

Abstract
Motivation: Outer membrane beta-barrels (OMBBs) are the proteins found in the outer membrane of bacteria, mitochondria and chloroplasts. There are thousands of beta-barrels reported in genomic databases with ∼2–3% of the genes in gram-negative bacteria encoding these proteins. These proteins have a wide variety of biological functions including active and passive transport, cell adhesion, catalysis and structural anchoring. Of the non-redundant OMBB structures in the Protein Data Bank, half have been solved during the past 5 years. This influx of information provides new opportunities for understanding the chemistry of these proteins. The distribution of charges in proteins in the outer membrane has implications for how the mechanism of outer membrane protein insertion is understood. Understanding the distribution of charges might also assist in organism selection for the heterologous expression of mitochondrial OMBBs.
Results: We find a strong asymmetry in the charge distribution of these proteins. For the outward-facing residues of the beta-barrel within regions of similar amino acid density for both membrane leaflets, the external side of the outer membrane contains almost three times the number of charged residues as the internal side of the outer membrane. Moreover, the lipid bilayer of the outer membrane is asymmetric, and the overall preference for amino acid types to be in the external leaflet of the membrane correlates roughly with the hydrophobicity of the membrane lipids. This preference is demonstrably related to the difference in lipid composition of the external and internal leaflets of the membrane.
Contact: joanna.slusky@fccc.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The proteins of the outer membrane make up 2–3% of gram-negative bacteria proteome (Martelli et al., 2002; Wimley, 2002; Zhai and Saier, 2002). These proteins are responsible for transport, cell surface enzymatic functions and structural anchoring (Wimley, 2003). Outer membrane beta-barrels (OMBBs) of bacteria are remotely homologous to the OMBBs of mitochondria (Jimenez-Morales and Liang, 2011). The structure of outer membrane proteins are almost exclusively antiparallel beta-barrels (Fig. 1a).
Fig. 1.
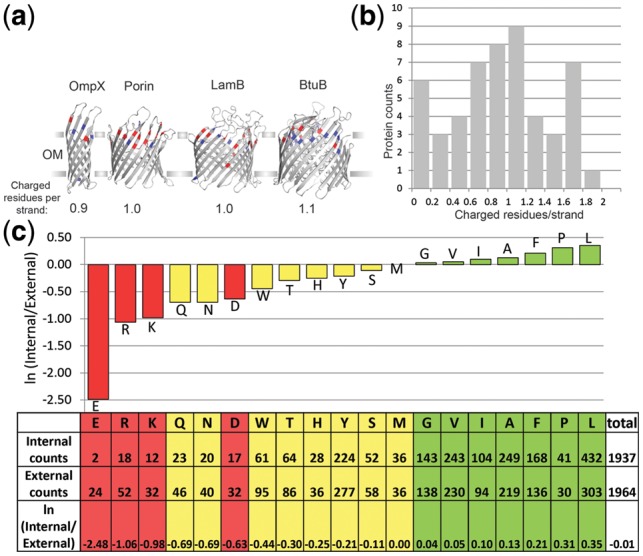
Charged residues prefer the external side of beta-barrels. (a) Four OMBBs with roughly average numbers of external-half charged residues per strand are shown in gray. The strands of the barrel are shown as with sheet secondary structure and the rest of the protein as loops. OmpX (PDBID 1QJ8) is an 8-stranded OMBB; Porin (PDBID 2POR) is a 16-stranded OMBB; LamB (PDBID 1AF6) is an 18-stranded OMBB; BtuB (PDBID 2GUF) is a 22-stranded OMBB. Outward-facing charged residues in the barrel are colored, glutamate and aspartate are red, lysine and arginine are blue. Proteins drawn using pymol (http://www.pymol.org/). (b) Histogram showing the distribution of the number of external charges among the OMBB proteins studied herein. The numbers of external-half outward-facing charged residues in the barrel per strand of the barrel are shown. Bins are inclusive of the bin minimum. (c) External region preference by amino acid. A preference score based on the frequency of occurrence in the external versus the internal region of the outer membrane is calculated for each amino acid. Red amino acids are charged, yellow are polar and green are aliphatic. The more hydrophilic an amino acid is the more it prefers the external side of the outer membrane
Outer membrane proteins are more hydrophilic than inner membrane proteins. Inner membrane proteins, which are mostly multipass transmembrane helical bundles, have lipid-facing exteriors that are extremely hydrophobic and protein-facing interiors that are similarly hydrophobic to the hydrophobic cores of soluble proteins (Rees et al., 1989; Rees and Eisenberg, 2000). The structures of OMBBs, on the other hand, are such that they are more like inside-out soluble proteins. The lipid-facing exterior is highly hydrophobic, but the interior is often accessible to water and is composed of small hydrophilic residues (Wimley, 2002).
The study of the lipid-facing positions of OMBBs is of particular interest as it may lead to a clearer understanding of the folding mechanism of OMBBs. OMBBs have long been known to spontaneously insert into lipid vesicles (Surrey and Jähnig, 1992). In vivo, protein insertion cannot occur without the β-Barrel Assembly Machinery (BAM). It has been hypothesized that OMBBs can insert by themselves in vivo but that the kinetics are too slow to allow for the insertion to occur at the speed and frequency necessary for OMBBs to promote cell viability (Tamm et al., 2004).
Previous studies on the folding of OMBBs have used symmetric lipid vesicles or computational models that make the outer membrane a symmetric lipid bilayer. Reconstitution of mutated outer membrane phospholipase in lipid bilayer vesicles demonstrated that substitution of leucine for native residues in lipid-facing positions stabilizes OMBBs and substitution of those same positions to arginine destabilizes OMBBs. The effect of these mutations on stabilization is stronger toward the center of the vesicle bilayer and is less pronounced closer to the aqueous interface (Moon and Fleming, 2011).
Previous bioinformatic assessment of lipid-facing OMBB amino acid composition was accomplished by symmetrizing the outer membrane such that the center of the membrane was defined as z = 0; 1 Å in either the internal direction or the external direction was defined as z = 1. By symmetrizing the membrane, the authors could double the counts for their study and provide a statistically significant picture of the preferences of all 20 amino acids for each 1.5 Å segment of the membrane. Aliphatic amino acids were shown to prefer the interior of the membrane, polar and charged amino acids were shown to prefer the exterior of the membrane and aromatics were shown to prefer the interfacial region (Hsieh et al., 2012). These results are similar to earlier studies of inner membrane amino acid preferences (Senes et al., 2007).
More recent asymmetric assessment of inner membrane alpha helical proteins (Schramm et al., 2012) demonstrated the positive-inside rule (von Heijne and Gavel, 1988) in the symmetric inner membrane, as arginine and lysine have a strong propensity for localizing on the cytoplasmic-facing loops of the inner membrane. This positive-inside rule determines the topology of inner membrane proteins and is believed to be a result of at least three factors: the voltage across the inner membrane (Andersson and von Heijne, 1994), the charges in the Sec translocon (Goder et al., 2004) and the presence of negatively charged phospholipids (van Klompenburg et al., 1997; Xie et al., 2006).
Of these factors, the only one to be relevant to the outer membrane is that of voltage across the membrane. The outer membrane is known to have an electrical potential of similar magnitude and direction as the electrical potential across the inner membrane (Sen et al., 1988; Stock et al., 1977). However, of the other two factors, one is not present, and one is lessened. Outer membrane proteins are inserted into the membrane using the unrelated BAM machinery rather than the Sec translocon. Moreover, although the inner membrane and the outer membrane are composed of the same phospholipids [phosphatidylethanolamine (PE), phosphatidylglycerol (PG) and cardiolipin], there is evidence that the percentage of negatively charged phospholipids is one-half to one-quarter of that in the inner membrane (Osborn et al., 1972).
The outer membrane itself is highly asymmetric with an inner leaflet of phospholipid and an outer leaflet of lipopolysaccharides (LPS) (Kamio and Nikaido, 1976). It has been determined that a group of proteins coordinates the maintenance of this lipid asymmetry (Malinverni and Silhavy, 2009). Moving charges in the loops of OMBBs does not change their topology (Koebnik, 1999), and previous analysis has indicated that there is a preference for positive charges on the lipid-facing external region of OMBBs but no difference in the prevalence of negatively charged amino acids between the lipid-facing internal and external regions of the outer membrane (Jackups and Liang, 2005).
Since then, the number of non-homologous crystal structures has more than doubled (http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html). In this work, we determine that charged and polar amino acids are disproportionately favored in the external side of the outer membrane. The localization of the charged amino acids correlates well with the structure of the lipids of the outer membrane, to the extent that differences in lipids between organisms are correlated with a difference in localizations of charges in those organisms. The description herein of OMBBs’ asymmetry with respect to all charged amino acids, not just positive charge, demonstrates the importance of the asymmetry of the outer membrane itself with respect to understanding the stability and insertion of OMBBs.
2 METHODS
2.1 Database construction
Fifty-five protein structures of OMBBs were used (Supplementary Table S1) with a homology ≤50%. A maximum number of structures were generated by searching through PDBfam (Xu and Dunbrack, 2012) using the Pfam (Finn et al., 2010) clan code CL0193 as well as the Pfam family codes PF07017 and PF11924. Refinement of the dataset by homology cutoff of 50% and resolution of 3.5 Å was determined using PISCES (Wang and Dunbrack, 2003). PISCES selected the structures of highest resolution with no two sequences at >50% sequence identity. For the final dataset, some structure substitutions and omissions were made based on mutations, length of protein resolved, topology, organism or publication status (Supplemental Information).
2.2 Barrel determination
2.2.1 Strand determination
To better reflect the characteristics of the beta-barrel, we wanted to include all residues that would conform to be within a barrel even if there is a break of a few residues in the hydrogen-bonding pattern. Because this definition is more expansive than that of DSSP or other secondary structure assignment algorithms, a method for strand determination was implemented as follows. For each structure, hydrogens were generated for backbone nitrogens. Strands were determined by a distance of ≤2.75 Å between generated hydrogens and backbone oxygens. A residue was assigned to a strand if the residue’s amino hydrogen was within hydrogen-bonding distance of another residue’s carbonyl oxygen (or vice versa) and if either the hydrogen donor or the hydrogen acceptor had a β-sheet like φ and ψ (if φ < −100° and ψ < −100° or ψ > 50°; or if φ > 150° and ψ < −50° or ψ > 100°). To maximize continuous strands, strand identity was then conferred based on sequence proximity to other strand-belonging residues and based on whether the strands formed a circular barrel structure. Circularity was resolved based on ensuring that each strand had at least one hydrogen-bonding partner in both the previous and subsequent strand.
2.2.2 Inward/outward
Once the strands are identified, the barrel axis is determined. First, two best-fit ellipses are identified using the positions of the Cα atoms at the topmost and bottommost amino acids for each strand. The vector between the centroid of the two ellipses is then defined as the barrel axis. An amino acid’s directional vector was defined as the direction from the midpoint of its amino nitrogen and its carbonyl carbon to its carbon alpha. If the angle between the barrel axis and this directional vector was <90°, the residue was considered to be facing inward, otherwise it was defined as outward-facing.
2.2.3 z offset
The internal and external sides were identified using the previously determined rule that both the N and C termini of OMBBs are internal (Schulz, 2002).The barrel axis was then used to create a rotation matrix so that the barrel could be rotated such that the barrel axis was on the z-axis. The midpoint of the membrane (z = 0) was defined as the location at which the phospholipid layer meets the LPS layer. The barrel is then translated so that the centroid of the bottom ellipse is at −12 Å. This z offset was determined through assessment of the placement of the outward-facing aromatic residues. Tryptophans and tyrosines are known to prefer the interfacial region of OMBBs (Wimley, 2002) and to stabilize the structures when they are located in that region (Hong et al., 2007). At a z offset of −12 Å, the outward-facing aromatic residues were well distributed at the interfacial region with a peak at −9 Å—corresponding to previous calculations (Hsieh et al., 2012) and corresponding to the location of the interfacial region of the membrane as has been previously modeled (Shroll and Straatsma, 2002).
2.3 Error calculation and significance tests
As described below, S is a score of how much an amino acid prefers the external region of the membrane over the internal region of the membrane. Significance for the S score was assessed by a binomial test where the null hypothesis was that the share of each amino acid on each side of z = 0 was the same as the total share of amino acids on that side. Only amino acids where the significance of the amino acid distribution on both sides was better than 5% were used for comparison to the water-octanol hydrophobicity scale or the outer membrane insertion scale (Supplementary Figs S1 and S2).
Pearson’s chi-squared test was used for assessing the significance of the curve for each amino acid proportion distribution across the z-axis of the membrane (Fig. 2). The null hypothesis was that each amino acid should be equally distributed across the z-axis. All amino acids shown were better than 5% significance.
Fig. 2.
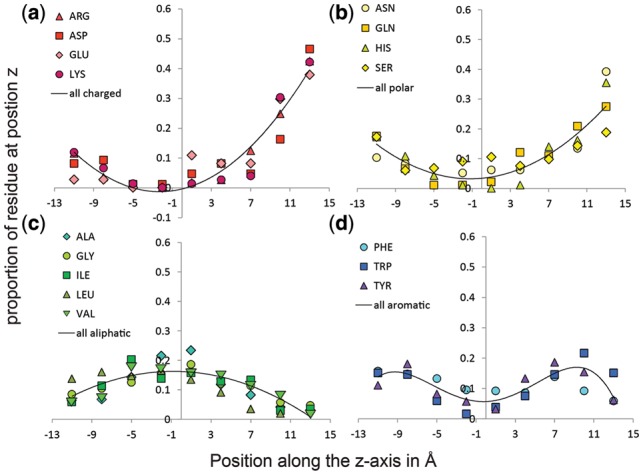
Amino acid location preference within the membrane. The distribution of the proportion of each residue at nine positions in the outer membrane demonstrates how different types of amino acids localize in the outer membrane. (a) Charged amino acids Arg, Asp, Glu and Lys are shown. The average of the four charged types was fit to a polynomial curve of order 2 with an R2 of 0.96. (b) Polar amino acids Asn, Gln, His and Ser are shown. The average of the four polar types was fit to a polynomial curve of order 2 with an R2 of 0.97. (c) Aliphatic amino acids Ala, Gly, Ile, Leu and Val are shown. The average of the five aliphatic types was fit to a polynomial curve of order 2 with an R2 of 0.92. (d) Aromatic amino acids Phe, Trp and Tyr are shown. The average of the three aromatic types was fit to a polynomial curve of order 4 with an R2 of 0.91. The curve of each residue sums to 1
Standard error measurements were calculated for average hydrophobicity (Figs 3 and 4) as the square root of the bin variance weighted by the count of each amino acid in that bin divided by the square root of the total counts in that bin.
Fig. 3.
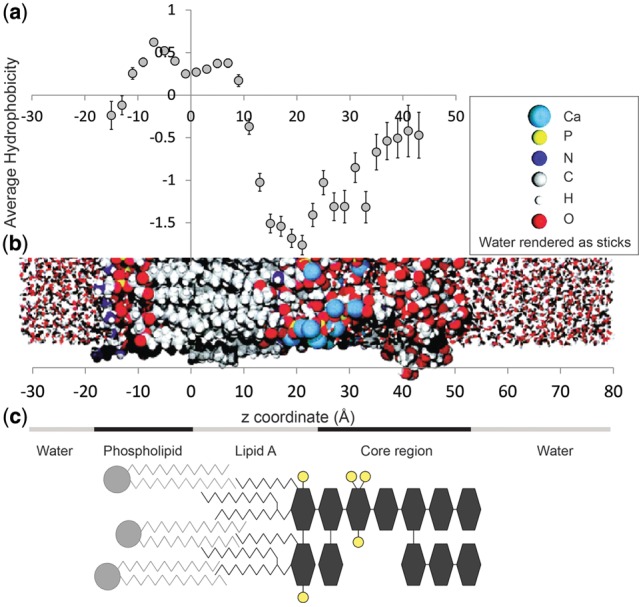
Average hydrophobicity correlates with the structure of the membrane. (a) The average hydrophobicity for each 2 Å of outward-facing barrel amino acids as a function of position in the membrane ±1 SEM. (b) A MDs simulation of the outer membrane (Shroll and Straatsma, 2002) displaying how the membrane is configured as a function of z. (c) Schematic of the composition of the outer membrane. Phospholipid shown in light gray—head groups are circles and acyl chains are zigzag lines. Most of the LPS is shown in dark gray except for the phosphates, which are shown in yellow. Sugars are shown as hexagons and acyl chains as zigzag lines
Fig. 4.

Correlation between a shorter lipid A and a left-shifted drop of average hydrophobicity at the core region. (a) Average hydrophobicity ± 1 SEM for each 2 Å of the outer membrane divided by organism. Gray circles for E.coli, black diamonds for P.aeruginosa. Curves shown are a local polynomial fit with a bandwidth of 2 Å, gray for E.coli, black for P.aeruginosa. (b) Molecular structure of lipid A in E.coli. (c) Molecular structure of lipid A in P.aeruginosa
3 RESULTS
3.1 Charge outside rule
We compared each amino acid type’s occurrence in the external region of the membrane (z > 0) with its occurrence at the internal region of the membrane (z < 0). All non-loop outward-facing residues with a Cα position of |z| ≤ 12 Å were used. The ±12 Å cutoff was set because the bottom centroid was placed at −12 Å (methods) and thus the density of amino acids in the membrane falls off dramatically below that point. This range of z is fully buried within the membrane and does not consider regions of the proteins outside the membrane itself. The locations of 3901 amino acids were surveyed—1937 internal and 1964 external.
To quantify an amino acid’s preference for the external or internal region of the membrane, a preference score was calculated.
Positive score values indicate preference for the internal region and negative score values indicate preference for the external region of the membrane. The results demonstrate that polar residues, and in particular charged residues, strongly prefer the external half of the membrane (Fig. 1c). Glutamic acid is the most enriched amino acid in the external region of the membrane followed by arginine and lysine, and then glutamine, asparagine and aspartic acid. The aliphatic residues show a mild preference for the internal region. This progression of preference for all 20 amino acid types correlates roughly with hydrophobicity.
The calculated score can be compared with other hydrophobicity scales. For the 10 amino acids whose preference for the external region are significant at 5% or better (see Methods), S correlates well with a water-to-octanol hydrophobicity scale (Wimley et al., 1996) (R2 = 0.58) (Supplementary Fig. S1) and correlates even better with the whole-protein OMBB hydrophobicity scale (Moon and Fleming, 2011) (R2 = 0.86) (Supplementary Fig. S2 with the removal of the two basic residues). Thus the more hydrophilic an amino acid the more its presence is enriched in the external region of the outer membrane.
3.2 Charges distributed asymmetrically
To get a finer-grained understanding of amino acid preference in the membrane, we divided the membrane beyond the two bins of external side and internal side of the membrane. By dividing each amino acid into 9 bins of 3 Å each, a more subtle picture of amino acid preference emerges (Fig. 2).
Charged amino acids increase strongly the further they get into the external region of the membrane (Fig. 2a). Polar amino acids also increase on the external side but not as dramatically as the charged amino acids. Of the polar amino acids, serine’s distribution is more symmetric between the internal region and the external region than the others (Fig. 2b) consistent with it being less polar. Aliphatics prefer the internal region and the center of the membrane with alanine preferring the center of the membrane somewhat more than the others and leucine having a stronger preference for the internal region than the other aliphatic amino acids (Fig. 2c). Aromatic amino acids prefer the interfacial regions and so the distribution of the aromatic amino acids is shaped like an upside-down ‘w’ (Fig. 2d). Consistent with the differences in their hydrophobicity, tryptophan prefers the external region slightly more than the others and phenylalanine prefers the external region slightly less. Because of fewer counts and more uniform distribution, the distribution of Cys, Pro, Met and Thr were not significant to 5% (see Methods) and are therefore not shown. A 3 Å bin size was chosen such that all other amino acids distributions would be significant at 5%.
3.3 The charge distribution correlates well with membrane structure
A clearer picture of the charge asymmetry emerges by comparing the average amino acid hydrophobicity to the outer membrane structure. The outer membrane of bacterial cells is highly asymmetric. The inner half of the bilayer is composed of phospholipid and the outer layer of the bilayer is composed of LPS. The LPS is composed of lipid A and a core region of monosaccharides, and in ‘smooth LPS’, a third region called the O-region, which is composed of repeating groups of monosaccharides. Generally speaking, lipid A is composed of two glucosamine units, each with a phosphate group. Around six acyl chains are attached to these two carbohydrates (Caroff and Karibian, 2003) (Fig. 4b and c).The structure of all three components of the LPS varies from organism to organism and even within an organism depending on the organism’s environment.
A molecular dynamics (MD) simulation of an outer membrane bilayer was undertaken by Shroll and Straatsma (2002). In this simulation, the phospholipid PE was used for the inner leaflet of the bilayer and for the outer leaflet, an LPS that is similar in structure to the rough LPS of Pseudomonas aeruginosa—though with slightly shorter and differently branched acyl chains on the lipid A (Nikaido, 2003) (Fig. 3b). Overall, the length of the acyl chain of the phospholipid tends to be longer than the acyl chain of lipid A. Pseudomonas aeruginosa’s lipid A is among the shortest of bacterial lipids and yet the region of phospholipid alone is small. As demonstrated by the simulation, because of its bulk, lipid A tends to pack less well than the phosopholipid bilayer, and the acyl chains of the phospholipid thus intercalate into the LPS layer.
The average hydrophobicity by membrane depth of the beta-barrel amino acids was compared with the previously solved outer membrane structure (Shroll and Straatsma, 2002). Average hydrophobicity of all outward-facing barrel amino acids was calculated for every 2 Å bin from a membrane depth of −16 Å to +44 Å. Bins of 2 Å could be used in this case because of the greater number of counts produced by combining all 20 amino acids. The hydrophobicity of each amino acid in each bin was assigned using the water-to-octanol hydrophobicity scale (Wimley et al., 1996). Standard errors were calculated and are shown in Figure 3a.
A comparison of the structure of the simulated membrane and the average hydrophobicity at each point in the membrane indicates a structural explanation for the amino acid charge asymmetry. Although the LPS layer is in general thicker than the phospholipid layer, the MD simulation demonstrates that calcium ions intercalate deeply into the LPS, interdigitating with the headgroups of lipid A. The localization of this ion intercalation was later demonstrated experimentally (Schneck, et al., 2010). The intercalation seems to happen because of the phosphates in the lipid A glucosamines. The presence of charged phosphates and calcium ions in the region of z = 15 to 30 Å in the membrane strongly suggests that the presence of charged and polar amino acids at that depth stabilize the structure in that particular orientation in the membrane. Indeed, it has been previously shown that positively charged residues in that region create lipid A binding sites (Ferguson et al., 2000).
3.4 Average hydrophobicity varies by membrane structure
The available membrane protein structures were then analyzed by organism to identify differences between outer leaflet charge content of beta-barrels. The average hydrophobicity by membrane depth was calculated separately for the 17 Escherichia coli monomer structures and the 12 P.aeruginosa monomer structures (Fig. 4a). For each of these two organisms, the average hydrophobicity by membrane depth of the outward-facing beta-barrel amino acids was calculated for every 2 Å from a membrane depth of −16 Å to +18 Å. The results are shown in Figure 4a. The data for the two organisms were also fit using a local polynomial with a bandwidth of 2 Å. These regressions show similar results. There is a difference in the hydrophobicity with the introduction of different lipid As. The major drop in hydrophobicity for P.aeruginosa barrels occurs at bin 10–12 Å, while for E.coli barrels the major drop is at the 12–14 Å bin, a difference of ∼2 Å.
This large decrease in hydrophobicity was shown in Figure 3a to correspond to the free hydroxyl region of the lipid A’s acyl chains and the lipid A glucosamines, the most hydrophilic region of the LPS leaflet of the bilayer. Thus a 2-Å difference in its location is likely related to structural differences between the lipid A of E.coli and P.aeruginosa. Indeed, the lipid A of P.aeruginosa (Kulshin et al., 1991) is 2 carbons shorter than the lipid A of E.coli (Raetz, 1990) as shown in Figure 4b. Thus, the P.aeruginosa lipid A glucosamines are likely at a lower z value than that of the E.coli lipid A glucosamines. Another contributing factor to this difference in average hydrophobicity profiles is that the lipid A of P.aeruginosa contains more, free hydroxyl groups than the lipid A of E.coli. The increase of free hydroxyl groups would increase the polarity of the local environment, thus correlating with lower hydrophobicity. This is precisely what is seen in the lower hydrophobicity values for P.aeruginosa over E.coli in the top region of the lipid A acyl chain. It is important to note that the difference is not likely due to differences in barrel length between E.coli and P.aeruginosa. There is little difference between the average barrel axis length of the P.aeruginosa barrels (33.5 Å) and the E.coli barrels (34.6 Å).
4 DISCUSSION
There is a significant asymmetry in the distribution of outward-facing charged and polar amino acids in OMBBs. Charged amino acids are strongly preferred in the external-membrane side of OMBBs and are much more rarely found on the internal side (Fig. 1). The preference of amino acid types by location within the membrane correlates well with previously determined hydrophobicity scales (Supplementary Figs S1 and S2). These preferences may be useful for OMBB structure prediction, which is evolving away from template-based models (Randall et al., 2008) and moving more toward knowledge-based potential functions (Naveed et al., 2011) and structure predictors that use an explicit z-potential (Hayat and Elofsson, 2012).
The asymmetric charge distribution is correlated with the asymmetric structure of the outer membrane. Low hydrophobicites are seen at the outermost edge of the internal region where the phospholipid head group is situated. The hydrophobicity increases and then plateaus where the fatty acyl chains of the phospholipid group interdigitate with the fatty acyl chains of lipid A of the outer leaflet. The hydrophobicity then drops precipitously at the free hydroxyl region of the acyl chains of lipid A and reaches a minimum at the site of the phosphatidyl glucosamines of lipid A. The average hydrophobicity then rises slightly in the saccharide region of the LPS outer leaflet becoming less hydrophobic as it gets closer to the water interface (Fig. 3).
The differences in outer membranes among organisms are reflected in the distribution of the hydrophobicity of their amino acids. The only two organisms for which there is currently enough structural data to produce statistically significant results are P.aeruginosa and E.coli. When comparing the average hydrophobicities of the two, the most notable difference is at z in the range of 10–12. In this location of the membrane, the average hydrophobicities of the two organisms sharply diverge. The E.coli hydrophobicity is still in line with the hydrophobicities in the fatty acyl chain region, whereas the P.aeruginosa hydrophobicity is indicative of the free hydroxyl/glucosamine region of lipid A. This difference and the subsequent slightly lower hydrophobicities of P.aeruginosa are consistent with the structural differences of the membrane in these two organisms in precisely those regions. The lipid A of P.aeruginosa is both shorter and more polar than the lipid A of E.coli. Thus the change in hydrophobicity would be predicted to occur at a lower z value, and then the subsequent hydrophobicities in that region would continue to be slightly lower as is consistent with the structure of the LPS and the hydrophobicities corresponding to it (Fig. 3).
The observation that differences in the structure of an organism’s lipid A is manifested in the amino acid composition of its OMBBs could be useful for heterologous expression of recombinant mammalian mitochondrial OMBB proteins. To date only one mitochondrial OMBB’s structure has been solved (Bayrhuber et al., 2008; Ujwal et al., 2008). A major bottleneck to membrane protein characterization is overexpression, which can sometimes be overcome by careful selection of an appropriate heterologous expression system (Bernaudat et al., 2011; Tate, 2001). Stabilization of mitochondrial OMBBs might be accomplished by expression in organisms with lipid A structures that are more similar to the mitochondrial outer membrane external leaflet—i.e. a lipid A that is more similar to a phospholipid. Alternatively, stabilization might be achieved by mutating selected outward-facing residues of the desired mammalian protein at critical z positions that correspond with the phosphate locations in lipid A of the desired host. Thus mitochondrial proteins might be more easily expressed in prokaryotic membranes and greater structural knowledge of mitochondrial proteins might be ascertained.
Spontaneous insertion of OMBBs both in vivo and in vitro should be reexamined in light of the demonstrated charge asymmetry of OMBBs. It seems unlikely that the highly charged part of the protein that is bound for the external region of the membrane in vivo passes all the way through the lipid bilayer of vesicles. Although the exact energetic cost is unclear, certainly the high energetic cost of temporarily stripping waters from the polar region and burying charges in a hydrophobic environment would be unnecessarily high. Theoretical calculations of moving even a single protonated arginine from water to a membrane environment have estimated the cost at 38 kcal/mol (Honig and Hubbell, 1984). More recent MD simulations have suggested that in the inner membrane an arginine attached to a helix would cost 17 kcal/mol (Dorairaj and Allen, 2007), whereas experiments using the translocon machinery suggest the cost is 2.5 kcal/mol (Hessa et al., 2005). Even with the most conservative estimate, with an average of one external charged residue per strand for OMBBs (Fig. 1b) and a minimum of eight strands, this sums to a minimum of ≈20 kcal/mol.
However, OMBBs can still insert into vesicles in a manner that allows them to have a native fold—backward, with the hydrophobic end first. Inserting backward from the outside of the vesicle would allow the OMBBs to be topologically identical to how OMBBs are inserted into the cellular outer membrane from the inside, with the charged end facing outward with respect to the vesicle and the less charged end facing inward.
Spontaneous insertion of OMBBs into vesicles has been demonstrated for at least 11 OMBBs (Burgess et al., 2008; Mahalakshmi et al., 2007; Pocanschi et al., 2006; Shanmugavadivu et al., 2007; Surrey and Jähnig, 1992; Surrey et al., 1996) under a large variety of conditions—including differences in vesicle composition, temperature and pH. However, the forward insertion mechanism is the only directional mechanism that has been demonstrated for outer membrane protein insertion into vesicles (Kleinschmidt et al., 1999; Kleinschmidt and Tamm, 1999). This mechanism was established using time-dependent fluorescence quenching of tryptophan. It is notable that this mechanism was demonstrated for the OMBB outer membrane protein A (OmpA), which has a large periplasmic domain that might encourage forward insertion. It is also notable that OmpA has only 5 external-region outward-facing charged residues in its barrel (0.625 per strand) compared with the average OMBB, which has 17 similarly located charged residues (1 per strand) (Fig. 1b). Thus OmpA’s mechanism might be an anomaly as a result of an easier forward insertion mechanism owing to less charge hindrance. Moreover, when comparing spontaneous OMBB insertion in vesicles, each protein preferred a different lipid composition, temperature and pH (most of which were non-physiological) (Burgess et al., 2008), possibly supporting a heterogeneity of in vitro insertion mechanism.
In contrast to the vesicle experiments, OMBBs cannot insert spontaneously in vivo. This is perhaps because the charged region would have to pass through the entire outer membrane to be oriented correctly. In light of this, the BAM complex might not only speed up insertion, as has previously been posited, but likely also has a mechanism to shield the charged region from the apolar region of the outer membrane. Many proposed mechanisms (Kim et al., 2012) do not take into account this necessary function of the BAM complex and will need to be reexamined in light of the extreme charge asymmetry.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Gunnar von Heijne and William F. DeGrado for helpful discussions. We gratefully acknowledge Cynthia B. Myers of the Fox Chase organic synthesis facility and Qifang Xu and Benjamin H. North of the Dunbrack laboratory for technical assistance.
Funding: National Institutes of Health [T32CA-009035-37 (to J.S.G.S.); R01-GM84453 (to R.L.D.)]
Conflict of Interest: None declared.
REFERENCES
- Andersson H, von Heijne G. Membrane protein topology: effects of delta mu H+ on the translocation of charged residues explain the ‘positive inside’ rule. EMBO J. 1994;13:2267–2272. doi: 10.1002/j.1460-2075.1994.tb06508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayrhuber M, et al. Structure of the human voltage-dependent anion channel. Proc. Natl Acad. Sci. 2008;105:15370–15375. doi: 10.1073/pnas.0808115105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernaudat F, et al. Heterologous expression of membrane proteins: choosing the appropriate host. PLoS One. 2011;6:e29191. doi: 10.1371/journal.pone.0029191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess NK, et al. β-Barrel proteins that reside in the Escherichia coli outer membrane in vivo demonstrate varied folding behavior in vitro. J. Biol. Chem. 2008;283:26748–26758. doi: 10.1074/jbc.M802754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caroff M, Karibian D. Structure of bacterial lipopolysaccharides. Carbohydr. Res. 2003;338:2431–2447. doi: 10.1016/j.carres.2003.07.010. [DOI] [PubMed] [Google Scholar]
- Dorairaj S, Allen TW. On the thermodynamic stability of a charged arginine side chain in a transmembrane helix. Proc. Natl Acad. Sci. 2007;104:4943–4948. doi: 10.1073/pnas.0610470104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson AD, et al. A conserved structural motif for lipopolysaccharide recognition by procaryotic and eucaryotic proteins. Structure. 2000;8:585–592. doi: 10.1016/s0969-2126(00)00143-x. [DOI] [PubMed] [Google Scholar]
- Finn RD, et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goder V, et al. Sec61p contributes to signal sequence orientation according to the positive-inside rule. Mol. Biol. Cell. 2004;15:1470–1478. doi: 10.1091/mbc.E03-08-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayat S, Elofsson A. Ranking models of transmembrane β-barrel proteins using Z-coordinate predictions. Bioinformatics. 2012;28:i90–i96. doi: 10.1093/bioinformatics/bts233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessa T, et al. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 2005;433:377–381. doi: 10.1038/nature03216. [DOI] [PubMed] [Google Scholar]
- Hong H, et al. Role of aromatic side chains in the folding and thermodynamic stability of integral membrane proteins. J. Am. Chem. Soc. 2007;129:8320–8327. doi: 10.1021/ja068849o. [DOI] [PubMed] [Google Scholar]
- Honig BH, Hubbell WL. Stability of “salt bridges” in membrane proteins. Proc. Natl Acad. Sci. 1984;81:5412–5416. doi: 10.1073/pnas.81.17.5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh D, et al. A knowledge-based potential highlights unique features of membrane α-helical and β-barrel protein insertion and folding. Protein Sci. 2012;21:50–62. doi: 10.1002/pro.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackups R, Jr, Liang J. Interstrand pairing patterns in β-barrel membrane proteins: the positive-outside rule, aromatic rescue, and strand registration prediction. J. Mol. Biol. 2005;354:979–993. doi: 10.1016/j.jmb.2005.09.094. [DOI] [PubMed] [Google Scholar]
- Jimenez-Morales D, Liang J. Pattern of amino acid substitutions in transmembrane domains of β-barrel membrane proteins for detecting remote homologs in bacteria and mitochondria. PLoS One. 2011;6:e26400. doi: 10.1371/journal.pone.0026400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamio Y, Nikaido H. Outer membrane of Salmonella typhimurium: accessibility of phospholipid head groups to phospholipase C and cyanogen bromide activated dextran in the external medium. Biochemistry. 1976;15:2561–2570. doi: 10.1021/bi00657a012. [DOI] [PubMed] [Google Scholar]
- Kim KH, et al. The bacterial outer membrane β-barrel assembly machinery. Protein Sci. 2012;21:751–768. doi: 10.1002/pro.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinschmidt JH, Tamm LK. Time-resolved distance determination by tryptophan fluorescence quenching: probing intermediates in membrane protein folding. Biochemistry. 1999;38:4996–5005. doi: 10.1021/bi9824644. [DOI] [PubMed] [Google Scholar]
- Kleinschmidt JH, et al. Outer membrane protein a of Escherichia coli inserts and folds into lipid bilayers by a concerted mechanism. Biochemistry. 1999;38:5006–5016. doi: 10.1021/bi982465w. [DOI] [PubMed] [Google Scholar]
- Koebnik R. Structural and functional roles of the surface-exposed loops of the beta-barrel membrane protein ompa from Escherichia coli. J. Bacteriol. 1999;181:3688–3694. doi: 10.1128/jb.181.12.3688-3694.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulshin VA, et al. Structural characterization of the lipid A component of Pseudomonas aeruginosa wild-type and rough mutant lipopolysaccharides. Eur. J. Biochem. 1991;198:697–704. doi: 10.1111/j.1432-1033.1991.tb16069.x. [DOI] [PubMed] [Google Scholar]
- Mahalakshmi R, et al. NMR structural studies of the bacterial outer membrane protein OmpX in oriented lipid bilayer membranes. Biochim. Biophys. Acta. 2007;1768:3216–3224. doi: 10.1016/j.bbamem.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinverni JC, Silhavy TJ. An ABC transport system that maintains lipid asymmetry in the Gram-negative outer membrane. Proc. Natl Acad. Sci. 2009;106:8009–8014. doi: 10.1073/pnas.0903229106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelli PL, et al. A sequence-profile-based HMM for predicting and discriminating β barrel membrane proteins. Bioinformatics. 2002;18:S46–S53. doi: 10.1093/bioinformatics/18.suppl_1.s46. [DOI] [PubMed] [Google Scholar]
- Moon CP, Fleming KG. Side-chain hydrophobicity scale derived from transmembrane protein folding into lipid bilayers. Proc. Natl Acad. Sci. 2011;108:10174–10177. doi: 10.1073/pnas.1103979108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naveed H, et al. Predicting three-dimensional structures of transmembrane domains of β-barrel membrane proteins. J. Am. Chem. Soc. 2011;134:1775–1781. doi: 10.1021/ja209895m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn MJ, et al. Mechanism of assembly of the outer membrane of Salmonella typhimurium: isolation and characterization of cytoplasmic and outer membrane. J. Biol. Chem. 1972;247:3962–3972. [PubMed] [Google Scholar]
- Pocanschi CL, et al. The major outer membrane protein of fusobacterium nucleatum (FomA) folds and inserts into lipid bilayers via parallel folding pathways. J. Mol. Biol. 2006;355:548–561. doi: 10.1016/j.jmb.2005.10.060. [DOI] [PubMed] [Google Scholar]
- Raetz CRH. Biochemistry of endotoxins. Annu. Rev. Biochem. 1990;59:129–170. doi: 10.1146/annurev.bi.59.070190.001021. [DOI] [PubMed] [Google Scholar]
- Randall A, et al. TMBpro: secondary structure, β-contact and tertiary structure prediction of transmembrane β-barrel proteins. Bioinformatics. 2008;24:513–520. doi: 10.1093/bioinformatics/btm548. [DOI] [PubMed] [Google Scholar]
- Rees DC, Eisenberg D. Turning a reference inside-out: commentary on an article by stevens and arkin entitled: “Are membrane proteins ‘inside-out’ proteins?” (Proteins 1999;36:135–143) Proteins. 2000;38:121–122. [PubMed] [Google Scholar]
- Rees DC, et al. Hydrophobic organization of membrane proteins. Science. 1989;245:510–513. doi: 10.1126/science.2667138. [DOI] [PubMed] [Google Scholar]
- Schramm CA, et al. Knowledge-based potential for positioning membrane-associated structures and assessing residue-specific energetic contributions. Structure. 2012;20:924–935. doi: 10.1016/j.str.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneck E, et al. Quantitative determination of ion distributions in bacterial lipopolysaccharide membranes by grazing-incidence X-ray fluorescence. Proceedings of the National Academy of Sciences. 2010;107:9147–9151. doi: 10.1073/pnas.0913737107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz GE. The structure of bacterial outer membrane proteins. Biochim. Biophys. Acta. 2002;1565:308–317. doi: 10.1016/s0005-2736(02)00577-1. [DOI] [PubMed] [Google Scholar]
- Sen K, et al. Porin channels in intact cells of Escherichia coli are not affected by Donnan potentials across the outer membrane. J. Biol. Chem. 1988;263:1182–1187. [PubMed] [Google Scholar]
- Senes A, et al. Ez, a depth-dependent potential for assessing the energies of insertion of amino acid side-chains into membranes: derivation and applications to determining the orientation of transmembrane and interfacial helices. J. Mol. Biol. 2007;366:436–448. doi: 10.1016/j.jmb.2006.09.020. [DOI] [PubMed] [Google Scholar]
- Shanmugavadivu B, et al. Correct folding of the β-barrel of the human membrane protein VDAC requires a lipid bilayer. J. Mol. Biol. 2007;368:66–78. doi: 10.1016/j.jmb.2007.01.066. [DOI] [PubMed] [Google Scholar]
- Shroll RM, Straatsma TP. Molecular structure of the outer bacterial membrane of Pseudomonas aeruginosa via classical simulation. Biopolymers. 2002;65:395–407. doi: 10.1002/bip.10279. [DOI] [PubMed] [Google Scholar]
- Stock JB, et al. Periplasmic space in Salmonella typhimurium and Escherichia coli. J. Biol. Chem. 1977;252:7850–7861. [PubMed] [Google Scholar]
- Surrey T, Jähnig F. Refolding and oriented insertion of a membrane protein into a lipid bilayer. Proc. Natl Acad. Sci. USA. 1992;89:7457–7461. doi: 10.1073/pnas.89.16.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surrey T, et al. Folding and membrane insertion of the trimeric β-barrel protein OmpF. Biochemistry. 1996;35:2283–2288. doi: 10.1021/bi951216u. [DOI] [PubMed] [Google Scholar]
- Tamm LK, et al. Folding and assembly of [beta]-barrel membrane proteins. Biochim. Biophys. Acta. 2004;1666:250–263. doi: 10.1016/j.bbamem.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Tate CG. Overexpression of mammalian integral membrane proteins for structural studies. FEBS Lett. 2001;504:94–98. doi: 10.1016/s0014-5793(01)02711-9. [DOI] [PubMed] [Google Scholar]
- Ujwal R, et al. The crystal structure of mouse VDAC1 at 2.3 Å resolution reveals mechanistic insights into metabolite gating. Proc. Natl Acad. Sci. 2008;105:17742–17747. doi: 10.1073/pnas.0809634105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Klompenburg W, et al. Anionic phospholipids are determinants of membrane protein topology. EMBO J. 1997;16:4261–4266. doi: 10.1093/emboj/16.14.4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G, Gavel Y. Topogenic signals in integral membrane proteins. Eur. J. Biochem. 1988;174:671–678. doi: 10.1111/j.1432-1033.1988.tb14150.x. [DOI] [PubMed] [Google Scholar]
- Wang G, Dunbrack RL. PISCES: a protein sequence culling server. Bioinformatics. 2003;19:1589–1591. doi: 10.1093/bioinformatics/btg224. [DOI] [PubMed] [Google Scholar]
- Wimley WC. Toward genomic identification of β-barrel membrane proteins: composition and architecture of known structures. Protein Sci. 2002;11:301–312. doi: 10.1110/ps.29402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimley WC. The versatile [beta]-barrel membrane protein. Curr. Opin. Struct. Biol. 2003;13:404–411. doi: 10.1016/s0959-440x(03)00099-x. [DOI] [PubMed] [Google Scholar]
- Wimley WC, et al. Solvation energies of amino acid side chains and backbone in a family of host–guest pentapeptides. Biochemistry. 1996;35:5109–5124. doi: 10.1021/bi9600153. [DOI] [PubMed] [Google Scholar]
- Xie J, et al. Phosphatidylethanolamine and monoglucosyldiacylglycerol are interchangeable in supporting topogenesis and function of the polytopic membrane protein lactose permease. J. Biol. Chem. 2006;281:19172–19178. doi: 10.1074/jbc.M602565200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Dunbrack RL. Assignment of protein sequences to existing domain and family classification systems: Pfam and the PDB. Bioinformatics. 2012;28:2763–2772. doi: 10.1093/bioinformatics/bts533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Y, Saier MH. The β-barrel finder (BBF) program, allowing identification of outer membrane β-barrel proteins encoded within prokaryotic genomes. Protein Sci. 2002;11:2196–2207. doi: 10.1110/ps.0209002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.