

Abstract
Autophagy and mitophagy are important cellular processes that are responsible for breaking down cellular contents, preserving energy and safeguarding against accumulation of damaged and aggregated biomolecules. This graphic review gives a broad summary of autophagy and discusses examples where autophagy is important in controlling protein degradation. In addition we highlight how autophagy and mitophagy are involved in the cellular responses to reactive species and mitochondrial dysfunction. The key signaling pathways for mitophagy are described in the context of bioenergetic dysfunction.
Keywords: Neurodegeneration, Alpha-synuclein, Lysosomes, Fission, Fusion, Reactive species, Cellular bioenergetics, Pharmacological agents
Highlights
► Autophagy is a lysosomal-mediated intracellular protein degradation pathway. ► Autophagy is important for turnover and quality control of proteins and organelles. ► Autophagy and mitophagy are highly regulated. ► Autophagy and mitophagy play important roles in aging and diseases.
Introduction
Cellular damage occurs in response to genetic perturbations, nutrient deprivation, aging, and environmental toxins. The task of managing general and specific cellular damage is largely under the control of the highly regulated process called autophagy. The term autophagy is used to describe lysosomal-mediated degradation of intracellular contents, which can be divided into 3 basic mechanisms: (1) chaperone-mediated autophagy, (2) microautophagy, and (3) macroautophagy (Fig. 1). Chaperone-mediated autophagy, initiated by chaperone Hsc70, recognizes one protein at a time, and Hsc70 carries the protein to the lysosomes via binding to the lysosomal associated membrane protein (LAMP2A). The proteins recognized by Hsc70 contain the KFERQ consensus sequence [1]. Whether additional chaperones and lysosomal receptors participate in chaperone-mediated autophagy is unknown. Microautophagy is achieved by invagination of lysosomal membranes. Lipid, protein or organelles can be degraded through this pathway. Recent studies have shown that proteins containing the KFERQ consensus sequence may also be recruited to the lysosomes via phosphatidylserine, and degraded by microautophagy [1]. Whether lipid, organelles and other proteins are marked by specific modifications to be recognized by the lysosomes is highly likely but the majority of these have yet to be defined.
Fig. 1.
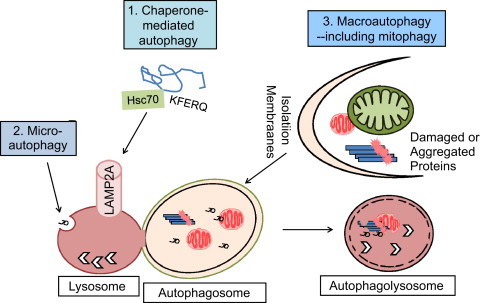
Autophagic and mitophagic degradation of proteins and organelles. Autophagy is a term used to describe lysosomal-mediated degradation of proteins, lipids, and organelles. Three major autophagy pathways have been described. (1) Chaperone-mediated autophagy involves chaperone protein heat shock protein, Hsc70 recognizing target proteins that have a KFERQ consensus sequence, followed by binding to lysosomal-associated membrane protein LAMP-2A, and transport of the targeted protein to the lysosomes to be degraded. (2) Microautophagy involves invagination of lysosomal membranes to encircle cellular contents that may include proteins and lipids. (3) Macroautophagy is the most extensively studied autophagy, which involves formation of double membrane structures that encircle proteins, lipids, and organelles. Degradation of mitochondria through the macroautophagy pathway is also termed mitophagy. Degradation of other cellular structures, such as fragments of the nucleus, lipid droplets, peroxisomes, ribosomes and endoplasmic reticulum, have also been called, nucleophagy, lipophagy, pexophagy, ribophagy, and reticulophagy. The macroautophagy pathway can perform bulk degradation of cellular contents in response to starvation and more than 35 Autophagy-Related Proteins (ATGs) are involved in this process. Fusion of the double membrane autophagosomes with the lysosomes resulted in autophagolysosomes that degrade the inner membrane of the autophagosomes and the contents inside the autophagosomes.
Macroautophagy is the most extensively studied autophagy process [2]. It was first described using electron microscopy as unique morphological structures with double membranes encircling amorphous or partially degraded materials including mitochondria and endoplasmic reticulum. Early studies noted that these structures are enriched in response to glucagon and starvation in the liver [3]. During the past 2 decades, more than 35 genes have been identified in yeast and most of the corresponding mammalian homologs have been identified [4]. The mTOR pathway plays a major role in sensing free amino acids, cellular bioenergetic deficits, hypoxia and DNA damage, and thereby regulate macroautophagy [5]. The sensing of free amino acids by mTOR seems to be dependent on localization of the mTOR complex to the lysosomes [5].
One major function of macroautophagy is the control of accumulation of over-produced, long-lived or damaged proteins. Deficiencies of macroautophagy may contribute to accumulation of protein aggregates, which are apparent in a number of neurodegenerative diseases, including Alexander disease, multiple system atrophy, amyloid lateral sclerosis, Alzheimer's, Parkinson's, and Huntington's diseases. Increased appearance of autophagosomes is a consistent feature in aging and Parkinson's disease brains [6,7]. The accumulation of these structures is consistent with a failure or overwhelming of the autophagy pathway. The accumulation of Lewy Bodies and protein aggregates is generally thought to indicate that autophagy is unable to meet the demands for protein clearance in the disease. One such prototype aggregation-prone protein is α-synuclein. α-synuclein gene triplication and mutation have been found to be responsible for a subset of familial Parkinson's disease [8]. Furthermore, α-synuclein accumulates in a majority of sporadic Parkinson's disease brains [8]. Both chaperone-mediated autophagy and macroautophagy have been shown to participate in α-synuclein degradation, along with proteasomes [9] (Fig. 2). However, α-synuclein that is phosphorylated at serine 129 as appears in Lewy Bodies in Parkinson's disease, and α-synuclein that is mutated in familial Parkinson's disease cannot be degraded by chaperone-mediated autophagy [10], and is thus presumably highly sensitive to blockade of macroautophagy. The proteases within the lysosome can be limiting in the degradation of aggregated proteins. We and others have shown that lysosomal cathepsin D deficient mice, sheep and patients exhibit α-synuclein accumulation, indicating that autophagy is important for α-synuclein turnover [11,12]. Additional examples of autophagy playing important roles in decrease protein accumulation include the involvement of cathepsin B in amyloid beta accumulation [13], and the involvement of autophagy protein Atg5 and Atg7 in ubiquitinated protein accumulation [14,15].
Fig. 2.
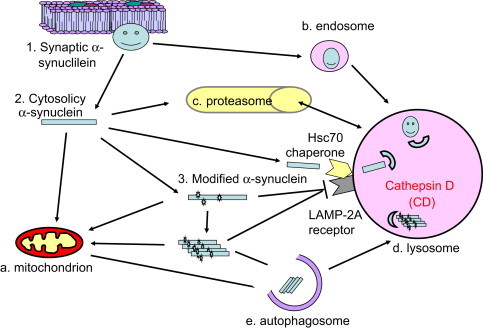
Autophagic clearance of aggregation-prone proteins. Due to either genetic predisposition, aging or environmental perturbations, specific proteins may become unfolded, abnormally modified, or mis-targeted. If accumulated, these protein species may cause further cellular damage and induce cell death. This is particularly important in post-mitotic cells which cannot divide and so dilute out these toxic species. Protein aggregates or inclusions accumulate in neurodegenerative diseases, including Alexander disease, multiple system atrophy, Alzheimer's, Parkinson's and Huntington's diseases. One such protein is α-synuclein, which may exist in 3 different forms: (1) synaptic vesicle-associated form that may be involved in synaptic neurotransmitter release, (2) cytosolic form which may target to the mitochondria and is able to inhibit mitochondrial complex I activity, and (3) α-synuclein modified by reactive species or phosphorylation. Plasma membrane associated protein can be degraded by the endosomes. Cytosolic unmodified α-synuclein can be degraded by both the proteasome pathway and the lysosomal-mediated autophagy pathway. Wildtype α-synuclein can also be recognized by chaperone protein Hsc70 and transported to the lysosomes for degradation through the chaperone-mediated autophagy. However, α-synuclein modified at serine 129 which accumulates in Parkinson's disease Lewy Bodies, and mutated α-synuclein as appears in familial Parkinson's disease, cannot be degraded through the chaperone-mediated autophagy. They further contribute to cytotoxicity by inhibition of chaperone-mediated autophagy preventing removal of α-synuclein and other protein targets. For this reason mutated, oligomerized and abnormally modified α-synuclein can only be degraded through macroautophagy. If the macroautophagy pathway is overwhelmed, then α-synuclein accumulates in the neurons. Recent work demonstrated that lysosomal cathepsin D is important for degradation of α-synuclein, and human patients with cathepsin D mutation, cathepsin D knockout mice, as well as sheep with cathepsin D mutation, exhibit pronounced α-synuclein accumulation in the brain. Other lysosomal cathepsins, such as cathepsin B, have been shown to be important for attenuating accumulation and toxicity of β amyloid and mutant huntingtin.
Cellular damage can arise both from accumulation of toxic species of proteins that may be detrimental to redox signaling or even directly inhibitory to mitochondrial function, and as a consequence of mitochondrial dysfunction. Indeed, mitochondria are major hub in the cell integrating energy demand, reactive species and apoptosis signaling. Mitochondrial dysfunction and accumulation of oxidative damage contribute to pathogenesis of a variety of diseases, including neurodegenerative diseases, liver steatosis, lung and cardiovascular diseases, and cancer [16–31] (Fig. 3). Specific examples include decreased complex I activity, increased mitochondrial and nuclear DNA damage, and decreased reduced glutathione [32–37]. Of particular interest to redox biology, oxidative stress can either be a signal to activate autophagy, or exert damage to the autophagy machinery to inhibit autophagy. For example, oxidative stress may increase DNA damage, activate p53 and AMPK which in turn inhibits mTOR and activates autophagy [38]. On the other hand, Atg4 is activated in reduced conditions, and inactivated in response to oxidative stress [39]. Reciprocally, autophagy may decrease cellular oxidative stress by clearance of reactive species generating organelles, reactive species damaged proteins, or alternatively, decrease specific antioxidants [40]. A similar relationship between mitochondrial activities and autophagy also exists. Mitochondria-deficient cells or cells treated with oligomycin or antimycin A, exhibit attenuated autophagic gene induction and autophagic flux in response to starvation [41]. Furthermore, mitochondrial contribution to activation of macroautophagy may include generation of reactive species [42], providing membranes for autophagosomal formation [43], or providing a platform for membrane-associated complexes to engage the autophagy process [44].
Fig. 3.
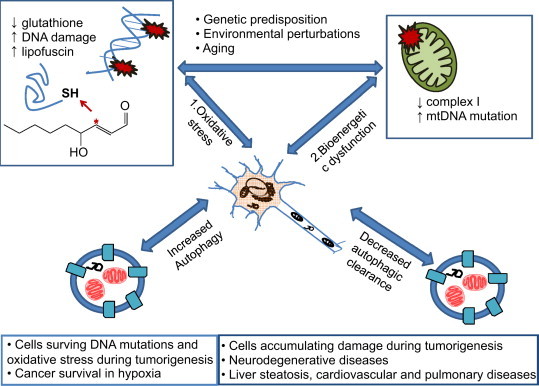
Autophagy and mitophagy in aging and diseases. In aging, or due to genetic predisposition and environmental perturbations, cellular damage occurs. Mitochondria dysfunction is also associated with oxidative stress. (1) Cellular oxidative and nitrative stress are mediators of the irreversible post-translational protein and DNA modification which increase hydrophobicity and so cause aggregates within the cells. Cellular reduced glutathione decreases resulting in higher levels of reactive species that can modify proteins. Lipid peroxidation is also promoted and further modifies proteins. An example of lipid peroxidation product is 4-hydroxynonemal shown in the diagram. (2). Mitochondrial damage occurs in heart, liver, lung, kidney and nervous system diseases. For example, in Parkinson's disease, mitochondrial complex I activity is decreased, and mitochondrial DNA mutations accumulate. This damage, if not controlled, may be detrimental to cell survival. To deal with accumulated protein aggregates, oxidized lipid and associated proteins and damaged mitochondria, autophagic activities are required. Both enhanced and inhibited autophagy may be involved in tumorigenesis to accumulate damage and to enhance cell survival in the presence of genomic mutations, genotoxic agents, and under hypoxic conditions. Decreased autophagy is also detrimental to homeostasis of post-mitotic tissues such as occurs in neurodegenerative diseases, liver steatosis, cardiovascular and pulmonary diseases. Understanding the mechanisms and regulation of autophagy and mitophagy in disease pathogenesis may aid in the development of new strategies for treatment of proliferative and degenerative diseases.
Autophagic removal of mitochondria is important for mitochondrial quality control. Poor quality mitochondria may enhance cellular oxidative stress, generate apoptosis signals, and induce cell death. The bioenergetic crisis may also be further exacerbated by reactive species damage to glycolytic and glutathione-mediated antioxidant pathways. Because healthy mitochondrial function is essential for cell survival, selective removal of a subset of dysfunctional mitochondria is a highly regulated process and requires coordinated functions of mitochondrial and cytosolic proteins (Fig. 4). This is controlled by a complex array of proteins which are constantly being revised and enhanced. For example, recent studies demonstrated that PINK1 is stabilized in the mitochondria in response to lowered membrane potential, recruits Parkin, which can ubiquitinate Mfn1 and 2, VDAC, TOMs, Fis1 and MIRO, and induce mitophagy [45]. Controlled mitophagy may coordinate with mitochondrial biogenesis to sustain cell survival and function [46].
Fig. 4.
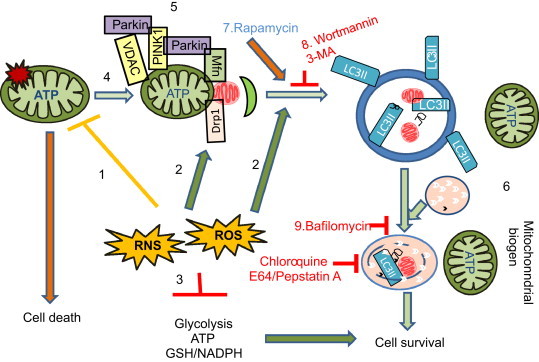
Mitophagy in mitochondrial quality control. (1) Reactive oxygen species (ROS) and reactive nitrogen species (RNS) can target the mitochondrion and induce damage to proteins, DNA and lipids. (2) ROS and RNS may also be part of the signaling mechanism for autophagy and mitophagy regulation. (3) Furthermore, ROS and RNS may lead to modification of the glycolytic pathway, and perturbation of the cellular redox status. (4) Mitochondrial damage results in a decrease in mitochondrial membrane potential or an increase in mitochondrial fission, and both have been shown to signal mitophagy. Elongated mitochondria or increase of mitochondrial fusion have been shown to protect mitochondria from mitophagy. (5) Mitochondrial PINK1 is unstable due to presenilin-associated rhomboid-like (PARL) protease activities. A decrease of mitochondrial membrane potential will inhibit PINK1 degradation by PARL. Stabilized PINK1 recruits Parkin to mitochondria, where Parkin has been shown to be able to ubiquitinate Mitofusin (Mfn1/2), VDAC, and TOM proteins, and lead to enhanced mitophagy. (6) Clearance of damaged mitochondria by mitophagy may facilitate mitochondrial biogenesis and enhance cell survival. (7) Activation of autophagy and mitophagy by rapamycin and other newly developed small chemical compounds have been investigated for their potential to enhance cell survival in response to ROS/RNS induced damage. Autophagy and mitophagy can be inhibited by (8) 3-methyladenine (3-MA) or Wortmannin that are PI3K inhibitors; by (9) bafilomycin and chloroquine that alter vacuolar and lysosomal pH, prevent autophagosomal–lysosomal fusion; or by E64 and pepstatin A that inhibit lysosomal protease activities. Inhibition of autophagy usually leads to enhanced cell death but in some circumstances autophagy can contribute to cytotoxicity. Identification and testing compounds that modulate autophagy and mitophagy is needed for treatment of a variety of diseases in which oxidative protein modification accumulates in the cell.
Pharmacological activators and inhibitors, such as rapamycin, 3-methyladenine (3-MA) and chloroquine have aided research into autophagy regulation and consequence of altered autophagy and mitophagy regulation in health and diseases. Derivatives or additional compounds identified through high throughput screening may provide new, safe and effective compounds that target to autophagy and mitophagy pathway as treatment of cancer, neurodegenerative diseases, and diseases in the liver, heart, lung, kidney and β cells [47].
Acknowledgments
This work was supported by NS064090 and a VA merit award (JZ).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Reference
- 1.Santambrogio L., Cuervo A.M. Chasing the elusive mammalian microautophagy. Autophagy. 2011;7(6):652–654. doi: 10.4161/auto.7.6.15287. [DOI] [PubMed] [Google Scholar]
- 2.Klionsky D.J., Abdalla F.C., Abeliovich H., Abraham R.T., cevedo-Arozena A., Adeli K. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Z., Klionsky D.J. Eaten alive: a history of macroautophagy. Nature Cell Biology. 2010;12(9):814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mizushima N., Yoshimori T., Ohsumi Y. The role of Atg proteins in autophagosome formation. Annual Review of Cell and Developmental Biology. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 5.Laplante M., Sabatini D.M. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anglade P., Vyas S., Javoy-Agid F., Herrero M.T., Michel P.P., Marquez J. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histology and Histopathology. 1997;12(1):25–31. [PubMed] [Google Scholar]
- 7.Anglade P., Vyas S., Hirsch E.C., Agid Y. Apoptosis in dopaminergic neurons of the human substantia nigra during normal aging. Histology and Histopathology. 1997;12(3):603–610. [PubMed] [Google Scholar]
- 8.Martin I., Dawson V.L., Dawson T.M. Recent advances in the genetics of Parkinson's disease. Annual Review of Genomics and Human Genetics. 2011;12:301–325. doi: 10.1146/annurev-genom-082410-101440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Webb J.L., Ravikumar B., Atkins J., Skepper J.N., Rubinsztein D.C. Alpha-Synuclein is degraded by both autophagy and the proteasome. Journal of Biological Chemistry. 2003;278(27):25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 10.Cuervo A.M., Stefanis L., Fredenburg R., Lansbury P.T., Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305(5688):1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 11.Qiao L., Hamamichi S., Caldwell K.A., Caldwell G.A., Yacoubian T.A., Wilson S. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Molecular Brain. 2008;1(1):17. doi: 10.1186/1756-6606-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cullen V., Lindfors M., Ng J., Paetau A., Swinton E., Kolodziej P. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Molecular Brain. 2009;2(1):5. doi: 10.1186/1756-6606-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mueller-Steiner S., Zhou Y., Arai H., Roberson E.D., Sun B., Chen J. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer's disease. Neuron. 2006;51(6):703–714. doi: 10.1016/j.neuron.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 14.Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 15.Komatsu M., Waguri S., Chiba T., Murata S., Iwata J.I., Tanida I. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 16.Lee J., Giordano S., Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochemical Journal. 2012;441(2):523–540. doi: 10.1042/BJ20111451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hill B.G., Dranka B.P., Zou L., Chatham J.C., Darley-Usmar V.M. Importance of the bioenergetic reserve capacity in response to cardiomyocyte stress induced by 4-hydroxynonenal. Biochemical Journal. 2009;424(1):99–107. doi: 10.1042/BJ20090934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Higdon A.N., Dranka B.P., Hill B.G., Oh J.Y., Johnson M.S., Landar A. Methods for imaging and detecting modification of proteins by reactive lipid species. Free Radical Biology and Medicine. 2009;47(3):201–212. doi: 10.1016/j.freeradbiomed.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hill B.G., Higdon A.N., Dranka B.P., Darley-Usmar V.M. Regulation of vascular smooth muscle cell bioenergetic function by protein glutathiolation. Biochimica et Biophysica Acta. 2010;1797(2):285–295. doi: 10.1016/j.bbabio.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dranka B.P., Benavides G.A., Diers A.R., Giordano S., Zelickson B.R., Reily C. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radical Biology and Medicine. 2011 doi: 10.1016/j.freeradbiomed.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giordano S., Lee J., Darley-Usmar V.M., Zhang J. Distinct effects of Rotenone, 1-methyl-4-phenylpyridinium and 6-hydroxydopamine on cellular bioenergetics and cell death. PLoS ONE. 2012;7(9):e44610. doi: 10.1371/journal.pone.0044610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chacko B.K., Reily C., Srivastava A., Johnson M.S., Ye Y., Ulasova E. Prevention of diabetic nephropathy in Ins2(+/)(AkitaJ) mice by the mitochondria-targeted therapy MitoQ. Biochemical Journal. 2010;432(1):9–19. doi: 10.1042/BJ20100308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zelickson B.R., Benavides G.A., Johnson M.S., Chacko B.K., Venkatraman A., Landar A. Nitric oxide and hypoxia exacerbate alcohol-induced mitochondrial dysfunction in hepatocytes. Biochimica Biophysica Acta. 2011;1807(12):1573–1582. doi: 10.1016/j.bbabio.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chacko B.K., Srivastava A., Johnson M.S., Benavides G.A., Chang M.J., Ye Y. Mitochondria-targeted ubiquinone (MitoQ) decreases ethanol-dependent micro and macro hepatosteatosis. Hepatology. 2011;54(1):153–163. doi: 10.1002/hep.24377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee S.J., Ryter S.W., Xu J.F., Nakahira K., Kim H.P., Choi A.M. Carbon monoxide activates autophagy via mitochondrial reactive oxygen species formation. American Journal of Respiratory Cell and Molecular Biology. 2011;45(4):867–873. doi: 10.1165/rcmb.2010-0352OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ryter S.W., Lam H.C., Chen Z.H., Choi A.M. Deadly triplex: smoke, autophagy and apoptosis. Autophagy. 2011;7(4):436–437. doi: 10.4161/auto.7.4.14501. [DOI] [PubMed] [Google Scholar]
- 27.Constantin M., Choi A.J., Cloonan S.M., Ryter S.W. Therapeutic potential of heme oxygenase-1/carbon monoxide in lung disease. International Journal of Hypertension. 2012;2012:859235. doi: 10.1155/2012/859235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gutierrez J., Ballinger S.W., Darley-Usmar V.M., Landar A. Free radicals, mitochondria, and oxidized lipids: the emerging role in signal transduction in vascular cells. Circulation Research. 2006;99(9):924–932. doi: 10.1161/01.RES.0000248212.86638.e9. [DOI] [PubMed] [Google Scholar]
- 29.Ni H.M., Bockus A., Boggess N., Jaeschke H., Ding W.X. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology. 2012;55(1):222–232. doi: 10.1002/hep.24690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding W.X., Li M., Chen X., Ni H.M., Lin C.W., Gao W. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010 Jul:23. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hill B.G., Benavides G.A., Lancaster J.R., Ballinger S., Dell’italia L., Zhang J. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Journal of Biological Chemistry. 2012 doi: 10.1515/hsz-2012-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schapira A.H., Gegg M. Mitochondrial contribution to Parkinson's disease pathogenesis. Parkinsons Disease. 2011;2011:159160. doi: 10.4061/2011/159160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ballinger S.W., Bouder T.G., Davis G.S., Judice S.A., Nicklas J.A., Albertini R.J. Mitochondrial genome damage associated with cigarette smoking. Cancer Research. 1996;56(24):5692–5697. [PubMed] [Google Scholar]
- 34.Bohr V.A. Repair of oxidative DNA damage in nuclear and mitochondrial DNA, and some changes with aging in mammalian cells. Free Radical Biology and Medicine. 2002;32(9):804–812. doi: 10.1016/s0891-5849(02)00787-6. [DOI] [PubMed] [Google Scholar]
- 35.Lebel M., de Souza-Pinto N.C., Bohr V.A. Metabolism, genomics, and DNA repair in the mouse aging liver. Current Gerontology and Geriatrics Research. 2011;2011:859415. doi: 10.1155/2011/859415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ballatori N., Krance S.M., Notenboom S., Shi S., Tieu K., Hammond C.L. Glutathione dysregulation and the etiology and progression of human diseases. Journal of Biological Chemistry. 2009;390(3):191–214. doi: 10.1515/BC.2009.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perry T.L., Godin D.V., Hansen S. Parkinson's disease: a disorder due to nigral glutathione deficiency? Neuroscience Letters. 1982;33(3):305–310. doi: 10.1016/0304-3940(82)90390-1. [DOI] [PubMed] [Google Scholar]
- 38.Feng Z., Zhang H., Levine A.J., Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proceedings of the National Academy of Sciences USA. 2005;102(23):8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scherz-Shouval R., Shvets E., Fass E., Shorer H., Gil L., Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO Journal. 2007;26(7):1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu L., Wan F., Dutta S., Welsh S., Liu Z., Freundt E. Autophagic programmed cell death by selective catalase degradation. Proceedings of the National Academy of Sciences USA. 2006;103(13):4952–4957. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graef M., Nunnari J. Mitochondria regulate autophagy by conserved signalling pathways. EMBO Journal. 2011;30(11):2101–2114. doi: 10.1038/emboj.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li L., Chen Y., Gibson S.B. Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell Signal. 2012;25(1):50–65. doi: 10.1016/j.cellsig.2012.09.020. [DOI] [PubMed] [Google Scholar]
- 43.Hailey D.W., Rambold A.S., Satpute-Krishnan P., Mitra K., Sougrat R., Kim P.K. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141(4):656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lei Y., Wen H., Yu Y., Taxman D.J., Zhang L., Widman D.G. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity. 2012;36(6):933–946. doi: 10.1016/j.immuni.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jin S.M., Youle R.J. PINK1 and Parkin-mediated mitophagy at a glance. Journal of Cell Science. 2012;125(Pt 4):795–799. doi: 10.1242/jcs.093849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Michel S., Wanet A., De P.A., Rommelaere G., Arnould T., Renard P. Crosstalk between mitochondrial (dys)function and mitochondrial abundance. Journal of Cell Physiology. 2012;227(6):2297–2310. doi: 10.1002/jcp.23021. [DOI] [PubMed] [Google Scholar]
- 47.Rubinsztein D.C., Codogno P., Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nature Reviews Drug Discovery. 2012;11(9):709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]