

Abstract
Alzheimer’s disease (AD) is the most common form of dementia among the elderly, affecting millions of people worldwide and representing a substantial economic burden. AD is a progressive disease associated with memory loss and impaired cognitive function. The neuropathology is characterized by cortical accumulation of amyloid plaques and neurofibrillary tangles (NFTs). Amyloid plaques are small, aggregated peptides called beta amyloid (Aβ) and NFTs are aggregates of hyperphosphorylated Tau protein. Because Aβ disrupts multiple intracellular signaling pathways, resulting in some of the clinical symptoms of AD, understanding the underlying molecular mechanisms has implications for the diagnosis and treatment of AD. Recent studies have demonstrated that Aβ regulates striatal-enriched protein tyrosine phosphatase (STEP) (PTPN5). Aβ accumulation is associated with increases in STEP levels and activity that in turn disrupts glutamate receptor trafficking to and from the neuronal membrane. These findings indicate that modulating STEP levels or inhibiting its activity may have beneficial effects for patients with AD, making it an important target for drug discovery. This article reviews the biology of STEP and its role in AD as well as the potential clinical applications.
I. Introduction
Alzheimer’s disease (AD) is a common neurodegenerative disorder in people aged 65 years and older and its prevalence is increasing as the population ages. It is characterized by irreversible and progressive loss of cognitive function. Clinical symptoms include mild to severe memory loss, problems with cognition and behavior, and gradual losses in the activities of daily living (Castellani et al., 2010). At cellular level, AD is associated with gradual synapse loss, followed by severe neurodegeneration in the brain areas related to cognitive functions. None of the available pharmacological treatments for AD provide more than temporary relief from the relentless decline in cognitive and daily function. It is critically important to understand the pathophysiology of this disease at the molecular level in order to develop new pharmacological treatments.
In AD, brain regions involved in cognitive functions such as hippocampus, cortex, and amygdala show pronounced pathological alterations. Postmortem studies of AD brains have established the neuropathological hallmark of this disease: the accumulation of amyloid plaques and neurofibrillary tangles (NFTs). Beta amyloid (Aβ) peptides accumulate during the course of the disease and contribute to synaptic dysfunction (Hardy & Selkoe, 2002; Haass & Selkoe, 2007). Transgenic mice that overproduce Aβ (Philipson et al., 2010) show that the Aβ produced at the onset of the illness disrupts synaptic function and contributes to cognitive impairment early in the disease process (Hsiao et al., 1996; Jacobsen et al., 2006). The toxic effect (Terry et al., 1991) of Aβ on synapse function is confirmed by its ability to inhibit long-term potentiation (LTP), induce aberrant changes in the synaptic networks, cause synapse loss, and disrupt cognitive functions in animal models (Lacor et al., 2007; Palop & Mucke, 2010; Shankar et al., 2008; Walsh et al., 2002).
Striatal-enriched protein tyrosine phosphatase (STEP) is a brain-enriched tyrosine phosphatase (Lombroso et al., 1991). Accumulating evidence implicates STEP in the pathophysiology of AD. STEP regulates several synaptic events including glutamate receptor trafficking, which plays a crucial role in learning and memory (Baum et al., 2010; Fitzpatrik & Lombroso, 2011a; Goebel-Goody et al., 2012). Recent findings indicate that Aβ peptides generated during the course of disease regulate the function of STEP by up-regulating its activity and protein levels through different mechanisms. Increased STEP activity and protein levels lead to excessive internalization of glutamate receptors both, NMDARs (N-methyl-D-aspartate receptors) and AMPARs (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) from the neuronal membrane, which is thought to be responsible for the synaptic changes associated with cognitive and memory deficits in AD (Kurup et al., 2010a; Snyder et al., 2005; Zhang et al., 2010). The role of STEP in these events has been confirmed in AD mouse models by several in vitro and in vivo studies, as well as behavioral and electrophysiological studies. This chapter reviews what we know about STEP beginning with its discovery and ending with recent demonstrations of its role in the pathophysiology of AD.
II. Striatal-Enriched Protein Tyrosine Phosphatase (STEP)
Protein kinases and protein phosphatases regulate a great variety of cellular pathways including cell division and higher order brain functions including learning and memory (Mayford, 2007). A major class of protein kinases is those that phosphorylate their substrates at tyrosine residues to initiate or modulate intracellular events. Protein tyrosine phosphatases (PTPs) oppose these activities by dephosphorylating the tyrosine residues, thus playing a major role in cellular signaling. Although STEP was initially discovered as a protein enriched in the striatum (Lombroso et al., 1991), it is distributed in other regions of brain including the cortex and hippocampus (Lombroso et al., 1993). STEP is not present in the cerebellum; here it is substituted by a homologous PTP called STEP-like PTP (Shiozuka et al., 1995). Another closely related PTP expressed in immune cells termed “HePTP” (Hematopoietic Protein Tyrosine Phosphatase) shares sequence homology with STEP (Adachi et al., 1992).
STEP is an intracellular tyrosine phosphatase encoded by the ptpn5 gene. It exists as two major isoforms, STEP61 and STEP46, named after their protein mobility in SDS-PAGE (Boulanger et al., 1995; Bult et al., 1997) (Fig. 1). The distribution of these two STEP isoforms varies within different brain regions. STEP61 is present in the cortex, hippocampus, and striatum, whereas all isoforms are present in the striatum (Boulanger et al., 1995; Bult et al., 1996). The expression pattern of STEP isoforms changes during development (Raghunathan, Matthews, Lombroso, & Naegele, 1996). Rodent studies indicate that STEP61 is expressed at birth and its expression continues throughout adulthood, whereas STEP46 first appears at postnatal day 6 and progressively increases until adulthood, indicating that the expression of STEP is developmentally regulated, although a specific role of STEP during later stages of life or in cognitive deficits that occur with aging has not yet been examined (Okamura et al., 1997; Raghunathan et al., 1996). STEP46 is primarily a cytosolic protein, whereas STEP61 is targeted to membrane compartments (e.g., endoplasmic reticulum, Golgi bodies, and endosomes) and postsynaptic densities (Goebel-Goody et al., 2009; Oyama et al., 1995).
FIGURE 1. Domain structure of STEP isoforms.
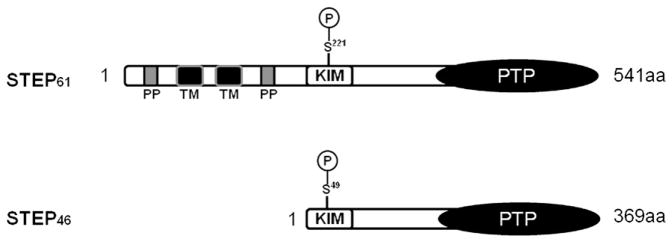
STEP occurs as two major isoforms: STEP61 (541 amino acid residues) and STEP46 (369 amino acid residues). Both these isoforms have conserved PTP catalytic domain at C-terminal and KIM (kinase interacting motif) domain. KIM domain has a conserved serine residue, which gets phosphorylated by protein kinase A (PKA), denoted as Ser221 in STEP61 and Ser49 in STEP46. STEP61 isoform has additional N-terminal region (172 amino acids) containing polyproline-rich domains (PP) and transmembrane domains (TM).
A. Domain Structure
Both STEP61 and STEP46 isoforms share a common conserved PTP catalytic domain at their C-terminal region consisting of a conserved sequence ([I/V]HCxAGxxR[S/T]G) that contains a critical cysteine residue required for phosphatase activity. Mutation of the cysteine residue results in a catalytically inactive variant. All STEP isoforms contain a kinase interacting motif (KIM), which is required for the interactions between STEP and its physiological substrates (Bult et al., 1996). The KIM contains a critical serine residue; phosphorylation of this residue by protein kinase A (PKA) prevents STEP from interacting with and dephosphorylating its substrates (Paul et al., 2000; Paul et al., 2003).
STEP61 differs from STEP46 by an additional 172 amino acid residues in the N-terminal region. This sequence contains two hydrophobic domains that are essential for targeting STEP61 to the neuronal membrane, including postsynaptic densities. The N-terminal region of STEP61 also contains two polyproline-rich domains and PEST motifs, which are rich in proline (P), glutamic acid (E), serine (S), and threonine (T) (Boulanger et al., 1995; Bult et al., 1996; Oyama et al., 1995). The N-terminal polyproline domain is required for the association of STEP61 with Fyn kinase (Nguyen, et al., 2002), while the second polyproline domain is necessary for the interaction of STEP61 with Pyk2 (Xu et al., 2012). The PEST sequences in several proteins are known to mediate rapid degradation (Shumway et al., 1999; Spencer et al., 2004), they may also serve as recognition motifs for proteolytic cleavage or ubiquitination of STEP under certain physiological conditions. Additional STEP members like (STEP38 and STEP20 exists, but their functions are not known. These isoforms do not contain conserved PTP domain and they are catalytically inactive (Bult et al., 1996; Sharma, et al., 1995). The recently resolved crystal structure of STEP shows some distinctive features compared to other PTPs (Eswaran et al., 2006), including a unique open conformation that is critical for PTP catalysis (WPD loop). This structure may prove useful in the search for small, specific STEP inhibitors.
B. STEP Regulation
STEP is regulated by several mechanisms including phosphorylation, ubiquitination, proteolytic cleavage, oligomerization, and local translation (Deb, et al., 2011; Kurup et al., 2010a; Paul et al., 2000; Xu et al., 2009; Zhang et al., 2008). Recent work shows that PKA phosphorylation and ubiquitination of STEP play a role in AD (Kurup et al., 2010a; Snyder et al., 2005). Both events decrease STEP activity in neurons. PKA phosphorylation of a regulatory serine residue within the KIM domain interferes with the ability of STEP to interact with its substrates (Paul et al., 2000). Ubiquitination rapidly removes STEP61 from synaptic sites and promotes degradation by the proteasome (Kurup et al., 2010a; Xu et al., 2009). A model has thus emerged that STEP normally opposes the development of synaptic strengthening by inactivating enzymes that facilitates this process. STEP must be inactivated at synaptic sites, either by phosphorylation within the KIM domain or by rapid degradation for synaptic plasticity and learning to take place (Braithwaite et al., 2006b; Fitzpatrick & Lombroso, 2011; Goebel-Goody et al., 2012). Thus, events that disrupt STEP inactivation would oppose synaptic strengthening.
1. Phosphorylation
Phosphorylation is an important form of posttranslational modification that regulates various intracellular signaling pathways. Both STEP61 and STEP46 isoforms are phosphorylated by PKA. PKA-mediated STEP phosphorylation was initially discovered after dopamine receptor (D1R) activation (Paul et al., 2000). Dopamine (DA) D1 receptor (D1R) stimulation activates PKA leading to phosphorylation of both STEP61 and STEP46 at a conserved serine residue (designated ser221 in STEP61 and ser49 in STEP46). Phosphorylation at these serine residues results in steric interference, preventing STEP from interaction with its substrates (Paul et al., 2000). PKA also opposes the dephosphorylation of STEP by inhibiting the phosphatase PP1 through a DARPP-32-mediated pathway. PKA phosphorylates DARPP-32 at Thr34, and this phosphorylated form of DARPP-32 acts as a potent inhibitor for PP1 and blocks its activity (Greengard et al., 1999). By initiating these parallel events PKA stabilizes the phosphorylated and inactive forms of STEP (Valjent et al., 2005). PKA phosphorylates another unique serine residue ser160 in STEP61 at its N-terminal region, although the functional significance is not known.
STEP61 phosphorylation at ser221 is reduced in AD mouse models and in neuronal cultures treated with Aβ (Kurup et al., 2010a; Snyder et al., 2005). Such conditions would increase the ability of STEP to interact with and dephosphorylate its substrates. Dephosphorylation of these serine residues is mediated by calcineurin (PP2B)/PP1 pathway, favoring its interaction with substrates (Snyder et al., 2005; Valjent et al., 2005). As discussed in the following sections, Aβ peptide binds to α7 nicotinic acetylcholine receptors (α7 nAChRs) that in turn lead to the activation of PP2B and STEP dephosphorylation (Snyder et al., 2005).
2. Ubiquitination
The ubiquitination of target proteins involves the covalent attachment of ubiquitin to the substrate and often leads to proteasomal degradation of the protein. The ubiquitin–proteasome system (UPS) plays an important role in cellular protein recycling and has been implicated in several pathological processes including cancer and neurodegenerative disease (Hegde & Upadhya, 2011; Yi & Ehlers, 2007). STEP61 is a target for ubiquitin-mediated proteasomal degradation. STEP61 levels are increased in cortical neurons treated with Aβ (Kurup et al., 2010a). The increase in STEP61 is insensitive to transcription or translation inhibitors, suggesting that STEP61 accumulation occurs when normal degradation is blocked. This work led to the isolation of STEP-ubiquitin conjugates from cells treated with proteasome inhibitors, suggesting that STEP61 is a direct substrate for ubiquitin conjugation and proteasomal degradation. Together, this work indicates that Aβ-mediated inhibition of the proteasome leads to STEP61 accumulation (Kurup et al., 2010a).
STEP is differentially regulated by synaptic and extrasynaptic NMDARs (Xu et al., 2009). These receptors are localized in distinct compartments on the neuronal membrane where they initiate signaling pathways when activated by glutamate (Hardingham & Bading, 2010; Ivanov et al., 2006). Synaptic NMDAR activation is coupled to extracellular-regulated kinase (ERK) activation and is involved in synaptic strengthening and neuronal survival (Hardingham et al., 2002). In contrast, extrasynaptic NMDAR activation is linked to p38 activation and cell death pathways. When synaptic NMDARs are stimulated, STEP61 is ubiquitinated and rapidly degraded from synaptic sites by the UPS pathway (Xu et al., 2009). STEP degradation is required for sustained ERK activation. Activated ERK phosphorylates several synaptic and cytoplasmic proteins, and is translocated to the nucleus where it phosphorylates and activates transcription factors such as CREB and Elk-1 that are involved in spine remodeling (Thiels & Klann, 2001).
C. STEP Substrates
1. Mitogen-Activated Protein Kinase
The mitogen-activated protein kinase (MAPK) family of proteins consists of several enzymes that activate signaling pathways to regulate cellular differentiation, cell survival and synaptic plasticity (Sweatt, 2004; Thomas & Huganir, 2004). The MAPK family of extracellular signal-regulated kinases (ERK1/2) and p38 are both STEP substrates. STEP dephosphorylates regulatory tyrosine residues in the activation loop of ERK1/2 (Tyr204 in ERK1 and Tyr187 in ERK2) and p38 (Tyr182), leading to inactivation of these proteins (Munoz et al., 2003; Paul et al., 2003; Xu et al., 2009).
The ERK1/2 signaling pathway regulates synaptic plasticity by post-translational modification of synaptic proteins and by initiating nuclear transcription in neurons (Davis, Vanhoutte, Pages, Caboche, & Laroche, 2000). Activation of ERK1/2 also initiates the local translation of mRNAs targeted to synapses, as well as promoting neurotransmitter release from presynaptic axon terminals (Gelinas et al., 2007; Jovanovic et al., 2000). These events lead to changes in dendritic morphology required for the induction and maintenance of synaptic plasticity (Thomas & Huganir, 2004). Both STEP61 and STEP46 dephosphorylate the regulatory tyrosine residue of ERK2, thereby playing a role in regulating the duration of ERK signaling (Paul et al., 2003).
The impact of STEP on these mechanisms has been demonstrated with a membrane-permeable TAT (transactivator of transcription)-STEP-cysteine to serine isoform. TAT-STEP (CS) is an inactive variant of STEP, which binds to but does not release its substrates, as release depends on dephosphorylation (Snyder et al., 2005; Tashev et al., 2009). Infusion of TAT-STEP (CS) into the lateral amygdale of rats had no effect on the acquisition of Pavlovian fear conditioning but blocked the consolidation of these memories, suggesting that the inhibition of ERK-mediated downstream events is required for memory consolidation (Paul et al., 2007). Further insights into the role of STEP in regulating ERK2 are provided by studies involving the STEP knock out mouse (STEP KO). The hippocampus of STEP KOs show increased activation of ERK1/2 and its downstream phosphorylation targets, CREB and ELK transcription factors (Venkitaramani et al., 2011; Venkitaramani et al., 2009). Furthermore, STEP KO mice perform better in hippocampus-dependent memory tasks compared to wild-type littermates, which is consistent with prolonged ERK1/2 activation (Venkitaramani et al., 2011).
p38 is a second member of the MAPK family that is dephosphorylated and inactivated by STEP (Munoz et al., 2003; Poddar et al., 2010; Xu et al., 2009). p38 activation plays a role in NMDAR-mediated neuronal excitotoxicity and initiates cell death pathways (Bossy-Wetzel et al., 2004; Semenova et al., 2007). Excess glutamate release results in p38 phosphorylation by preferential activation of extrasynaptic GluN2B-containing NMDARs, and p38 in turn phosphorylates several key proteins involved in cell death pathways (Poddar et al., 2010; Xu et al., 2009). Extrasynaptic NMDAR activation results in STEP61 cleavage by calpain, resulting in a nonfunctional isoform, STEP33. STEP33 lacks an intact KIM domain and does not interact with its substrates, including p38. This leads to the sustained activation of p38 and favors p38-mediated cell death pathways. Preventing STEP61 cleavage with a peptide corresponding to the calpain cleavage site protects neurons from glutamate-mediated excitotoxicity (Xu et al., 2009). This neuroprotective effect is accompanied by decreased STEP61 cleavage and decreased p38 phosphorylation at its regulatory tyrosine residue.
In summary, ERK2 and p38 are differentially regulated by STEP61, depending on whether synaptic or extrasynaptic NMDARs are activated. Stimulation of synaptic NMDARs results in the degradation of STEP61 by the UPS, favoring the development of synaptic plasticity and neuronal survival. Extrasynaptic NMDAR stimulation results in STEP61 cleavage by calpain, p38 activation, and promotes cell death. Both of these events are implicated at different stages of in AD. The role of STEP in regulating p38-mediated pathway and neuronal death in later stage of AD is under investigation.
2. Fyn
Fyn is a member of Src family of tyrosine kinases that was originally identified as a protooncogene regulating cellular growth (Semba et al., 1986). It is a nonreceptor tyrosine kinase associated in part with the cytoplasmic side of the plasma membrane. Fyn is targeted to the postsynaptic density and regulates neuronal signaling, including synaptic plasticity (Ali & Salter, 2001; Husi et al., 2000; Walikonis et al., 2000). Fyn activity is regulated by its own tyrosine phosphorylation; it is activated by autophosphorylation at a tyrosine residue (Tyr420), whereas phosphorylation by C-terminal Src kinase (Tyr531) leads to Fyn inactivation (Sun et al., 1998; Superti-Furga, et al., 1993). One role that Fyn plays in synaptic strengthening is to participate in the trafficking of NMDARs (Kohr & Seeburg, 1996; Lau & Huganir, 1995). Fyn phosphorylates GluN2B subunit at Y1472 residue in a conserved (YEKL) motif of, resulting in NMDAR insertion into membrane (Nakazawa, Komai, & Tezuka et al., 2001; Roche et al., 2001).
STEP binds to Fyn by interacting with the first polyproline domain and the KIM domain. STEP opposes Fyn activation by dephosphorylating the tyrosine residue (Tyr420) (Nguyen et al., 2002). STEP KO mice have increased Fyn tyrosine phosphorylation at Tyr420 and increased phosphorylation of NR2B GluN2B subunit at Tyr1472. Moreover, STEP KO mice show increased surface NMDAR levels, enhanced theta-burst LTP in hippocampal slices, and improved hippocampus-dependent memory (Venkitaramani et al., 2011; Zhang et al., 2010). Together, these findings suggest that STEP regulation of Fyn contributes to suppression of synaptic plasticity and memory consolidation.
3. Glutamate Receptors
Glutamate is the most abundant excitatory neurotransmitter in the central nervous system. It is involved in several physiological processes including synaptic plasticity, learning and memory, and several pathological processes that promote excitotoxicity through excessive glutamate release (Lau & Tymianski, 2010; Riedel et al., 2003). Glutamate binding to metabotropic glutamate receptors (mGluR) mediates cellular signaling via G-protein coupled pathways. Glutamate activation of ionotropic glutamate receptors leads to ion influx and changes in the postsynaptic membrane potential, which in turn activates signaling cascades inside neurons (Mayer & Armstrong, 2004; Traynelis et al., 2010). As mentioned earlier, two major classes of ionotropic receptors regulated by STEP include the NMDARs and AMPARs, both of which play major roles in synaptic plasticity and learning and memory (Pelkey et al., 2002; Snyder et al., 2005; Zhang et al., 2008). In AD, Aβ-mediated synaptotoxicity is associated with decreased NMDARs and AMPARs-dependent excitatory synaptic transmission, decreased surface receptor levels, and spine loss. These changes in the glutamatergic function may eventually lead to synaptic depression, alterations in synaptic networks, and cognitive deficits associated with the progression of AD (Palop & Mucke, 2010).
(a) NMDARs are tetramers composed of two GluN1 (formerly known as NR1) subunits and two GluN2 subunits (GluN2A–GluN2D), and less commonly, the GluN3 subunit. NMDARs are ligand-gated ion channels activated by a selective pharmacological agonist called NMDA. NMDARs require the co-agonist glycine for full activation. A distinctive feature of NMDAR activation is the requirement for strong postsynaptic depolarization. Activation requires both glutamate/glycine binding and strong post-synaptic membrane depolarization to remove an internal Mg2+ from blocking the channel pore. NMDARs are selectively permeable to Ca2+ ions, which activate numerous signaling molecules including the protein kinases and protein phosphatases required for LTP and long-term depression (LTD) (Cull-Candy et al., 2001; Rebola et al., 2010).
Surface expression and channel function of NMDARs is modulated by the Src family kinases such as Fyn (Nakazawa et al., 2001; Nakazawa et al., 2006; Roche et al., 2001). Fyn phosphorylates the GluN2B subunit at a conserved motif, leading to exocytosis of the GluN1/GluN2B receptor complex. STEP opposes NMDAR surface expression by two parallel pathways: it inactivates Fyn and dephosphorylates the Tyr1472 of the GluN2B subunit (Braithwaite et al., 2006a; Nguyen et al., 2002; Pelkey et al., 2002; Snyder et al., 2005). Dephosphorylated Tyr1472 of GluN2B is a docking site for adaptor protein AP-2 and promotes the internalization of NMDA receptor by a clathrin-mediated endocytic pathway (Lavezzari et al., Roche, 2003).
Dysregulation of NMDAR function and trafficking is involved in several neuropsychiatric disorders including AD (Lau & Zukin, 2007). STEP represents one mechanism by which Aβ regulates NMDAR trafficking (Selkoe, 2008; Venkitaramani et al., 2007). Aβ binds to α7 nAChRs with high affinity and activates calcineurin (PP2B), which leads to STEP dephosphorylation within the KIM domain. Activated STEP dephosphorylates GluN2B, leading to the internalization of surface NMDARs. Application of synthetic oligomeric Aβ peptides or Aβ oligomers (derived from 7PA2 conditioned medium) to primary cortical neurons or cortical slices leads to STEP activation (Kurup et al., 2010a; Snyder et al., 2005). Reduced STEP phosphorylation is associated with decreased surface of GluN1 and GluN2B complexes. In Aβ overexpressing AD mouse models, STEP phosphorylation is significantly decreased at its KIM domain as determined by a phospho-specific antibody against STEP ser221 (Kurup et al., 2010a; Snyder et al., 2005). The surface expression of NMDARs is significantly elevated in STEP KO mice. Cortical cultures derived from STEP KO mice are insensitive to the affects of Aβ in mediating NMDAR receptor internalization, suggesting a critical role of STEP in regulation of NMDAR trafficking by Aβ (Kurup et al., 2010b).
(b) AMPARs are ligand-gated ion channels composed of the hetero-oligomeric subunits GluA1 to GluA4. AMPARs are permeable to cations such as Na+ and K+ and to a lesser extent Ca2+. The presence of the GluA2 subunit in the channel makes it less permeable to calcium ions. AMPARs mediate fast synaptic transmission leading to depolarization of postsynaptic membranes and the removal of the Mg2+ block from NMDARs. AMPARs thus play an important role in synaptic plasticity and long-term memory (Santos, et al., 2009; Traynelis et al., 2010).
LTP and LTD are synaptic plasticity events, which are suggested to play a role in the regulation of synaptic strength. Trafficking of AMPARs play a vital role in LTP and LTD, and is regulated by different kinases and phosphatases (Anggono & Huganir, 2012). Recent studies show that tyrosine phosphatases are implicated in LTD (Moult et al., 2006). STEP is locally translated at the synapse during mGluR-dependent LTD and regulates AMPAR trafficking. Activation of mGluRs by agonist DHPG (S-3,5-dihydroxyphenylglycine) is correlated with increased STEP translation, decreased tyrosine phosphorylation of GluA2 subunit and internalization of AMPAR subunits form neuronal surface (Zhang et al., 2008). DHPG-induced AMPAR endocytosis and GluA2 dephosphorylation in hippocampal cultures and slices are blocked by a substrate trapping dominant negative STEP protein [TAT-STEP (CS)]. In addition, STEP KO cultures fail to show AMPAR internalization upon stimulation with DHPG, but are rescued by the addition of WT STEP protein (TAT-STEP WT) suggesting the role of STEP activity in AMPA receptor internalization (Zhang et al., 2008). These results suggest STEP activity is required to regulate AMPAR trafficking. The identity of the tyrosine residue(s) in GluA2 that are dephosphorylated by STEP is not known. AMPAR trafficking is modulated by Aβ. Adding Aβ to cortical cultures or slices causes synaptic depression and is associated with the loss of dendritic spines and the removal of AMPARs from the membrane (Almeida et al., 2005; Hsieh et al., 2006; Parameshwaran et al., 2007). Recent studies shed light on the role of STEP in Aβ-mediated endocytosis of AMPARs. STEP61 levels and activity are increased when Aβ is added to cortical cultures or slices and is associated with decreases in surface AMPAR subunits GluA1/GluA2, as well as NMDAR subunits GluN1/GluN2B (Zhang et al., 2011). This effect is specific to Aβ oligomers but not monomers. Adding Aβ oligomers leads to STEP activation by dephosphorylation of the regulatory serine residue within the KIM domain. The catalytic activity of immunoprecipitated STEP has been analyzed by in vitro phosphatase assays that use a phospho-substrate corresponding to the GluA2 C-terminal region. Together, these results indicate that STEP activation contributes to the Aβ-mediated endocytosis of both NMDARs and AMPARs (Kurup et al., 2010b; Zhang et al., 2011).
D. Regulation of STEP by Beta Amyloid
Aβ peptide is derived from amyloid precursor protein (APP) by the sequential action of β and γ secretases (Turner, et al., 2003; Wolfe, 2010). The Aβ peptide that is generated by this process slowly accumulates in the brain and is thought to contribute to the pathophysiology of AD. Recent studies indicate that soluble Aβ oligomers formed in the initial stages of AD, even before amyloid plaques formation, disrupt synaptic function (Palop & Mucke, 2010). This idea is supported by studies showing that soluble Aβ oligomers inhibit LTP (Walsh et al., 2002), induce synapse loss (Lacor et al., 2007), and cause cognitive defects in animal models (Shankar et al., 2008).
STEP opposes synaptic strengthening by down regulating several enzymes involved in synaptic plasticity (Braithwaite et al., 2006b; Goelbel-Goody et al., 2012). Under normal conditions, STEP is either removed or inactivated to favor synaptic strengthening. In contrast, several neurological disorders involve STEP accumulation and overactivation. For example, Aβ activates STEP by two mechanisms: (1) dephosphorylating the KIM domain of STEP by activating PP2B (Snyder et al., 2005) and (2) blocking efficient STEP degradation by inhibiting the proteasome system (Kurup et al., 2010a). Aβ binds to the α7 nAChRs through a critical aromatic residue (Tyr188) present in the agonist binding domain and activates the calcium-dependent phosphatase PP2B (Snyder et al., 2005; Tong, Arora, White, & Nichols, 2011). Activation of the PP2B/PP1 pathway dephosphorylates STEP at the KIM domain, thereby increasing the ability of STEP to interact with its substrates. As previously mentioned, PKA phosphorylation of STEP within its KIM domain inhibits the affinity of STEP for its substrates, whereas STEP dephosphorylation by PP2B/PP1 increases its affinity. PP2B-mediated STEP activation leads to increased binding and dephosphorylation of GluN2B and subsequently enhanced endocytosis of the NMDA receptors. Furthermore, Aβ-mediated NMDAR endocytosis is blocked by the α7 nAChR antagonist, α-bungarotoxin, and by the PP2B inhibitor, cyclosporine, as well as a membrane permeable TAT-STEP (CS), which preferentially binds to STEP substrates and competes with endogenous STEP protein (Snyder et al., 2005).
Aβ also regulates STEP levels by an independent mechanism involving the UPS (Kurup et al., 2010a). Aβ inhibits proteasome activity and causes accumulation of several proteins that are normally degraded by the proteasomal pathway (Keller et al., 2000; Tseng et al., 2008). In human AD brains, decreased proteasomal activity is associated with an accumulation of ubiquitin-immunoreactive inclusion bodies (Lam et al., 2000; Mori, et al., 1987). AD mouse models and exogenous Aβ treated cultures show an accumulation of several UPS substrates, suggesting a defect in the clearance of these proteins by proteasomes (Almeida, et al., 2006; David et al., 2002; Oh et al., 2005; Qing et al., 2004). In support of this hypothesis, an increase in STEP levels is observed in cortical cultures treated with Aβ-enriched condition medium (derived from APP expressing 7PA2 cell lines) (Walsh et al., 2002). The increase in STEP levels is insensitive to translation or transcriptional inhibitors, and this effect is specific to Aβ in the conditioned medium. Immunodepletion of Aβ from conditioned medium prior to adding it to cultures blocks the increase in STEP levels. Aβ-stimulation of cortical cultures leads to dose-dependent increases in STEP levels and decreases in membrane bound NMDAR receptors. Similarly, mutant APP mouse models that express high levels of Aβ show a progressive increase in STEP levels with age that correlates with increases in Aβ species in the cortex. In this mouse model of AD, increased STEP levels are associated with decreased expression of NMDAR and AMPAR subunits in the membrane (Kurup et al., 2010a; Zhang et al., 2011). Together, these findings highlight STEP regulation by Aβ species through two parallel pathways (Fig. 2). Aβ binding to α7 nAChRs activates STEP through a PP2B/PP1-dependent dephosphorylation of STEP. Aβ also blocks STEP degradation through inhibition of the proteasome, leading to increased STEP expression. The kinetics of these events show that application of Aβ to cortical slices initially decreases phospho-STEP levels, which is followed by a gradual increase in STEP levels and then subsequent loss of surface GluN2B (Kurup et al., 2010a). Further studies with STEP KO cultures confirm the direct implication of STEP on Aβ-mediated glutamate receptor endocytosis. Treatment of wild-type cultures with Aβ-containing conditioned medium decreases the number of GluN1/GluN2B and GluA1/GluA2 subunits in the membrane as examined by surface biotinylation experiments. In contrast, Aβ-containing conditioned medium does not recapitulate the decrease in GluN1/GluN2B and GluA1/GluA2 subunits in STEP KO cultures, which clearly explained the role of STEP in mediating glutamate receptor endocytosis through Aβ (Kurup et al., 2010b; Zhang et al., 2011).
FIGURE 2. Role of STEP in Alzheimer’s disease.
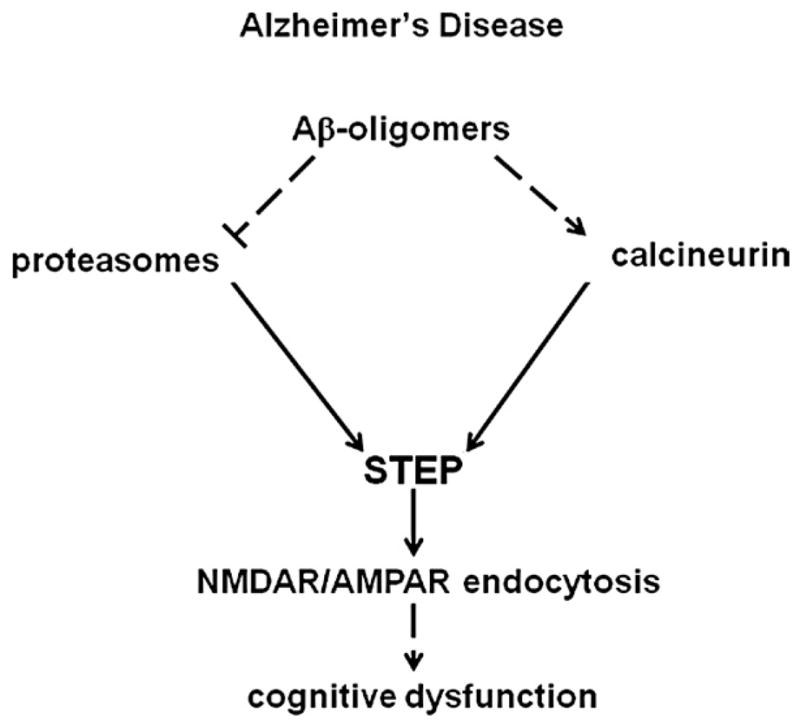
In Alzheimer’s disease, Aβ-oligomers activate STEP by two parallel pathways. (1) Aβ-oligomers bind to α7-nicotinic receptors and cause calcinuerin-dependent dephosphorylation of STEP at its KIM domain (2) Aβ-oligomers block the proteasome-mediated degradation of STEP and increase STEP levels. Both these events lead to increase in STEP function and increased endocytosis of NMDA and AMPA receptors causing cognitive dysfunction.
E. Transgenic AD Mouse Models
Transgenic mouse models have made important contributions to our understanding of AD (German & Eisch, 2004). These animal models carry one or more human mutant gene that is implicated in familial AD such as: APP, presenelin-1, or Tau. These models recapitulate some, but not all of the features of the disease phenotype, but have been useful for the exploration of underlying pathological mechanisms of the disease, disease progression, and to identify new therapeutic strategies (Ashe & Zahs, 2010). Although several AD mouse models have been characterized, this review focuses mainly on two widely used transgenic AD mouse models, Tg2576 and the triple transgenic mice (3×Tg-AD), for which data for STEP exist.
1. Tg2576 Mouse Model
The Tg2576 mouse model carries a mutant APP (APP695SWE) found in human familial AD and produces excess Aβ. The mice show synaptic and cognitive defects in the early stages of the disease, and amyloid plaques accumulate as the disease progresses (Hsiao et al., 1996). This model has been used to test the effect of soluble Aβ in the early stages of the disease and its effects on synaptic plasticity and cognitive function. The mice show significant cognitive defects associated with reduced spine density as early as 4 months of age, decreased hippocampal neurotransmission, and decreased LTP (Jacobsen et al., 2006). These changes occur even before the apparent accumulation amyloid plaques, supporting the idea that early effects of Aβ result in synaptic perturbations.
STEP61 levels in Tg2576 mouse brains increase progressively with age from 6 months onwards, and soluble Aβ increases in the cortex at the same time. The examination of NMDARs and AMPARs in synaptic membrane fractions of Tg2576 cortex show a significant decrease in GluN1, and GluN2B subunits of the NMDA receptor, and decreases in Tyr1472 phosphorylation of the GluN2B subunit (Kurup et al., 2010a). A reduction in AMPAR subunits GluA1/GluA2 is also observed in the cortical membrane fraction of Tg2576 compared to wild type (Zhang et al., 2011). The increase in STEP61 levels is associated with decreases in STEP phosphorylation at KIM domain, suggesting that STEP is overactive and causes excessive internalization of NMDARs and AMPARs. To directly access the catalytic activity of accumulated STEP, it was immunoprecipitated from the membrane fractions of Tg2576 brain tissue and subjected to an in vitro phosphatase assay. STEP immunoprecipitated from Tg2576 brain shows increased phosphatase activity of a phospho-GluN2B substrate (Kurup et al., 2010a; Zhang et al., 2011).
Work in the Tg2576 AD mouse model confirms the role of STEP in glutamate receptor trafficking. This was tested in STEP knockout mice that replace the genomic STEP phosphatase domain with the neomycin gene in embryonic stem (ES) cells by homologous recombination (Venkitaramani et al., 2009). These mice are viable and show no obvious phenotypic abnormalities. Biochemical characterization of STEP KO brains show a absence of STEP expression, increased tyrosine phosphorylation of STEP substrates, and increased membrane expression of glutamate receptors, including NMDARs (Venkitaramani et al., 2009, 2011; Zhang et al., 2010) and AMPARs in synaptosomal membrane fractions (Zhang et al., 2008). The potential effect of genetically lowering STEP levels in Tg2576 mice was examined by crossing STEP KOs with Tg2576 mice, resulting in Tg2576 progeny that have high levels of Aβ but are null for STEP. Glutamate receptor levels (GluN1/GluN2B and GluA1/GluA2) were analyzed in the cortical tissue of this double transgenic mouse. As predicted, membrane expression of glutamate receptors (GluN1/GluN2B and GluA1/GluA2) was increased, suggesting that the STEP elimination is sufficient to rescue the biochemical defects in the cortex of Tg2576 mice (Kurup et al., 2010a, 2010b; Zhang et al., 2011). Aβ levels are comparable in Tg2576 and double transgenic mice in the absence of STEP, suggesting that the rescue is not due to altered Aβ metabolism or clearance.
2. Triple-Transgenic Mouse Model
The triple-transgenic mouse model (3×Tg-AD) of AD carries three transgenes: PS1M146V, APPswe, and tauP301L (Oddo et al., 2003a). All three genes are implicated in human AD and the accumulation of characteristic Aβ plaques (composed of Aβ peptides) and NFTs (composed of hyperphosphorylated Tau). 3×Tg-AD mice show synaptic dysfunction and LTP defects even before the plaques and tangles are apparent. This supports the amyloid cascade hypothesis, suggesting that synaptic dysfunction caused by soluble Aβ is responsible for cognitive impairment in early stages of AD, and is independent of plaques or tangles (Oddo, et al., 003b).
The 3×Tg-AD mouse model mimics human AD in several ways, including steady-state expression of APP and the Tau transgene, which are present in the hippocampus and cortex, whereas other brain regions such as the cerebellum show the least expression (Oddo et al., 2003a). Recent studies with 3×Tg-AD (Oddo et al., 2003a) and STEP KO (Venkitaramani et al., 2009) provide direct evidence for the role of STEP in AD pathophysiology. Zhang and colleagues crossed STEP KO mice with 3×Tg-AD to produce progeny of 3×Tg-AD that are null for STEP (Zhang et al., 2010). The progeny of these double mutants were tested with behavioral tasks to assess cognitive function. At 6 months of age, 3×Tg-AD mice show significant impairment in spatial reference memory, spatial working memory, and memory tasks mediated by the hippocampus. In addition, NMDAR subunits (GluN1/GluN2B) are significantly reduced in the hippocampal synaptosomal fractions of 3×Tg-AD compared to wild type, which is associated with increases in STEP levels. Interestingly, the double mutants (3×Tg-AD-STEP KO) showed cognitive rescue in similar behavioral paradigms at 6 months of age, indicating that lowering STEP is sufficient to rescue the cognitive defects observed in the 3×Tg-AD mice.
In addition to cognitive rescue, the double mutant hippocampal synaptosomal fractions showed restored GluN1/GluN2B subunit levels, which were similar to wild-type receptor levels. Electrophysiological studies show that theta-burst LTP is significantly enhanced in double mutants compared to 3×Tg-AD. The attenuation of cognitive deficits in the double mutants occurred despite the continued elevation of Aβ and phospho-tau. This finding suggests that reducing STEP levels in the early stages of AD was beneficial (Zhang et al., 2010). Further study is needed to determine whether the cognitive rescue is restricted to early stages of AD or persists to later stages when amyloid accumulation and NFTs are increased. Nonetheless, these findings suggest that STEP is a link between the toxic effects of Aβ, synaptic dysfunction, and cognitive deficits in AD.
III. STEP Inhibitors
The integral role STEP plays in synaptic function and the striking implications for its role in AD point to this molecule as an important target for drug discovery. Identifying small molecules that inhibit STEP activity has potential therapeutic value for the treatment of AD. Tyrosine phosphatase catalysis occurs within a highly conserved phosphatase domain. Most existing PTP inhibitors have a tyrosine phosphate-mimicking group that interacts with a highly conserved phosphate-binding loop in the catalytic center (reviewed by Blaskovich, 2009). It is also possible that small molecules that stabilize PTPs in the open inactive conformation of PTP may be useful for identifying STEP inhibitors. In the active state, the flexible WPD (Try-Pro-Asp) loop plays an important role in PTP catalysis (Barr, 2010). The WPD loop is more flexible in STEP and contains an atypical open conformation that is dominated by charged residues such as glutamine; it is located further away from the catalytic site, thereby creating a large binding pocket in the WPD loop (Eswaran et al., 2006). This binding pocket might be an interacting site for small molecules that increase the specificity for STEP compared to other PTPs. Strategies to identify STEP inhibitors are in progress; hopefully STEP inhibitors will be available in the market in near future.
IV. Conclusion
Recent advances have helped to clarify the regulation of STEP, identify its substrates, and explore its contribution to AD. STEP dephosphorylates and inactivates specific substrates including ERK1/2, p38, and Fyn that begin to explain its role in neuronal signaling. STEP down regulates membrane expression of NMDARs and AMPARs, and thereby opposes the development of synaptic strengthening. STEP KO mice show enhanced theta-burst LTP in the hippocampus and perform better in some hippocampus-dependent memory tasks. The synaptic and cognitive changes that occur in STEP KO mice are associated with increased NMDAR and AMPARs at synaptic membrane. STEP activity and function are both upregulated in AD. Increased STEP levels are found in human AD brains and in several AD mouse models. The two mechanisms that result in the upregulation of STEP activity are increased dephosphorylation as well as decreased degradation by the proteasome. Both of these events contribute to increased STEP activity and result in excessive internalization of NMDARs and AMPARs. Lowering STEP levels attenuates the biochemical and cognitive deficits observed in AD mouse models and validate STEP as a potential target for drug discovery.
Acknowledgments
This work was supported by the National Institutes of Health, Grants [MH091037, MH052711] to P.J.L and Grant [AG09464] to A.C.N. We thank Dr. Marilee Ogren for critically reading and editing the manuscript. We also thank all members of the Lombroso laboratory for helpful discussions during the manuscript preparation.
Abbreviations
- AMPARs
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- ERK
extracellular regulated kinase
- NMDARs
N-methyl-D-aspartate receptors
- PKA
protein kinase A
- PTPN5
protein tyrosine phosphatase nonreceptor type five
- TAT
transactivator of transcription
Footnotes
Conflict of Interest: The authors declare no conflict of interest regarding the work reported here.
References
- Adachi M, Sekiya M, Isobe M, Kumura Y, Ogita Z, Hinoda Y, et al. Molecular cloning and chromosomal mapping of a human protein-tyrosine phosphatase LC-PTP. Biochemical and Biophysical Research Communication. 1992;186:1607–1615. doi: 10.1016/s0006-291x(05)81592-x. [DOI] [PubMed] [Google Scholar]
- Ali DW, Salter MW. NMDA receptor regulation by Src kinase signalling in excitatory synaptic transmission and plasticity. Current Opinion in Neurobiology. 2001;11:336–342. doi: 10.1016/s0959-4388(00)00216-6. [DOI] [PubMed] [Google Scholar]
- Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, et al. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiology of Disease. 2005;20:187–198. doi: 10.1016/j.nbd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Almeida CG, Takahashi RH, Gouras GK. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. Journal of Neuroscience. 2006;26:4277–4288. doi: 10.1523/JNEUROSCI.5078-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anggono V, Huganir RL. Regulation of AMPA receptor trafficking and synaptic plasticity. Current Opinion in Neurobiology. 2012 doi: 10.1016/j.conb.2011.12.006. (Epub ahead of print) http://dx.doi.org/10.1016/j.bbr.2011.03.031. [DOI] [PMC free article] [PubMed]
- Ashe KH, Zahs KR. Probing the biology of Alzheimer’s disease in mice. Neuron. 2010;66:631–645. doi: 10.1016/j.neuron.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr AJ. Protein tyrosine phosphatases as drug targets: Strategies and challenges of inhibitor development. Future Medicinal Chemistry. 2010;2:1563–1576. doi: 10.4155/fmc.10.241. [DOI] [PubMed] [Google Scholar]
- Baum ML, Kurup P, Xu J, Lombroso PJ. A STEP forward in neural function and degeneration. Communictive & Integretive Biology. 2010;3:419–422. doi: 10.4161/cib.3.5.12692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaskovich MA. Drug discovery and protein tyrosine phosphatases. Current Medicinal Chemistry. 2009;16:2095–2176. doi: 10.2174/092986709788612693. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Talantova MV, Lee WD, Scholzke MN, Harrop A, Mathews E, et al. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron. 2004;41:351–365. doi: 10.1016/s0896-6273(04)00015-7. [DOI] [PubMed] [Google Scholar]
- Boulanger LM, Lombroso PJ, Raghunathan A, During MJ, Wahle P, Naegele JR. Cellular and molecular characterization of a brain-enriched protein tyrosine phosphatase. Journal of Neuroscience. 1995;15:1532–1544. doi: 10.1523/JNEUROSCI.15-02-01532.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braithwaite SP, Paul S, Nairn AC, Lombroso PJ. Synaptic plasticity: One STEP at a time. Trends in Neurosciences. 2006a;29:452–458. doi: 10.1016/j.tins.2006.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braithwaite SP, Adkisson M, Leung J, Nava A, Masterson B, Urfer R, et al. Regulation of NMDA receptor trafficking and function by striatal-enriched tyrosine phosphatase (STEP) European Journal of Neuroscience. 2006b;23:2847–2856. doi: 10.1111/j.1460-9568.2006.04837.x. [DOI] [PubMed] [Google Scholar]
- Bult A, Zhao F, Dirkx R, Jr, Sharma E, Lukacsi E, Solimena M, et al. STEP61: A member of a family of brain-enriched PTPs is localized to the endoplasmic reticulum. Journal of Neuroscience. 1996;16:7821–7831. doi: 10.1523/JNEUROSCI.16-24-07821.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bult A, Zhao F, Dirkx R, Jr, Raghunathan A, Solimena M, Lombroso PJ. STEP: A family of brain-enriched PTPs. Alternative splicing produces trans-membrane, cytosolic and truncated isoforms. European Journal of Cell Biology. 1997;72:337–344. [PubMed] [Google Scholar]
- Castellani RJ, Rolston RK, Smith MA. Alzheimer disease. Disease-a-Month. 2010;56:484–546. doi: 10.1016/j.disamonth.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Current Opinion Neurobiology. 2001;11:327–335. doi: 10.1016/s0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- David DC, Layfield R, Serpell L, Narain Y, Goedert M, Spillantini MG. Proteasomal degradation of tau protein. Journal of Neurochemistry. 2002;83:176–185. doi: 10.1046/j.1471-4159.2002.01137.x. [DOI] [PubMed] [Google Scholar]
- Davis S, Vanhoutte P, Pages C, Caboche J, Laroche S. The MAPK/ERK cascade targets both Elk-1 and cAMP response element-binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. Journal of Neuroscience. 2000;20:4563–4572. doi: 10.1523/JNEUROSCI.20-12-04563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb I, Poddar R, Paul S. Oxidative stress-induced oligomerization inhibits the activity of the non-receptor tyrosine phosphatase STEP61. Journal of Neurochemistry. 2011;116:1097–1111. doi: 10.1111/j.1471-4159.2010.07165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswaran J, von Kries JP, Marsden B, Longman E, Debreczeni JE, Ugochukwu E, et al. Crystal structures and inhibitor identification for PTPN5, PTPRR and PTPN7: A family of human MAPK-specific protein tyrosine phosphatases. Biochemical Journal. 2006;395:483–491. doi: 10.1042/BJ20051931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick CJ, Lombroso PJ. The role of Striatal-Enriched Protein Tyrosine Phosphatase (STEP) in cognition. Frontiers in Neuroanatomy. 2011;5:47. doi: 10.3389/fnana.2011.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German DC, Eisch AJ. Mouse models of Alzheimer’s disease: insight into treatment. Reviews in the Neuroscience. 2004;15:353–369. doi: 10.1515/revneuro.2004.15.5.353. [DOI] [PubMed] [Google Scholar]
- Goebel-Goody SM, Baum M, Paspalas CD, Fernandez SM, Carty NC, Kurup P, et al. Therapeutic implications for striatal-enriched protein tyrosine phosphatase (STEP) in neuropsychiatric disorders. Pharmacological Reviews. 2012;64:65–87. doi: 10.1124/pr.110.003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel-Goody SM, Davies KD, Alvestad Linger RM, Freund RK, Browning MD. Phospho-regulation of synaptic and extrasynaptic N-methyl-d-aspartate receptors in adult hippocampal slices. Neuroscience. 2009;158:1446–1459. doi: 10.1016/j.neuroscience.2008.11.006. [DOI] [PubMed] [Google Scholar]
- Greengard P, Allen PB, Nairn AC. Beyond the dopamine receptor: The DARPP-32/protein phosphatase-1 cascade. Neuron. 1999;23:435–447. doi: 10.1016/s0896-6273(00)80798-9. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nature Reviews. Molecular Cell Biology. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nature Reviews Neuroscience. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nature of Neuroscience. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hegde AN, Upadhya SC. Role of ubiquitin-proteasome-mediated proteolysis in nervous system disease. Biochimica et Biophysica Acta. 2011;1809:128–140. doi: 10.1016/j.bbagrm.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, et al. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nature of Neuroscience. 2000;3:661–669. doi: 10.1038/76615. [DOI] [PubMed] [Google Scholar]
- Ivanov A, Pellegrino C, Rama S, Dumalska I, Salyha Y, Ben-Ari Y, et al. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. Journal of Physiology. 2006;572:789–798. doi: 10.1113/jphysiol.2006.105510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, et al. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5161–5166. doi: 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Czernik AJ, Fienberg AA, Greengard P, Sihra TS. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nature Neuroscience. 2000;3:323–329. doi: 10.1038/73888. [DOI] [PubMed] [Google Scholar]
- Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer’s disease. Journal of Neurochemistry. 2000;75:436–439. doi: 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- Kohr G, Seeburg PH. Subtype-specific regulation of recombinant NMDA receptor-channels by protein tyrosine kinases of the src family. Journal of Physiology. 1996;492(Pt 2):445–452. doi: 10.1113/jphysiol.1996.sp021320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurup P, Zhang Y, Xu J, Venkitaramani DV, Haroutunian V, Greengard P, et al. Abeta-mediated NMDA receptor endocytosis in Alzheimer’s disease involves ubiquitination of the tyrosine phosphatase STEP61. Journal of Neuroscience. 2010a;30:5948–5957. doi: 10.1523/JNEUROSCI.0157-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurup P, Zhang Y, Venkitaramani DV, Xu J, Lombroso PJ. The role of STEP in Alzheimer’s disease. Channels (Austin) 2010b;4:347–350. doi: 10.4161/chan.4.5.12910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, et al. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. Journal of Neuroscience. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam YA, Pickart CM, Alban A, Landon M, Jamieson C, Ramage R, et al. Inhibition of the ubiquitin-proteasome system in Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:9902–9906. doi: 10.1073/pnas.170173897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Archiv. 2010;460:525–542. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nature Reviews Neuroscience. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- Lau LF, Huganir RL. Differential tyrosine phosphorylation of N-methyl-D-aspartate receptor subunits. Journal of Biological Chemistry. 1995;270:20036–20041. doi: 10.1074/jbc.270.34.20036. [DOI] [PubMed] [Google Scholar]
- Lavezzari G, McCallum J, Lee R, Roche KW. Differential binding of the AP-2 adaptor complex and PSD-95 to the C-terminus of the NMDA receptor subunit NR2B regulates surface expression. Neuropharmacology. 2003;45:729–737. doi: 10.1016/s0028-3908(03)00308-3. [DOI] [PubMed] [Google Scholar]
- Lombroso PJ, Murdoch G, Lerner M. Molecular characterization of a protein-tyrosine-phosphatase enriched in striatum. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:7242–7246. doi: 10.1073/pnas.88.16.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombroso PJ, Naegele JR, Sharma E, Lerner M. A protein tyrosine phosphatase expressed within dopaminoceptive neurons of the basal ganglia and related structures. Journal of Neuroscience. 1993;13:3064–3074. doi: 10.1523/JNEUROSCI.13-07-03064.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Armstrong N. Structure and function of glutamate receptor ion channels. Annual Review of Physiology. 2004;66:161–181. doi: 10.1146/annurev.physiol.66.050802.084104. [DOI] [PubMed] [Google Scholar]
- Mayford M. Protein kinase signaling in synaptic plasticity and memory. Current Opinion of Neurobiology. 2007;17:313–317. doi: 10.1016/j.conb.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Mori H, Kondo J, Ihara Y. Ubiquitin is a component of paired helical filaments in Alzheimer’s disease. Science. 1987;235:1641–1644. doi: 10.1126/science.3029875. [DOI] [PubMed] [Google Scholar]
- Moult PR, Gladding CM, Sanderson TM, Fitzjohn SM, Bashir ZI, Molnar E, et al. Tyrosine phosphatases regulate AMPA receptor trafficking during metabotropic glutamate receptor-mediated long-term depression. Journal of Neuroscience. 2006;26:2544–2554. doi: 10.1523/JNEUROSCI.4322-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz JJ, Tarrega C, Blanco-Aparicio C, Pulido R. Differential interaction of the tyrosine phosphatases PTP-SL, STEP and HePTP with the mitogen-activated protein kinases ERK1/2 and p38alpha is determined by a kinase specificity sequence and influenced by reducing agents. Biochemical Journal. 2003;372:193–201. doi: 10.1042/BJ20021941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa T, Komai S, Tezuka T, Hisatsune C, Umemori H, Semba K, et al. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-D-aspartate receptor. Journal of Biological Chemistry. 2001;276:693–699. doi: 10.1074/jbc.M008085200. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Komai S, Watabe AM, Kiyama Y, Fukaya M, Arima-Yoshida F, et al. NR2B tyrosine phosphorylation modulates fear learning as well as amygdaloid synaptic plasticity. EMBO Journal. 2006;25:2867–2877. doi: 10.1038/sj.emboj.7601156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TH, Liu J, Lombroso PJ. Striatal enriched phosphatase 61 dephosphorylates Fyn at phosphotyrosine 420. Journal of Biological Chemistry. 2002;277:24274–24279. doi: 10.1074/jbc.M111683200. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron. 2003a;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiology of Aging. 2003b;24:1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Oh S, Hong HS, Hwang E, Sim HJ, Lee W, Shin SJ, et al. Amyloid peptide attenuates the proteasome activity in neuronal cells. Mechanisms of Ageing Development. 2005;126:1292–1299. doi: 10.1016/j.mad.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Okamura A, Goto S, Nishi T, Yamada K, Yoshikawa M, Ushio Y. Postnatal ontogeny of striatal-enriched protein tyrosine phosphatase (STEP) in rat striatum. Experimental Neurology. 1997;145:228–234. doi: 10.1006/exnr.1997.6435. [DOI] [PubMed] [Google Scholar]
- Oyama T, Goto S, Nishi T, Sato K, Yamada K, Yoshikawa M, et al. Immunocytochemical localization of the striatal enriched protein tyrosine phosphatase in the rat striatum: A light and electron microscopic study with a complementary DNA-generated polyclonal antibody. Neuroscience. 1995;69:869–880. doi: 10.1016/0306-4522(95)00278-q. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nature of Neuroscience. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parameshwaran K, Sims C, Kanju P, Vaithianathan T, Shonesy BC, Dhanasekaran M, et al. Amyloid beta-peptide Abeta(1–42) but not Abeta(1–40) attenuates synaptic AMPA receptor function. Synapse. 2007;61:367–374. doi: 10.1002/syn.20386. [DOI] [PubMed] [Google Scholar]
- Paul S, Snyder GL, Yokakura H, Picciotto MR, Nairn AC, Lombroso PJ. The dopamine/D1 receptor mediates the phosphorylation and inactivation of the protein tyrosine phosphatase STEP via a PKA-dependent pathway. Journal of Neuroscience. 2000;20:5630–5638. doi: 10.1523/JNEUROSCI.20-15-05630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Nairn AC, Wang P, Lombroso PJ. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nature of Neuroscience. 2003;6:34–42. doi: 10.1038/nn989. [DOI] [PubMed] [Google Scholar]
- Paul S, Olausson P, Venkitaramani DV, Ruchkina I, Moran TD, Tronson N, et al. The striatal-enriched protein tyrosine phosphatase gates long-term potentiation and fear memory in the lateral amygdala. Biological Psychiatry. 2007;61:1049–1061. doi: 10.1016/j.biopsych.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipson O, Lord A, Gumucio A, O’Callaghan P, Lannfelt L, Nilsson LN. Animal models of amyloid-beta-related pathologies in Alzheimer’s disease. FEBS Journal. 2010;277:1389–1409. doi: 10.1111/j.1742-4658.2010.07564.x. [DOI] [PubMed] [Google Scholar]
- Poddar R, Deb I, Mukherjee S. Paul S NR2B-NMDA receptor mediated modulation of the tyrosine phosphatase STEP regulates glutamate induced neuronal cell death. Journal of Neurochemistry. 2010;115:1350–1362. doi: 10.1111/j.1471-4159.2010.07035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing H, Zhou W, Christensen MA, Sun X, Tong Y, Song W. Degradation of BACE by the ubiquitin-proteasome pathway. FASEB Journal. 2004;18:1571–1573. doi: 10.1096/fj.04-1994fje. [DOI] [PubMed] [Google Scholar]
- Raghunathan A, Matthews GA, Lombroso PJ, Naegele JR. Transient compartmental expression of a family of protein tyrosine phosphatases in the developing striatum. Brain Research. Developmental Brain Research. 1996;91:190–199. doi: 10.1016/0165-3806(95)00176-x. [DOI] [PubMed] [Google Scholar]
- Rebola N, Srikumar BN, Mulle C. Activity-dependent synaptic plasticity of NMDA receptors. Journal of Physiology. 2010;588:93–99. doi: 10.1113/jphysiol.2009.179382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel G, Platt B, Micheau J. Glutamate receptor function in learning and memory. Behavioural Brain Research. 2003;140:1–47. doi: 10.1016/s0166-4328(02)00272-3. [DOI] [PubMed] [Google Scholar]
- Roche KW, Standley S, McCallum J, Dune Ly C, Ehlers MD, Wenthold RJ. Molecular determinants of NMDA receptor internalization. Nature of Neuroscience. 2001;4:794–802. doi: 10.1038/90498. [DOI] [PubMed] [Google Scholar]
- Santos SD, Carvalho AL, Caldeira MV, Duarte CB. Regulation of AMPA receptors and synaptic plasticity. Neuroscience. 2009;158:105–125. doi: 10.1016/j.neuroscience.2008.02.037. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behavioural Brain Research. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semba K, Nishizawa M, Miyajima N, Yoshida MC, Sukegawa J, Yamanashi Y, et al. Yes-related protooncogene, syn, belongs to the protein-tyrosine kinase family. Proceedings of the National Academy of Sciences of the United States of America. 1986;83:5459–5463. doi: 10.1073/pnas.83.15.5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova MM, Maki-Hokkonen AM, Cao J, Komarovski V, Forsberg KM, Koistinaho M, et al. Rho mediates calcium-dependent activation of p38alpha and subsequent excitotoxic cell death. Nature of Neuroscience. 2007;10:436–443. doi: 10.1038/nn1869. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nature Medicine. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma E, Zhao F, Bult A, Lombroso PJ. Identification of two alternatively spliced transcripts of STEP: A subfamily of brain-enriched protein tyrosine phosphatases. Brain Research Molecular Brain Research. 1995;32:87–93. doi: 10.1016/0169-328x(95)00066-2. [DOI] [PubMed] [Google Scholar]
- Shiozuka K, Watanabe Y, Ikeda T, Hashimoto S, Kawashima H. Cloning and expression of PCPTP1 encoding protein tyrosine phosphatase. Gene. 1995;162:279–284. doi: 10.1016/0378-1119(95)00306-q. [DOI] [PubMed] [Google Scholar]
- Shumway SD, Maki M, Miyamoto S. The PEST domain of IkappaBalpha is necessary and sufficient for in vitro degradation by mu-calpain. Journal of Biological Chemistry. 1999;274:30874–30881. doi: 10.1074/jbc.274.43.30874. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nature of Neuroscience. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Spencer ML, Theodosiou M, Noonan DJ. NPDC-1, a novel regulator of neuronal proliferation, is degraded by the ubiquitin/proteasome system through a PEST degradation motif. Journal of Biological Chemistry. 2004;279:37069–37078. doi: 10.1074/jbc.M402507200. [DOI] [PubMed] [Google Scholar]
- Sun G, Sharma AK, Budde RJ. Autophosphorylation of Src and Yes blocks their inactivation by Csk phosphorylation. Oncogene. 1998;17:1587–1595. doi: 10.1038/sj.onc.1202076. [DOI] [PubMed] [Google Scholar]
- Superti-Furga G, Fumagalli S, Koegl M, Courtneidge SA, Draetta G. Csk inhibition of c-Src activity requires both the SH2 and SH3 domains of Src. EMBO Journal. 1993;12:2625–2634. doi: 10.1002/j.1460-2075.1993.tb05923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Current Opinion in Neurobiology. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Tashev R, Moura PJ, Venkitaramani DV, Prosperetti C, Centonze D, Paul S, et al. A substrate trapping mutant form of striatal-enriched protein tyrosine phosphatase prevents amphetamine-induced stereotypies and long-term potentiation in the striatum. Biological Psychiatry. 2009;65:637–645. doi: 10.1016/j.biopsych.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Annals of Neurology. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Thiels E, Klann E. Extracellular signal-regulated kinase, synaptic plasticity, and memory. Reviews in the Neuroscience. 2001;12:327–345. doi: 10.1515/revneuro.2001.12.4.327. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nature Reviews Neuroscience. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Tong M, Arora K, White MM, Nichols RA. Role of key aromatic residues in the ligand-binding domain of alpha7 nicotinic receptors in the agonist action of beta-amyloid. Journal of Biological Chemistry. 2011;286:34373–34381. doi: 10.1074/jbc.M111.241299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacological Reviews. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng BP, Green KN, Chan JL, Blurton-Jones M, LaFerla FM. Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiology of Aging. 2008;29:1607–1618. doi: 10.1016/j.neurobiolaging.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner PR, O’Conner K, Tate WP, Abraham WC. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Progress in Neuobiology. 2003;70:1–32. doi: 10.1016/s0301-0082(03)00089-3. [DOI] [PubMed] [Google Scholar]
- Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol JC, et al. Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:491–496. doi: 10.1073/pnas.0408305102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitaramani DV, Moura PJ, Picciotto MR, Lombroso PJ. Striatal-enriched protein tyrosine phosphatase (STEP) knockout mice have enhanced hippocampal memory. European Journal of Neuroscience. 2011;33:2288–2298. doi: 10.1111/j.1460-9568.2011.07687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitaramani DV, Chin J, Netzer WJ, Gouras GK, Lesne S, Malinow R, et al. Beta-amyloid modulation of synaptic transmission and plasticity. Journal of Neuroscience. 2007;27:11832–11837. doi: 10.1523/JNEUROSCI.3478-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitaramani DV, Paul S, Zhang Y, Kurup P, Ding L, Tressler L, et al. Knockout of striatal enriched protein tyrosine phosphatase in mice results in increased ERK1/2 phosphorylation. Synapse. 2009;63:69–81. doi: 10.1002/syn.20608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walikonis RS, Jensen ON, Mann M, Provance DW, Jr, Mercer JA, Kennedy MB. Identification of proteins in the postsynaptic density fraction by mass spectrometry. Journal of Neuroscience. 2000;20:4069–4080. doi: 10.1523/JNEUROSCI.20-11-04069.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wolfe MS. Structure, mechanism and inhibition of gamma-secretase and presenilin-like proteases. Biological Chemistry. 2010;391:839–847. doi: 10.1515/BC.2010.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Kurup P, Zhang Y, Goebel-Goody SM, Wu PH, Hawasli AH, et al. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. Journal of Neuroscience. 2009;29:9330–9343. doi: 10.1523/JNEUROSCI.2212-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Kurup P, Bartos JA, Patriarchi T, Hell JW, Lombroso PJ. STriatal-enriched protein tyrosine phosphatase (STEP) regulates Pyk2 activity. Journal of Biological Chemistry. 2012 doi: 10.1074/jbc.M112.3686542012. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JJ, Ehlers MD. Emerging roles for ubiquitin and protein degradation in neuronal function. Pharmacological Reviews. 2007;59:14–39. doi: 10.1124/pr.59.1.4. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Kurup P, Xu J, Carty N, Fernandez SM, Nygaard HB, et al. Genetic reduction of striatal-enriched tyrosine phosphatase (STEP) reverses cognitive and cellular deficits in an Alzheimer’s disease mouse model. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:19014–19019. doi: 10.1073/pnas.1013543107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kurup P, Xu J, Anderson GM, Greengard P, Nairn AC, et al. Reduced levels of the tyrosine phosphatase STEP block beta amyloid-mediated GluA1/GluA2 receptor internalization. Journal of Neurochemistry. 2011;119:664–672. doi: 10.1111/j.1471-4159.2011.07450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Venkitaramani DV, Gladding CM, Kurup P, Molnar E, Collingridge GL, et al. The tyrosine phosphatase STEP mediates AMPA receptor endocytosis after metabotropic glutamate receptor stimulation. Journal of Neuroscience. 2008;28:10561–10566. doi: 10.1523/JNEUROSCI.2666-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]