

Abstract
Motivation: One of the major challenges for contemporary bioinformatics is the analysis and accurate annotation of genomic datasets to enable extraction of useful information about the functional role of DNA sequences. This article describes a novel genome-wide statistical approach to the detection of specific DNA sequence motifs based on similarities between the promoters of similarly expressed genes. This new tool, cisExpress, is especially designed for use with large datasets, such as those generated by publicly accessible whole genome and transcriptome projects. cisExpress uses a task farming algorithm to exploit all available computational cores within a shared memory node. We demonstrate the robust nature and validity of the proposed method. It is applicable for use with a wide range of genomic databases for any species of interest.
Availability: cisExpress is available at www.cisexpress.org.
Contact: tatiana.tatarinova@usc.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The amount of raw data available in publicly accessible genomic databases is growing exponentially in the form of massive linear arrays of DNA sequences. Robust analysis and accurate annotation of these datasets are needed to extract useful information about the functional role of the DNA sequences. One of the key goals of genome analysis is the identification of putative genome function, especially the detection of potential DNA cis-regulatory elements, with high confidence. Identifying the regulatory motifs bound by transcription factors can provide crucial insights into the mechanisms of transcriptional regulation. By correctly identifying regulatory motifs, it is possible to predict the expression of the genes under specific circumstances or in specific tissues. Furthermore, molecular mechanisms of genetic diseases, caused by the incorrect expression of some genes, can be discovered. It has been recently demonstrated that in the case of the mouse genome, many in vitro-derived motifs performed similarly to motifs derived from in vivo data (Weirauch et al., 2013). However, major problems in studying the specificity of the binding of transcription factors to DNA motifs include a lack of data and deficiencies in predictive models for motif detection. Several efforts have been made to compare existing methods for computational identification of regulatory patterns (Sandve and Drablos, 2006; Tompa et al., 2005; Troukhan et al., 2009). These benchmarking studies suggested that prediction of regulatory elements remains a challenge for computational biologists, and that more work is required to optimize the algorithms used. Although different programs may perform better for individual datasets, no single method takes all relevant elements into consideration. Users are advised to use a combination of several motif-finding tools for best results. For this reason, we decided to develop a novel tool, called cisExpress, which is designed to achieve more effective analysis of large datasets in a manner that is both cost effective and highly robust in its predictive capacity.
2 METHODS
The core element of cisExpress is a significantly improved and enhanced adaptation of an earlier method, Motifer (Troukhan et al., 2009) (not publicly available), specifically modified to process large datasets. cisExpress is based on two important assumptions: (i) the function of promoter motifs is position specific, and (ii) microarray data provide reasonable measurements of transcript abundance and reflect promoter activity.
2.1 Data preparation
The majority of gene expression data used to validate our algorithm was taken from AtGenExpress developmental experiment (Schmid et al., 2005). In addition, we used abiotic stress experiment data (Kilian et al., 2007; von Koskull-Doring et al., 2007) and nitrate response experiments (Wang et al. 2003). Gene expression intensities were log transformed. For the variability dataset, we used standard deviation of gene expression values in the log scale across all analyzed conditions, and for the strength dataset, we used geometric average of gene expression variables, log transformed. Promoter sequences were obtained using the TSSer algorithm (Troukhan et al., 2009), using 188 506 5′ expressed sequence tag (EST) sequences and 25 026 full-length mRNA sequences of Arabidopsis thaliana, available at GenBank. Genomic sequences (version 9) were downloaded from The Arabidopsis Information Resource Web site. We aligned EST and mRNA sequences against the genomic sequence using Washington University Blast, storing only the best match per EST. Each EST and mRNA match was assigned to one locus, and frequencies of hits per genomic position were computed for each locus. The genomic position of the most representative transcription start site (TSS) per locus corresponds to the mode of EST/mRNA hits distribution. For each locus, we have extracted 1000 nt in positions [TSS-500, TSS+500].
Method
cisExpress divides the motif-finding problem into two general stages:
Stage 1: Detecting ‘seed’ motifs. This stage determines consensus sequences of motifs and their approximate position in the promoter region. It follows the steps of Motifer (Troukhan et al., 2009). The discovery of motifs is performed in overlapping windows of predefined size. For every word present in the observed window, we calculate Z-score according to Equation 1.9 in the Supplementary Material.
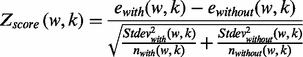 |
where ewith(w,k) and ewithout(w,k) are average gene expression values; Stdevwith(w,k) and Stdevwithout(w,k) are standard deviation of gene expression values; nwith(w,k) and nwithout(w,k) are the number of sequences of genes containing and not containing word w in the kth window.
Words with Z-scores above a predefined threshold are stored as primary motifs. Groups of similar motifs discovered within one window are then merged together resulting in longer and/or more ambiguous motifs. This part of the method outputs the motif in the form of a consensus sequence and includes the position of window where it was discovered.
Stage 2: ‘Optimizing the previously obtained motifs’. In this stage, a genetic algorithm (GA) (Holland, 1992; Wall, 2007) is applied to each of the motifs detected in Stage 1 to determine the best possible motif model and motif position (optimization process tries to maximize absolute value of Z-score of the motif). The output consists of an N×4 motif matrix (where N is the length of the motif), representing relative frequencies of nucleotides in the motif. For each position within the motif, there is a probability that each base occurs at that position. This representation also includes information about motif conservation and position. Specifications of used GA can be found in the Supplementary Material. This stage is unique to cisExpress.
Availability: cisExpress is available for use from www.cisexpress.org as a stand-alone open-source application or via a web interface. The web interface submits jobs to a high-performance computing system (currently HPC Wales, www.hpcwales.co.uk), where the efficient parallel implementation of cisExpress ensures easy and fast access to the tool without the requirement to install the software. In the near future, we aim to incorporate our novel improved method for promoter prediction into this interface. These tools will then form a pipeline.
3 RESULTS
The cisExpress algorithm was applied to 13 379 promoter regions of the classical genetic plant model, A.thaliana, using 11 gene expression datasets. Promoter sequences were randomly divided into training and testing sets (in a 50:50 ratio). Sequences in the training set were used to find the highest-scoring motifs for each experimental condition using the cisExpress and MatrixREDUCE (Foat et al., 2006) programs. The testing set promoters were examined for the presence of the motifs, and corresponding gene expression values were compared for genes whose promoters did and did not contain the motifs, using a t-test. A full description of the benchmark is given in the Supplementary Material. Most of the motifs identified by the two programs are those previously identified and well characterized in promoters of A.thaliana. For example, CACGTG is an abscisic acid responsive element-like binding site motif involved in both dehydration-related and low-temperature-responsive gene expression. The 5′-TATAAA-3′ DNA sequence (TATA)-box is typically associated with the variability of gene expression, including tissue and stress specificity (Troukhan et al., 2009). CATGC, detected in the SEEDS and FLOWERS sets, is a canonical RY (RY element, having a typical sequence CATGCATG, is conserved in the promoter regions of the genes of legume seed storage proteins) seed-specific motif. Motifs GGCCC in core promoter regions of activated genes were previously observed (Molina and Grotewold, 2005). Motif CTAGA, associated with the Huntingtin, Elongation factor 3, protein phosphatsase 2A, TOR1 repeats (HEAT) dataset, is a putative response element for multiprotein bridging factor 1 (MBF1), which controls the expression of 36 different transcripts during heat stress (Suzuki et al., 2011). Motif AGGCC (identified as important for gene expression under NITROGEN stress) is bound by a group of transcription factors known as the TCP [teosinte branched 1 (TB1), cycloidea (CYC), proliferating cell factors 1 and 2 (PCF1 and PCF2)] family (Walley et al., 2011). A comparison between MatrixREDUCE and cisExpress for 11 gene expression datasets is given in Table 1. For each experimental condition, Table 1 shows consensus sequence of the best 5-nt motif, position of the motif (for cisExpress) and P-value of the t-test. This shows that cisExpress is capable of detecting position weight matrices that are more specific and that result in lower t-test P-values across all analyzed conditions. cisExpress is able to outperform MatrixREDUCE owing to its use of motif position information and the addition of the GA optimization step to tune position weight matrices. The high performance computing back-end and efficient parallel implementation of the code also allows cisExpress to handle large datasets, such as those generated by publicly accessible whole genome and transcriptome projects. For the A.thaliana benchmark used in Table 1 (consisting of 98 discrete tasks to distribute among the available computational cores), the parallel strategy reduces execution time for the motif detecting part of the algorithm by a factor of 18.5 within a (64-core) node. For a larger dataset (with far more tasks than available cores), better parallel scaling would be expected. Complete parallel performance results for cisExpress are presented in Supplementary Material. In conclusion, we demonstrated the robust nature and validity of the cisExpress algorithm. cisExpress is an organism-independent tool and it is applicable for use with a wide range of genomic databases.
Table 1.
Comparison of cisExpress and MatrixREDUCE using 11 gene expression datasets of A.thaliana
| Condition |
cisExpress |
MatrixREDUCE |
|||
|---|---|---|---|---|---|
| Best 5-nt consensus | Position | P-value | Best 5-nt consensus | P-value | |
| Drought | CACGT | −110 … −60 | 10−14 | ACGTG | 10−13 |
| Heat | CTAGA | −70 … −50 | 10−2 | TCTAG | 10−4 |
| Cold | CTATA | −50 … −15 | 10−34 | TATAT | 10−4 |
| Roots | TCTAT | −40 … −20 | 10−21 | TATAA | 10−10 |
| Seeds | CATGC | −80 … −44 | 10−9 | CATGC | 10−5 |
| Nitrogen | AGGCC | −110 … −50 | 10−18 | AGGCC | 10−8 |
| Strength | GGCCC | −110 … −50 | 10−11 | GATCT | 10−10 |
| Variability | TATAA | −50 … −10 | 10−140 | TATAT | 10−4 |
| Flowers | CTATA | −40 … −20 | 10−14 | CATGC | 10−2 |
| Leaves | CTTAT | −40 … −20 | 10−20 | TAGGG | 10−9 |
| Light | CCGCG | −110 … −90 | 10−2 | AATAT | 10−2 |
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Professor Roger Jelliffe, USC, for helpful suggestions and for proof reading of the manuscript. This work made use of the facilities provided by the High Performance Computing Wales network, which is collaboration between Welsh universities, Government and Fujitsu Laboratories Europe.
Funding: EU Erasmus program (to M.T.). University of South Wales Research Investment Scheme (to T.T. and D.J.M.). NIH-NICHD: HD070996 and NIH: GM068968 grants (to T.T. in part).
Conflict of Interest: none declared.
References
- Foat BC, et al. Statistical mechanical modeling of genome-wide transcription factor occupancy data by MatrixREDUCE. Bioinformatics. 2006;22:e141–e149. doi: 10.1093/bioinformatics/btl223. [DOI] [PubMed] [Google Scholar]
- Holland JH. Adaptation in Natural and Artificial Systems. Cambridge, MA, USA: MIT Press; 1992. [Google Scholar]
- Kilian J, et al. The AtGenExpress global stress expression data set: protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. Plant J. 2007;50:347–363. doi: 10.1111/j.1365-313X.2007.03052.x. [DOI] [PubMed] [Google Scholar]
- Molina C, Grotewold E. Genome wide analysis of Arabidopsis core promoters. BMC Genomics. 2005;6:25. doi: 10.1186/1471-2164-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandve GK, Drablos F. A survey of motif discovery methods in an integrated framework. Biol. Direct. 2006;1:11. doi: 10.1186/1745-6150-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M, et al. A gene expression map of Arabidopsis thaliana development. Nat. Genet. 2005;37:501–506. doi: 10.1038/ng1543. [DOI] [PubMed] [Google Scholar]
- Suzuki N, et al. Identification of the MBF1 heat-response regulon of Arabidopsis thaliana. Plant J. 2011;66:844–851. doi: 10.1111/j.1365-313X.2011.04550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa M, et al. Assessing computational tools for the discovery of transcription factor binding sites. Nat. Biotechnol. 2005;23:137–144. doi: 10.1038/nbt1053. [DOI] [PubMed] [Google Scholar]
- Troukhan M, et al. Genome-wide discovery of cis-elements in promoter sequences using gene expression data. OMICS. 2009;13:139–151. doi: 10.1089/omi.2008.0034. [DOI] [PubMed] [Google Scholar]
- von Koskull-Doring P, et al. The diversity of plant heat stress transcription factors. Trends Plant Sci. 2007;12:452–457. doi: 10.1016/j.tplants.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Wall M. 2007 GAlib A C++ library of genetic algorithm components. Version 2.4.7. Available from: < http://lancet.mit.edu/ga/> (09 July 2013, date last accessed) [Google Scholar]
- Walley H, et al. Transcriptomic analysis reveals calcium regulation of specific promoter motifs in Arabidopsis. Plant Cell. 2011;23:4079–4095. doi: 10.1105/tpc.111.090480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, et al. Microarray analysis of the nitrate response in Arabidopsis roots and shoots reveals over 1,000 rapidly responding genes and new linkages to glucose, trehalose-6-phosphate, iron, and sulfate metabolism. Plant Physiol. 2003;132:556–567. doi: 10.1104/pp.103.021253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, et al. Evaluation of methods for modeling transcription factor sequence specificity. Nat. Biotechnol. 2013;31:126–134. doi: 10.1038/nbt.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.