 1994;126:127–138. doi: 10.1083/jcb.126.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galbiati F, Engelman JA, Volonte D, Zhang XL, Minetti C, Li M, Hou H, Jr, Kneitz B, Edelmann W, Lisanti MP. Caveolin-3 null mice show a loss of caveolae, changes in the microdomain distribution of the dystrophin-glycoprotein complex, and t-tubule abnormalities. J Biol Chem. 2001;276:21425–21433. doi: 10.1074/jbc.M100828200. [DOI] [PubMed] [Google Scholar]
- 16.Razani B, Combs TP, Wang XB, Frank PG, Park DS, Russell RG, Li M, Tang B, Jelicks LA, Scherer PE, Lisanti MP. Caveolin-1-deficient mice are lean, resistant to diet-induced obesity, and show hypertriglyceridemia with adipocyte abnormalities. J Biol Chem. 2002;277:8635–8647. doi: 10.1074/jbc.M110970200. [DOI] [PubMed] [Google Scholar]
- 17.Scherer PE, Lewis RY, Volonte D, Engelman JA, Galbiati F, Couet J, Kohtz DS, van Donselaar E, Peters P, Lisanti MP. Cell-type and tissue-specific expression of caveolin-2. Caveolins 1 and 2 co-localize and form a stable hetero-oligomeric complex in vivo. J Biol Chem. 1997;272:29337–29346. doi: 10.1074/jbc.272.46.29337. [DOI] [PubMed] [Google Scholar]
- 18.Cameron PL, Ruffin JW, Bollag R, Rasmussen H, Cameron RS. Identification of caveolin and caveolin-related proteins in the brain. J Neurosci. 1997;17:9520–9535. doi: 10.1523/JNEUROSCI.17-24-09520.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zschocke J, Manthey D, Bayatti N, van der Burg B, Goodenough S, Behl C. Estrogen receptor alpha-mediated silencing of caveolin gene expression in neuronal cells. J Biol Chem. 2002;277:38772–38780. doi: 10.1074/jbc.M205664200. [DOI] [PubMed] [Google Scholar]
- 20.Boulware MI, Kordasiewicz H, Mermelstein PG. Caveolin proteins are essential for distinct effects of membrane estrogen receptors in neurons. J Neurosci. 2007;27:9941–9950. doi: 10.1523/JNEUROSCI.1647-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D’Orlando C, Guzzi F, Gravati M, Biella G, Toselli M, Meneveri R, Barisani D, Parenti M. Retinoic acid- and phorbol ester-induced neuronal differentiation down-regulates caveolin expression in GnRH neurons. J Neurochem. 2008;104:1577–1587. doi: 10.1111/j.1471-4159.2007.05109.x. [DOI] [PubMed] [Google Scholar]
- 22.Head BP, Insel PA. Do caveolins regulate cells by actions outside of caveolae? Trends Cell Biol. 2007;17:51–57. doi: 10.1016/j.tcb.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 23.Head BP, Patel HH, Roth DM, Murray F, Swaney JS, Niesman IR, Farquhar MG, Insel PA. Microtubules and actin microfilaments regulate lipid raft/caveolae localization of adenylyl cyclase signaling components. J Biol Chem. 2006;281:26391–26399. doi: 10.1074/jbc.M602577200. [DOI] [PubMed] [Google Scholar]
- 24.Boeckers TM. The postsynaptic density. Cell Tissue Res. 2006;326:409–422. doi: 10.1007/s00441-006-0274-5. [DOI] [PubMed] [Google Scholar]
- 25.Perez AS, Bredt DS. The N-terminal PDZ-containing region of postsynaptic density-95 mediates association with caveolar-like lipid domains. Neurosci Lett. 1998;258:121–123. doi: 10.1016/S0304-3940(98)00846-5. [DOI] [PubMed] [Google Scholar]
- 26.Murata M, Peranen J, Schreiner R, Wieland F, Kurzchalia TV, Simons K. VIP21/caveolin is a cholesterol-binding protein. Proc Natl Acad Sci USA. 1995;92:10339–10343. doi: 10.1073/pnas.92.22.10339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mauch DH, Nagler K, Schumacher S, Goritz C, Muller EC, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- 28.Willmann R, Pun S, Stallmach L, Sadasivam G, Santos AF, Caroni P, Fuhrer C. Cholesterol and lipid microdomains stabilize the postsynapse at the neuromuscular junction. EMBO J. 2006;25:4050–4060. doi: 10.1038/sj.emboj.7601288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oshikawa J, Otsu K, Toya Y, Tsunematsu T, Hankins R, Kawabe J, Minamisawa S, Umemura S, Hagiwara Y, Ishikawa Y. Insulin resistance in skeletal muscles of caveolin-3-null mice. Proc Natl Acad Sci USA. 2004;101:12670–12675. doi: 10.1073/pnas.0402053101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kong MM, Hasbi A, Mattocks M, Fan T, O’Dowd BF, George SR. Regulation of D1 dopamine receptor trafficking and signaling by caveolin-1. Mol Pharmacol. 2007;72:1157–1170. doi: 10.1124/mol.107.034769. [DOI] [PubMed] [Google Scholar]
- 31.Shmuel M, Nodel-Berner E, Hyman T, Rouvinski A, Altschuler Y. Caveolin 2 regulates endocytosis and trafficking of the M1 muscarinic receptor in MDCK epithelial cells. Mol Biol Cell. 2007;18:1570–1585. doi: 10.1091/mbc.E06-07-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wyse BD, Prior IA, Qian H, Morrow IC, Nixon S, Muncke C, Kurzchalia TV, Thomas WG, Parton RG, Hancock JF. Caveolin interacts with the angiotensin II type 1 receptor during exocytic transport but not at the plasma membrane. J Biol Chem. 2003;278:23738–23746. doi: 10.1074/jbc.M212892200. [DOI] [PubMed] [Google Scholar]
- 33.Syme CA, Zhang L, Bisello A. Caveolin-1 regulates cellular trafficking and function of the glucagon-like peptide 1 receptor. Mol Endocrinol. 2006;20:3400–3411. doi: 10.1210/me.2006-0178. [DOI] [PubMed] [Google Scholar]
- 34.Lajoie P, Nabi IR. Regulation of raft-dependent endocytosis. J Cell Mol Med. 2007;11:644–653. doi: 10.1111/j.1582-4934.2007.00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pelkmans L, Helenius A. Endocytosis via caveolae. Traffic. 2002;3:311–320. doi: 10.1034/j.1600-0854.2002.30501.x. [DOI] [PubMed] [Google Scholar]
- 36.Hommelgaard AM, Roepstorff K, Vilhardt F, Torgersen ML, Sandvig K, van Deurs B. Caveolae: stable membrane domains with a potential for internalization. Traffic. 2005;6:720–724. doi: 10.1111/j.1600-0854.2005.00314.x. [DOI] [PubMed] [Google Scholar]
- 37.Becher A, McIlhinney RA. Consequences of lipid raft association on G-protein-coupled receptor function. Biochem Soc Symp. 2005;72:151–164. doi: 10.1042/bss0720151. [DOI] [PubMed] [Google Scholar]
- 38.de Weerd WF, Leeb-Lundberg LM. Bradykinin sequesters B2 bradykinin receptors and the receptor-coupled Galpha subunits Galphaq and Galphai in caveolae in DDT1 MF-2 smooth muscle cells. J Biol Chem. 1997;272:17858–17866. doi: 10.1074/jbc.272.28.17858. [DOI] [PubMed] [Google Scholar]
- 39.Whistler JL, Chuang HH, Chu P, Jan LY, von Zastrow M. Functional dissociation of mu opioid receptor signaling and endocytosis: implications for the biology of opiate tolerance and addiction. Neuron. 1999;23:737–746. doi: 10.1016/S0896-6273(01)80032-5. [DOI] [PubMed] [Google Scholar]
- 40.Bhatnagar A, Sheffler DJ, Kroeze WK, Compton-Toth B, Roth BL. Caveolin-1 interacts with 5-HT2A serotonin receptors and profoundly modulates the signaling of selected Galphaq-coupled protein receptors. J Biol Chem. 2004;279:34614–34623. doi: 10.1074/jbc.M404673200. [DOI] [PubMed] [Google Scholar]
- 41.Toran-Allerand CD. Minireview: a plethora of estrogen receptors in the brain: where will it end? Endocrinology. 2004;145:1069–1074. doi: 10.1210/en.2003-1462. [DOI] [PubMed] [Google Scholar]
- 42.Boulware MI, Weick JP, Becklund BR, Kuo SP, Groth RD, Mermelstein PG. Estradiol activates group I and II metabotropic glutamate receptor signaling, leading to opposing influences on cAMP response element-binding protein. J Neurosci. 2005;25:5066–5078. doi: 10.1523/JNEUROSCI.1427-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKenna NJ, Lanz RB, O’Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev. 1999;20:321–344. doi: 10.1210/er.20.3.321. [DOI] [PubMed] [Google Scholar]
- 44.McInerney EM, Weis KE, Sun J, Mosselman S, Katzenellenbogen BS. Transcription activation by the human estrogen receptor subtype beta (ER beta) studied with ER beta and ER alpha receptor chimeras. Endocrinology. 1998;139:4513–4522. doi: 10.1210/en.139.11.4513. [DOI] [PubMed] [Google Scholar]
- 45.Sheldahl LC, Shapiro RA, Bryant DN, Koerner IP, Dorsa DM. Estrogen induces rapid translocation of estrogen receptor beta, but not estrogen receptor alpha, to the neuronal plasma membrane. Neuroscience. 2008;153:751–761. doi: 10.1016/j.neuroscience.2008.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Francesconi A, Kumari R, Zukin RS. Regulation of group I metabotropic glutamate receptor trafficking and signaling by the caveolar/lipid raft pathway. J Neurosci. 2009;29:3590–3602. doi: 10.1523/JNEUROSCI.5824-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Couchoux H, Allard B, Legrand C, Jacquemond V, Berthier C. Loss of caveolin-3 induced by the dystrophy-associated P104L mutation impairs L-type calcium channel function in mouse skeletal muscle cells. J Physiol. 2007;580:745–754. doi: 10.1113/jphysiol.2006.124198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng X, Jaggar JH. Genetic ablation of caveolin-1 modifies Ca2+ spark coupling in murine arterial smooth muscle cells. Am J Physiol Heart Circ Physiol. 2006;290:H2309–H2319. doi: 10.1152/ajpheart.01226.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Daniel EE, Eteraf T, Sommer B, Cho WJ, Elyazbi A. The role of caveolae and caveolin 1 in calcium handling in pacing and contraction of mouse intestine. J Cell Mol Med. 2009;13:352–364. doi: 10.1111/j.1582-4934.2008.00667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. Localization of cardiac L-type Ca(2+) channels to a caveolar macromolecular signaling complex is required for beta(2)-adrenergic regulation. Proc Natl Acad Sci USA. 2006;103:7500–7505. doi: 10.1073/pnas.0503465103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Head BP, Patel HH, Tsutsumi YM, Hu Y, Mejia T, Mora RC, Insel PA, Roth DM, Drummond JC, Patel PM. Caveolin-1 expression is essential for N-methyl-d-aspartate receptor-mediated Src and extracellular signal-regulated kinase 1/2 activation and protection of primary neurons from ischemic cell death. FASEB J. 2008;22:828–840. doi: 10.1096/fj.07-9299com. [DOI] [PubMed] [Google Scholar]
- 52.Couet J, Sargiacomo M, Lisanti MP. Interaction of a receptor tyrosine kinase, EGF-R, with caveolins. Caveolin binding negatively regulates tyrosine and serine/threonine kinase activities. J Biol Chem. 1997;272:30429–30438. doi: 10.1074/jbc.272.48.30429. [DOI] [PubMed] [Google Scholar]
- 53.Ramirez CM, Gonzalez M, Diaz M, Alonso R, Ferrer I, Santpere G, Puig B, Meyer G, Marin R. VDAC and ERalpha interaction in caveolae from human cortex is altered in Alzheimer’s disease. Mol Cell Neurosci. 2009;42:172–183. doi: 10.1016/j.mcn.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 54.Ferrer I. Altered mitochondria, energy metabolism, voltage-dependent anion channel, and lipid rafts converge to exhaust neurons in Alzheimer’s disease. J Bioenerg Biomembr. 2009;41:425–431. doi: 10.1007/s10863-009-9243-5. [DOI] [PubMed] [Google Scholar]
- 55.Kanai Y, Hediger MA. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature. 1992;360:467–471. doi: 10.1038/360467a0. [DOI] [PubMed] [Google Scholar]
- 56.Gonzalez MI, Krizman-Genda E, Robinson MB. Caveolin-1 regulates the delivery and endocytosis of the glutamate transporter, excitatory amino acid carrier 1. J Biol Chem. 2007;282:29855–29865. doi: 10.1074/jbc.M704738200. [DOI] [PubMed] [Google Scholar]
- 57.Fecchi K, Volonte D, Hezel MP, Schmeck K, Galbiati F. Spatial and temporal regulation of GLUT4 translocation by flotillin-1 and caveolin-3 in skeletal muscle cells. FASEB J. 2006;20:705–707. doi: 10.1096/fj.05-4661fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Butchbach ME, Tian G, Guo H, Lin CL. Association of excitatory amino acid transporters, especially EAAT2, with cholesterol-rich lipid raft microdomains: importance for excitatory amino acid transporter localization and function. J Biol Chem. 2004;279:34388–34396. doi: 10.1074/jbc.M403938200. [DOI] [PubMed] [Google Scholar]
- 59.McFarland MJ, Porter AC, Rakhshan FR, Rawat DS, Gibbs RA, Barker EL. A role for caveolae/lipid rafts in the uptake and recycling of the endogenous cannabinoid anandamide. J Biol Chem. 2004;279:41991–41997. doi: 10.1074/jbc.M407250200. [DOI] [PubMed] [Google Scholar]
- 60.Samuvel DJ, Jayanthi LD, Bhat NR, Ramamoorthy S. A role for p38 mitogen-activated protein kinase in the regulation of the serotonin transporter: evidence for distinct cellular mechanisms involved in transporter surface expression. J Neurosci. 2005;25:29–41. doi: 10.1523/JNEUROSCI.3754-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harmey JH, Doyle D, Brown V, Rogers MS. The cellular isoform of the prion protein, PrPc, is associated with caveolae in mouse neuroblastoma (N2a) cells. Biochem Biophys Res Commun. 1995;210:753–759. doi: 10.1006/bbrc.1995.1723. [DOI] [PubMed] [Google Scholar]
- 62.Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science. 2000;289:1925–1928. doi: 10.1126/science.289.5486.1925. [DOI] [PubMed] [Google Scholar]
- 63.Pantera B, Bini C, Cirri P, Paoli P, Camici G, Manao G, Caselli A. PrPc activation induces neurite outgrowth and differentiation in PC12 cells: role for caveolin-1 in the signal transduction pathway. J Neurochem. 2009;110:194–207. doi: 10.1111/j.1471-4159.2009.06123.x. [DOI] [PubMed] [Google Scholar]
- 64.Pietri M, Caprini A, Mouillet-Richard S, Pradines E, Ermonval M, Grassi J, Kellermann O, Schneider B. Overstimulation of PrPC signaling pathways by prion peptide 106–126 causes oxidative injury of bioaminergic neuronal cells. J Biol Chem. 2006;281:28470–28479. doi: 10.1074/jbc.M602774200. [DOI] [PubMed] [Google Scholar]
- 65.Toni M, Spisni E, Griffoni C, Santi S, Riccio M, Lenaz P, Tomasi V. Cellular prion protein and caveolin-1 interaction in a neuronal cell line precedes fyn/erk 1/2 signal transduction. J Biomed Biotechnol. 2006;2006:69469. doi: 10.1155/JBB/2006/69469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Engelman JA, Chu C, Lin A, Jo H, Ikezu T, Okamoto T, Kohtz DS, Lisanti MP. Caveolin-mediated regulation of signaling along the p42/44 MAP kinase cascade in vivo. A role for the caveolin-scaffolding domain. FEBS Lett. 1998;428:205–211. doi: 10.1016/S0014-5793(98)00470-0. [DOI] [PubMed] [Google Scholar]
- 67.Zhang W, Razani B, Altschuler Y, Bouzahzah B, Mostov KE, Pestell RG, Lisanti MP. Caveolin-1 inhibits epidermal growth factor-stimulated lamellipod extension and cell migration in metastatic mammary adenocarcinoma cells (MTLn3). Transformation suppressor effects of adenovirus-mediated gene delivery of caveolin-1. J Biol Chem. 2000;275:20717–20725. doi: 10.1074/jbc.M909895199. [DOI] [PubMed] [Google Scholar]
- 68.Allen JA, Yu JZ, Dave RH, Bhatnagar A, Roth BL, Rasenick MM. Caveolin-1 and lipid microdomains regulate Gs trafficking and attenuate Gs/adenylyl cyclase signaling. Mol Pharmacol. 2009;76:1082–1093. doi: 10.1124/mol.109.060160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kang MJ, Seo JS, Park WY. Caveolin-1 inhibits neurite growth by blocking Rac1/Cdc42 and p21-activated kinase 1 interactions. Neuroreport. 2006;17:823–827. doi: 10.1097/01.wnr.0000220139.83671.60. [DOI] [PubMed] [Google Scholar]
- 70.Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- 71.Garcia-Cardena G, Martasek P, Masters BS, Skidd PM, Couet J, Li S, Lisanti MP, Sessa WC. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J Biol Chem. 1997;272:25437–25440. doi: 10.1074/jbc.272.41.25437. [DOI] [PubMed] [Google Scholar]
- 72.Sato Y, Sagami I, Shimizu T. Identification of caveolin-1-interacting sites in neuronal nitric-oxide synthase. Molecular mechanism for inhibition of NO formation. J Biol Chem. 2004;279:8827–8836. doi: 10.1074/jbc.M310327200. [DOI] [PubMed] [Google Scholar]
- 73.Shen J, Lee W, Li Y, Lau CF, Ng KM, Fung ML, Liu KJ. Interaction of caveolin-1, nitric oxide, and nitric oxide synthases in hypoxic human SK-N-MC neuroblastoma cells. J Neurochem. 2008;107:478–487. doi: 10.1111/j.1471-4159.2008.05630.x. [DOI] [PubMed] [Google Scholar]
- 74.Gaudreault SB, Chabot C, Gratton JP, Poirier J. The caveolin scaffolding domain modifies 2-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor binding properties by inhibiting phospholipase A2 activity. J Biol Chem. 2004;279:356–362. doi: 10.1074/jbc.M304777200. [DOI] [PubMed] [Google Scholar]
- 75.Massicotte G, Baudry M. Modulation of -alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA)/quisqualate receptors by phospholipase A2 treatment. Neurosci Lett. 1990;118:245–248. doi: 10.1016/0304-3940(90)90638-P. [DOI] [PubMed] [Google Scholar]
- 76.Catania MV, Hollingsworth Z, Penney JB, Young AB. Phospholipase A2 modulates different subtypes of excitatory amino acid receptors: autoradiographic evidence. J Neurochem. 1993;60:236–245. doi: 10.1111/j.1471-4159.1993.tb05843.x. [DOI] [PubMed] [Google Scholar]