

Abstract
This paper describes a photoredox palladium/iridium-catalyzed C–H arylation with diaryliodonium reagents. Details of the reaction optimization, substrate scope, and mechanism are presented along with a comparison to a related method in which aryldiazonium salts are used in place of diaryliodonium reagents. The unprecedentedly mild reaction conditions (25 ºC in methanol), the requirement for light and a photocatalyst, the inhibitory effect of radical scavengers, and the observed chemoselectivity trends are all consistent with a radical-thermal reaction with diaryliodonium reagents that is believed to proceed via an ‘ionic’ 2e− pathway and requires a much higher reaction temperature (100 ºC).
Keywords: C–H activation, diaryliodonium salts, palladium, photochemistry, radicals
Introduction
The merger of transition metal catalysis with radical chemistry has emerged as a powerful strategy for achieving high-yielding transformations under mild conditions.[1] The ability to reroute textbook metal-catalyzed reactions via alternative metal/radical-mediated pathways can lead to improvements in rate, functional group tolerance, and/or substrate scope. Manolikakes and Knochel recently reported a striking example of this strategy in the context of the Pd-catalyzed Kumada coupling.[2] They showed that the addition of i-PrI led to a remarkable rate acceleration in the coupling of aryl bromides with aryl Grignard reagents.[2a] This rate enhancement was rationalized based on a new ‘radical-catalyzed’ mechanistic manifold involving in situ generated alkyl and aryl radicals as key intermediates.
We hypothesized that a similar strategy could be used to accelerate Pd-catalyzed C–H arylation reactions with diaryliodonium reagents. As shown in Scheme 1a, Pd(OAc)2 is known to catalyze ligand-directed C–H arylation via an ‘ionic’ mechanism involving 2e− oxidation of a palladacycle intermediate by Ar2I+.[3–5] However, these transformations are extremely sluggish, typically requiring high temperatures (80–110 °C) over extended periods of time (8–24 h) in acetic acid.[6] We reasoned that the rates of these reactions could potentially be enhanced by rerouting them through radical pathways in which Ar2I+ is converted into Ar• in situ (Scheme 1b).
Scheme 1.
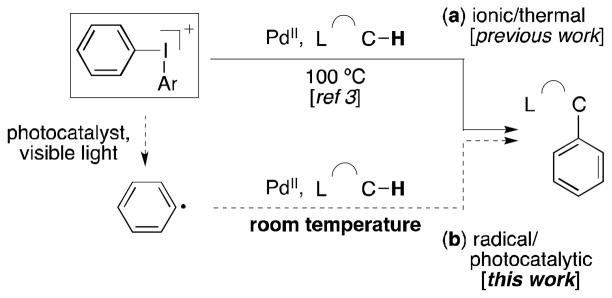
Two different pathways for Pd-catalyzed C–H arylation with Ph2I+.
Tentative support for the feasibility of this approach is provided by recent reports showing Pd-catalyzed C–H arylation using Ar• generated from aroyl peroxides[7a] or aryl diazonium salts.[7b] We report herein that this strategy enables C–H arylation with Ar2I+ under extremely mild conditions (room temperature in MeOH). We also present evidence supporting the proposal that two fundamentally different mechanisms are operating in the ionic (Scheme 1a) versus radical (Scheme 1b) systems. Finally, we compare this new transformation to a related method that uses aryl-N2+ as the arylating reagent.[7b]
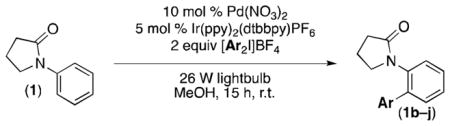
Results and Discussion
Ph2I+ can be converted to Ph• under mild conditions using visible light and a photocatalyst [Ru(bpy)3Cl2 or Ir(ppy)3; bpy = 2,2′-bipyridine, ppy = cyclometalated 2-phenylpyridine].[8,9] Thus, we initiated investigations of Pd-catalyzed C–H phenylation of 1 with [Ph2I]BF4 in combination with a photocatalyst. Visible light irradiation was provided by a 26 W household fluorescent light bulb, and the reactions were set up on the bench top with no precautions to exclude moisture or air. Remarkably, the use of Ru(bpy)3Cl2 resulted in a modest yield (18%) of the desired arylated product 1a after 15 h in MeOH at room temperature (Table 1, entry 1). Evaluation of several different PdII catalysts revealed that Pd(NO3)2 provides the best results, generating 1a in 23% yield (entry 2). Ru(bpy)3Cl2 and Ir(ppy)3 afforded comparable results (entries 2 and 3), but the cationic photocatalyst Ir(ppy)2(dtbbpy)PF6 (dtbbpy = 4,4′-ditertbutyl-2,2′-bipyridine)[10] provided a significant improvement (57% yield, entry 4). Replacing [Ph2I]BF4 with the corresponding triflate salt led to a further enhancement (66% yield, entry 5). Finally, briefly sparging the mixture with N2 prior to the start of the reaction resulted in 94% yield of 1a (entry 6).[11] Importantly, both of the metal catalysts, as well as visible light, are critical for efficient room temperature C–H phenylation.[12] Without any one of these three components, only traces of product 1a were formed (0–2%, entries 7–9).
Table 1.
Optimization of the room-temperature C–H phenylation reaction of 1 with Ph2I+.a

| ||||
|---|---|---|---|---|
| Entry | PdII | Photocatalyst | X | Yield (%)[b] |
| 1 | Pd(OAc)2 | Ru(bpy)3Cl2 | BF4− | 18 |
| 2 | Pd(NO3)2 | Ru(bpy)3Cl2 | BF4− | 23 |
| 3 | Pd(NO3)2 | Ir(ppy)3 | BF4− | 17 |
| 4 | Pd(NO3)2 | Ir(ppy)2(dtbbpy)PF6 | BF4− | 57 |
| 5 | Pd(NO3)2 | Ir(ppy)2(dtbbpy)PF6 | OTf− | 66 |
| 6[c] | Pd(NO3)2 | Ir(ppy)2(dtbbpy)PF6 | OTf− | 94 |
| 7[c] | none | Ir(ppy)2(dtbbpy)PF6 | OTf− | 2 |
| 8[c] | Pd(NO3)2 | none | OTf− | 2 |
| 9[c,d] | Pd(NO3)2 | Ir(ppy)2(dtbbpy)PF6 | OTf− | 0 |
General conditions: 1 (1 equiv), PdII (0.10 equiv), photocatalyst (0.05 equiv), [Ph2I]X (2 equiv), MeOH (0.2 M in 1), 26 W lightbulb, 15 h, rt.
Yields determined by GC.
Reaction was degassed by sparging with N2 for 1 min.
General conditions, but with no light.
A variety of aromatic substrates underwent room temperature C–H phenylation under these conditions (Table 2). In addition to pyrrolidinones 1 and 2, other N-aryl amides were effective directing groups (entries 3 and 4). C-Aryl amides, such as benzamides 5 and 6, also underwent room temperature C–H phenylation, albeit in moderate yields (40% and 54%). The N,N-disubstituted analog 7 provided phenylated product 7a in poor yield (9%), suggesting that C–H arylation is facilitated by the presence at least one N–H bond in this substrate class. 2-Arylpyridines 8 and 9 as well as ketoxime and aldoxime ethers 10 and 11 were also good substrates. The ability to use oxime ethers as directing groups is particularly notable, as these do not undergo C–H arylation with diaryliodonium reagents under the previously reported thermal reaction conditions.[3]
Table 2.
Substrate scope for the Pd/Ir-catalyzed C–H phenylation reaction with Ph2I+.[a]
| |||
|---|---|---|---|
| Entry | Substrate | Product | Isolated yield (%) |
| 1 |
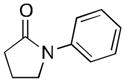 (1) |
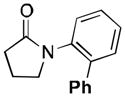 (1a) |
81 |
| 2 |
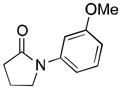 (2) |
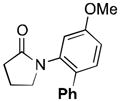 (2a) |
94 |
| 3[b] |
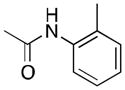 (3) |
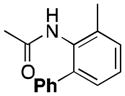 (3a) |
72 |
| 4[b,c,d] |
 (4) |
 (4a) |
44 |
| 5 |
 (5) |
 (5a) |
40 |
| 6 |
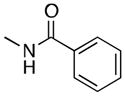 (6) |
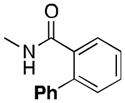 (6a) |
54 |
| 7 |
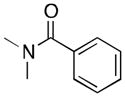 (7) |
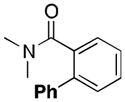 (7a) |
9 |
| 8 |
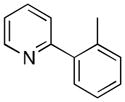 (8) |
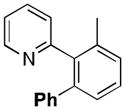 (8a) |
62 |
| 9 |
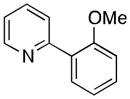 (9) |
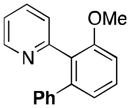 (9a) |
67 |
| 10 |
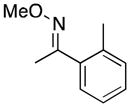 (10) |
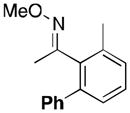 (10a) |
60 |
| 11 |
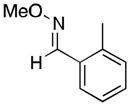 (11) |
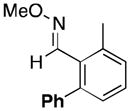 (11a) |
57 |
General conditions: substrate (1 equiv), [Ph2I]OTf (2 equiv), Pd(NO3)2 (0.10 equiv), Ir(ppy)2(dtbbpy)PF6 (0.05 equiv), MeOH (0.2 M in substrate), 26 W lightbulb, 15 h, r.t., degassed by sparging with N2.
[Ph2I]BF4 was the oxidant.
With 1 equiv MgO.
0.20 equiv Pd(NO3)2.
Diaryliodonium salts containing diverse arene substituents were evaluated in this photocatalytic C–H arylation. As shown in Table 3, the highest yields were obtained with those bearing relatively electron neutral substituents (e.g., p-Cl, p-Br, p-CH3, o-CH3, entries 4–7). Nonetheless, oxidants possessing more strongly electron-donating and electron-withdrawing substituents were also effective. For example, C–H arylation with p-methoxyphenyl (entry 9) as well as p-, m-, and o-trifluoromethylphenyl (entries 1–3) reagents proceeded in moderate to good yields. Remarkably, even the highly sterically hindered mesityl group could be transferred, albeit in low yield (11%, entry 8). Notably, the analogous thermal reaction of [Mes2I]OTf with 1 did not provide detectable quantities of 1i (as determined by GC).
Table 3.
Scope of Ar2I+ salts for Pd/Ir-catalyzed C–H arylation of 1.[a]
| |||
|---|---|---|---|
| Entry | Ar | Product | Isolated yield (%) |
| 1 | p-CF3C6H4 | 1b | 69 |
| 2 | m-CF3C6H4 | 1c | 56 |
| 3 | o-CF3C6H4 | 1d | 46 |
| 4 | p-ClC6H4 | 1e | 77 |
| 5 | p-BrC6H4 | 1f | 79 |
| 6 | p-CH3C6H4 | 1g | 87 |
| 7 | o-CH3C6H4 | 1h | 85 |
| 8[b] | Mes | 1i | 11 |
| 9 | p-CH3OC6H4 | 1j | 41 |
General conditions: 1 (1 equiv), [Ar2I]BF4 (2 equiv), Pd(NO3)2 (0.10 equiv), Ir(ppy)2(dtbbpy)PF6 (0.05 equiv), MeOH (0.2 M in 1), 26 W lightbulb, 15 h, r.t., degassed by
OTf salt of oxidant. sparging with N2.
We propose that the Ir/Pd-catalyzed photocatalytic C–H arylation proceeds via a fundamentally different mechanism than the analogous thermal reaction, despite the fact that the reagents and products are the same in both processes. A first piece of evidence to support this proposal is the reaction outcome in the presence of free radical scavengers. As shown in Table 4, the thermal C–H arylation reaction is not inhibited by the addition of 25 mol % galvinoxyl or 100 mol % of TEMPO. With both radical scavengers, the reactions proceed to complete conversion and afford comparable yield (entries 1–3). In contrast, the % conversion and the % yield of the photocatalytic reaction are suppressed in a dose-dependent manner by galvinoxyl and TEMPO (entries 4–8). These results are consistent with the intermediacy of radicals in the latter but not the former reaction.
Table 4.
Effect of radical scavengers on the Pd/Ir-catalyzed C–H arylation of 8.

| ||||
|---|---|---|---|---|
| Entry | Scavenger | Mol % | Conditions | Yield (%)[c] |
| 1 | none | -- | A | 79 ± 7 |
| 2 | galvinoxyl | 25 | A | 81 ± 1 |
| 3 | galvinoxyl | 100 | A | 74 ± 8 |
| 4 | none | -- | B | 52 ± 4 |
| 5 | galvinoxyl | 10 | B | 42 ± 2 |
| 6 | galvinoxyl | 25 | B | 20 ± 9 |
| 7 | TEMPO | 50 | B | 34 ±14 |
| 8 | TEMPO | 100 | B | 6 ± 2 |
Thermal conditions A: 8 (1 equiv), [Ph2I]BF4 (1.1 equiv), Pd(OAc)2 (0.10 equiv), AcOH (0.12 M in 8), 15 h, 100 ºC.
Photocatalytic conditions B: 8 (1 equiv), [Ph2I]BF4 (2 equiv), Pd(NO3)2 (0.10 equiv), Ir(ppy)2(dtbbpy)PF6 (0.05 equiv), MeOH (0.2 M in 8), 26 W lightbulb, 15 h, r.t., degassed by sparging with N2.
GC calibrated yield reported as % yield ± standard deviation.
In addition, the chemoselectivity of the reaction between 1 and unsymmetrical iodonium reagent 12 is highly dependent on the reaction conditions (eq 1). Under the thermal conditions, the less hindered phenyl group is transferred selectively, providing a 1: 0.2 ratio of 1a: 1d. In contrast, selective transfer of the o-CF3C6H4 group occurs under the photocatalytic conditions, affording a 1: 3.6 ratio of 1a: 1d. These differences in chemoselectivity provide further support for divergent mechanistic pathways.
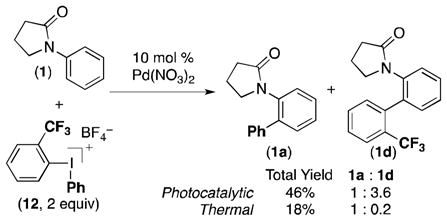 |
(1) |
We preliminarily propose a reaction pathway that merges PdII/IV and IrIII/IV catalytic cycles. As shown in Scheme 2, ground state Ir3+ undergoes photoexcitation by visible light (step i), and the resultant Ir3+* complex can reduce Ar2I+ to generate Ar•, ArI, and Ir4+ (step ii).[8,9] Ar• could then enter the Pd catalytic cycle by oxidizing cyclopalladated complex 13 (step iii). Complex 14 could then be further oxidized to 15 by Ir4+ (step iv), regenerating Ir3+. Finally, C–Ar bond-forming reductive elimination from 15 would afford product 16 and regenerate Pd2+ (step v). Importantly, a number of other pathways are also possible (for example, PdI/III catalysis) and cannot be distinguished based on the current data.
Scheme 2.
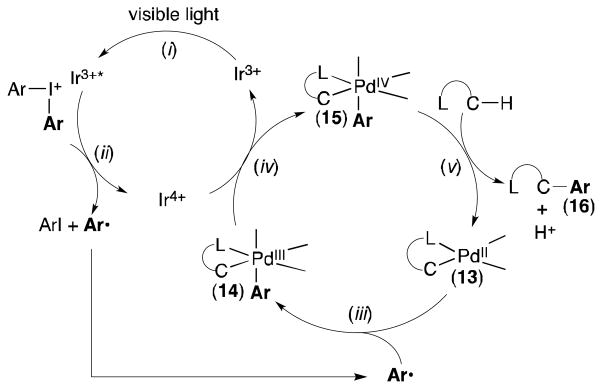
Possible mechanism for the Pd/Ir-catalyzed C– H arylation with Ar2I+.
We have recently reported a related room temperature photocatalytic C–H arylation reaction that was proposed to proceed via a mechanism similar to that shown in Scheme 1.[7b] However, this previously reported transformation used a different Ar• precursor (aryl diazonium salts), a different photocatalyst (Ru(bpy)Cl2), a different Pd catalyst (Pd(OAc)2) and slightly different optimized reaction conditions. Thus, a final set of studies was conducted to compare these two processes.
As illustrated in Table 5, the performance of the two systems is often comparable (e.g., for substrates 1 and 11). However, for benzamide substrates 5–7 and ketoxime ether 10, the Pd/Ir/Ph2I+ system provided better yields of C–H phenylation. Conversely, the Pd/Ru/PhN2+ system performed better for acetanilide 3, and it is effective for hydroxyl oxime 17, a substrate class that undergoes decomposition in the Pd/Ir/Ph2I+ system. Overall, the room-temperature Pd/photocatalyzed C–H arylation methods using Ar2I+ and ArN2+ reagents are often complementary in terms of substrate scope.[13]
Table 5.
Comparison of Pd/Ir-catalyzed C–H phenylation with Ph2I+ vs Pd/Rucatalyzed C–H phenylation with PhN2+.
| Substrate | Product | Yield (%) with Ph2I +[a,b] | Yield (%)with PhN2+[a,c] |
|---|---|---|---|
 (1) |
1a | 89 | 91 |
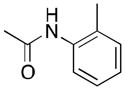 (3) |
3a | 69[d] | 89 |
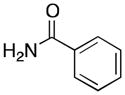 (5) |
5a | 54 | 25 |
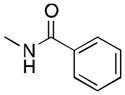 (6) |
6a | 52 | 38 |
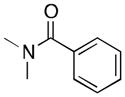 (7) |
7a | 11 | 8 |
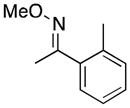 (10) |
10a | 52 | 23 |
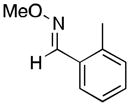 (11) |
11a | 63 | 68 |
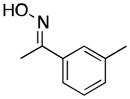 (17) |
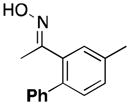
|
<1[e] | 66 |
GC calibrated yield.
General conditions: substrate (1 equiv), [Ph2I]OTf (2 equiv), Pd(NO3)2 (0.10 equiv), Ir(ppy)2(dtbbpy)PF6 (0.05 equiv), MeOH (0.2 M in substrate), 26 W lightbulb, 15 h, r.t., degassed by sparging with N2.
General conditions: substrate (1 equiv), [PhN2]BF4 (4 equiv), Pd(OAc)2 (0.10 equiv), Ru(bpy)3Cl2•6H2O (0.025 equiv), MeOH (0.1 M in substrate), 26 W lightbulb, r.t., 15 h, degassed by sparging with N2.
[Ph2I]BF4 was the oxidant.
Product 17a was not detected by GC, and only traces amounts of 17 and 3′-methylacetophenone were formed.
Conclusion
In summary, this paper describes a photoredox Pd/Ir-catalyzed C–H arylation with diaryliodonium reagents. The unusually low reaction temperature, the requirement for light and a photocatalyst, the inhibitory effect of radical scavengers, and the observed chemoselectivity trends are all consistent with a radical mechanism for this transformation. This stands in contrast to the analogous thermal reaction that requires dramatically higher temperature (100 ºC) and is believed to proceed via an ‘ionic’ 2e− pathway. This example adds to a growing body of work suggesting that re-routing traditional metal-catalyzed transformations via radical pathways can offer major advantages in terms of reaction rates, substrate scope and functional group tolerance.[1,2,14]
Experimental Section
Representative procedure for C–H phenylation of substrate 1
N-Phenylpyrrolidinone 1 (80.6 mg, 0.50 mmol, 1.0 equiv), [Ph2I]OTf (430 mg, 1.00 mmol, 2.0 equiv), Ir(ppy)2(dtbbpy)PF6 (22.8 mg, 0.025 mmol, 0.05 equiv), and Pd(NO3)2•2H2O (13.3 mg, 0.05 mmol, 0.10 equiv) were combined in MeOH (2.5 mL) in a 4 mL scintillation vial. The reaction mixture was cooled in an ice bath (to prevent solvent evaporation) and sparged with N2 using a submerged needle for 10 min, and the vial was then immediately sealed with a Teflon-lined cap. The vial was placed on a stir plate with two 26 W compact fluorescent light bulbs (one on either side of the vial about 5–8 cm away), and the reaction mixture was allowed to stir at room temperature for 15 h. The reaction mixture was diluted with EtOAc (50 mL) and washed with 10% aqueous Na2SO3 (2 × 25 mL) and brine (1 × 25 mL). The combined aqueous layers were extracted with EtOAc (3 × 10 mL), and the organic layers were then combined, dried over MgSO4, filtered, concentrated, and purified by column chromatography on silica gel (Rf = 0.17 in 20% hexanes/80% Et2O). Product 1a was obtained as a pale yellow oil (96.3 mg, 81% yield). 1H and 13C NMR data matched those reported in the literature.[6b]
Supplementary Material
Acknowledgments
This work was supported by the NIH NIGMS (GM073836).
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201######.
References
- 1.For recent reviews, see Ford L, Jahn U. Angew Chem. 2009;121:6504–6507.Angew Chem Int Ed. 2009;48:6386–6389. doi: 10.1002/anie.200901761.Jahn U. Top Curr Chem. 2012;320:121–189. doi: 10.1007/128_2011_261.Jahn U. Top Curr Chem. 2012;320:191–322. doi: 10.1007/128_2011_285.Jahn U. Top Curr Chem. 2012;320:323–451. doi: 10.1007/128_2011_288.
- 2.Manolikakes G, Knochel P. Angew Chem. 2009;121:211–215. doi: 10.1002/anie.200803730.Angew Chem Int Ed. 2009;48:205–209.For a related Negishi coupling see: Kienle M, Knochel P. Org Lett. 2010;12:2702–2705. doi: 10.1021/ol1007026.
- 3.a) Kalyani D, Deprez NR, Desai LV, Sanford MS. J Am Chem Soc. 2005;127:7330–7331. doi: 10.1021/ja051402f. [DOI] [PubMed] [Google Scholar]; b) Deprez NR, Sanford MS. J Am Chem Soc. 2009;131:11234–11241. doi: 10.1021/ja904116k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Other Pd-catalyzed C–H arylations using Ar2I+: Daugulis O, Zaitsev V. Angew Chem. 2005;117:4114–4116. doi: 10.1002/anie.200500589.Angew Chem Int Ed. 2005;44:4046–4048. doi: 10.1002/anie.200500589.Deprez NR, Kalyani D, Krause A, Sanford MS. J Am Chem Soc. 2006;128:4972–4973. doi: 10.1021/ja060809x.Spencer J, Chowdhry BZ, Mallet AI, Rathnam RP, Adatia T, Bashall A, Rominger F. Tetrahedron. 2008;64:6082–6089.Xiao B, Fu Y, Xu J, Gong TJ, Dai JJ, Yi J, Liu L. J Am Chem Soc. 2010;132:468–469. doi: 10.1021/ja909818n.Wagner A, Sanford MS. Org Lett. 2011;13:288–291. doi: 10.1021/ol102734g.Hickman AJ, Sanford MS. ACS Catal. 2011;1:170–174.
- 5.For Cu-catalyzed C–H arylation reactions with Ar2I+ see: Phipps RJ, Grimster NP, Gaunt MJ. J Am Chem Soc. 2008;130:8172–8174. doi: 10.1021/ja801767s.Phipps RJ, Gaunt MJ. Science. 2009;323:1593–1597. doi: 10.1126/science.1169975.Ciana CL, Phipps RJ, Brandt JR, Meyer FM, Gaunt MJ. Angew Chem. 2011;123:478–482. doi: 10.1002/anie.201004703.Angew Chem Int Ed. 2011;50:458–462. doi: 10.1002/anie.201004703.Duong HA, Gilligan RE, Cooke ML, Phipps RJ, Gaunt MJ. Angew Chem. 2011;123:483–486. doi: 10.1002/anie.201004704.Angew Chem Int Ed. 2011;50:463–466.
- 6.Arylation of substrates containing activated C–H bonds, such as the electron rich C–H bonds in indole, do not necessarily require forcing conditions (see ref 4b for an example). For one rare instance of room temperature ligand-directed arylation of unactivated aromatic C–H bonds with Ar2I+, see ref 4d. However, the latter transformation requires added TfOH and has not been demonstrated to be general for any directing groups beyond phenol esters.
- 7.a) Yu WY, Sit W, Zhou Z, Chan ASC. Org Lett. 2009;11:3174–3177. doi: 10.1021/ol900756g. [DOI] [PubMed] [Google Scholar]; b) Kalyani D, McMurtrey KB, Neufeldt SR, Sanford MS. J Am Chem Soc. 2011;133:18566–18569. doi: 10.1021/ja208068w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Lalevée J, Blanchard N, Tehfe MA, Morlet-Savary F, Fouassier JP. Macromolecules. 2010;43:10191–10195. [Google Scholar]; b) Lalevée J, Blanchard N, Tehfe MA, Peter M, Morlet-Savary F, Fouassier JP. Macromol Rapid Commun. 2011;32:917–920. doi: 10.1002/marc.201100098. [DOI] [PubMed] [Google Scholar]
- 9.Photolysis of Ar2I+ under ultraviolet irradiation (248–300 nm) in the presence or absence of organic triplet sensitizers is also known; see: Dektar JL, Hacker NP. J Org Chem. 1990;55:639–647.
- 10.a) Nagib DA, Scott ME, MacMillan DWC. J Am Chem Soc. 2009;131:10875–10877. doi: 10.1021/ja9053338. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Condie AG, González-Gómez JC, Stephenson CRJ. J Am Chem Soc. 2010;132:1464–1465. doi: 10.1021/ja909145y. [DOI] [PubMed] [Google Scholar]
- 11.Oxygen can quench photoexcited Ru2+ and Ir3+: Demas JN, Harris EW, McBride RPM. J Am Chem Soc. 1977;99:3547–3551.
- 12.Although one equivalent of H+ is generated over the course of the reaction, the growing acid concentration does not appear to influence the reaction rate. For example, when 20 mol % TfOH is added to the reaction of substrate 1, the GC calibrated yield of 1a after 2 h is 33%, compared to 32% in the absence of added TfOH.
- 13.The comparative advantages/drawbacks of diaryliodonium salts versus aryldiazonium salts is somewhat subjective and has been the topic of a recent review: Bonin H, Fouquet E, Felpin FX. Adv Synth Catal. 2011;353:3063–3084.
- 14.For another recent example from our group, see: Ye Y, Sanford MS. J Am Chem Soc. 2012;134:9034–9037. doi: 10.1021/ja301553c.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.