

Abstract
Motivation: Tumors exhibit numerous genomic lesions such as copy number variations, structural variations and sequence variations. It is difficult to determine whether a specific constellation of lesions observed across a cohort of multiple tumors provides statistically significant evidence that the lesions target a set of genes that may be located across different chromosomes but yet are all involved in a single specific biological process or function.
Results: We introduce the genomic random interval (GRIN) statistical model and analysis method that evaluates the statistical significance of the abundance of genomic lesions that overlap a specific locus or a pre-defined set of biologically related loci. The GRIN model retains certain biologically important properties of genomic lesions that are ignored by other methods. In a simulation study and two example analyses of leukemia genomic lesion data, GRIN more effectively identified important loci as significant than did three methods based on a permutation-of-markers model. GRIN also identified biologically relevant pathways with a significant abundance of lesions in both examples.
Availability: An R package will be freely available at CRAN and www.stjuderesearch.org/site/depts/biostats/software.
Contact: stanley.pounds@stjude.org
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Microarray and next-generation sequencing technologies have enabled biomedical researchers to characterize the genome of individual tissue samples at a high resolution. In cancer genomics, these technologies have been used to identify genomic lesions in which the DNA of the tumor has been altered relative to that of normal tissue from the same subject. Genomic lesions include copy number changes, sequence mutations and structural rearrangements. Genomic lesions may impact oncogenesis (Mullighan et al., 2007) and clinical prognosis (Mullighan et al., 2009). In this way, the development of the data collection technologies and associated data analysis methods has contributed profoundly to our understanding of the genomic basis of cancer development and prognosis.
Several analysis methods have been developed to identify and assign a statistical significance (e.g. P-value) to ‘hot spot’ loci that are affected by copy number alterations at a high frequency: the Genomic Identification of Significant Targets in Cancer (GISTIC; Beroukhim et al., 2007) algorithm and its extensions JISTIC (Sanchez-Garcia et al., 2010) and GISTIC 2.0 (Mermel et al., 2011); Significant Aberration in Cancer (SAIC; Yuan et al., 2012a); and TAGCNA (Yuan et al., 2012b). GISTIC is one of the most widely used methods. It computes a statistic that summarizes the frequency and amplitude of copy number alterations at each marker of a specific microarray platform. It then uses within-tumor permutation of the assignment of copy number status to marker locus as a null statistical model to evaluate statistical significance. In this way, GISTIC obtains a P-value for each microarray marker locus, and significant peaks are subsequently identified from this profile of P-values. The JISTIC method uses a modified algorithm for identifying peaks in the P-value profile. The GISTIC 2.0 algorithm modifies several components of the original GISTIC algorithm, but GISTIC 2.0 still relies on permutation of markers (POMs) or bins of markers to compute P-values. SAIC and TAGCNA compute statistics that describe the extent to which specific marker loci are affected by copy number alterations and use POMs as a null statistical model to determine statistical significance. Each of these methods has been successfully used for several applications and thus represent important contributions in computational cancer biology.
Nevertheless, each of these methods have some limitations that should be addressed. First of all, these methods use a biologically implausible statistical model of the null probability that a lesion affects a locus. GISTIC, JISTIC, GISTIC 2.0, SAIC and TAGCNA each use a POMs model for this purpose. POM permutes the assignment of copy number status to marker locus within each tumor. In this way, POM shatters single contigous lesions into numerous probabilistically independent entities. Consequently, many biologically important lesions are not identified as statistically significant (Mermel et al., 2011). Secondly, these methods are not readily applicable to genomic lesion data collected by whole-genome sequencing (WGS). These methods each require that the data be represented in the form of a marker-by-subject matrix. With WGS, every ‘mappable’ base pair in the genome is a ‘marker’ so the matrix will be large. Additionally, these methods were developed solely for copy number alterations and do not provide a way to incorporate other lesions, such as point mutations or structural rearrangements that may be detected with WGS (Wang et al., 2011). Finally, these methods only evaluate the statistical significance of the frequency that individual markers are affected by a lesion, but do not directly determine whether a given set of biologically related genes scattered across the genome (such as a particular pathway) have a statistically significant abundance of genomic lesions.
Therefore, we have developed the genomic random interval (GRIN) statistical model for statistical analysis of genomic lesion data. The GRIN model explicitly represents each genomic lesion as an entity that affects one point locus, a set of point loci or an interval locus along a chromosome. In this way, the GRIN model retains the continuity of the genomic lesions and naturally avoids the difficulties introduced by statistical models that do not retain lesion continuity. Additionally, the GRIN model can accommodate any genomic lesion that can be represented as a locus on the reference genome. Copy number alterations are represented by intervals with distinct start and end loci; point mutations are represented by their respective loci; and structural rearrangements are represented by the loci of the associated breakpoints. Finally, the GRIN model provides a computationally feasible approach to evaluate the statistical significance of the frequency that lesions affect a set of genes involved in a particular biological process. Furthermore, like other methods, GRIN also provides a mechanism to evaluate the significance of the frequency that lesions affect each point locus in the genome and the locus of each individual gene in the genome.
The remainder of this work is organized as follows. In Section 2, we describe the GRIN model in detail. Section 3 evaluates the performance of GRIN and other methods in a simulation study and two example analyses from leukemia studies. Section 4 provides discussion and concluding remarks.
2 METHODS
2.1 Genomic lesion data
Genomic lesion data give the type and locus of each genomic lesion observed for each tissue sample. Let 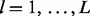 index the L genomic lesions and let
index the L genomic lesions and let 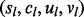 denote the subject sl, chromosome cl, start locus ul and end locus vl of lesion l. Table 1 gives an example of genomic lesion data from a study of early T-cell precursor (ETP; Zhang et al., 2012) leukemia and illustrates the mathematical notation of genomic lesion data.
denote the subject sl, chromosome cl, start locus ul and end locus vl of lesion l. Table 1 gives an example of genomic lesion data from a study of early T-cell precursor (ETP; Zhang et al., 2012) leukemia and illustrates the mathematical notation of genomic lesion data.
Table 1.
An example of genomic lesion data
| Lesion (l) | Subject (sl) | Chr (cl) | Start (ul) | End (vl) | Type |
|---|---|---|---|---|---|
| 1 | 1 | 1 | 235 201 | 8 847 300 | Loss |
| 2 | 1 | 1 | 8 905 051 | 12 761 104 | LOH |
 |
 |
 |
 |
 |
 |
| 10 | 1 | 1 | 211 055 963 | 211 055 963 | SB |
 |
 |
 |
 |
 |
 |
| 247 | 7 | 5 | 35 910 328 | 35 910 328 | Indel |
| 248 | 7 | 6 | 1 | 31 148 785 | Gain |
 |
 |
 |
 |
 |
 |
| 401 | 12 | X | 133 355 331 | 133 355 331 | PM |
The example data are a subset of that observed in a study of ETP leukemia (Zhang et al., 2012).
PM, non-silent point mutation; SB, structural breakpoint; LOH, loss of heterozygosity.
We wish to determine whether the lesions are significantly concentrated at any particular locus in the genome, within any particular gene in the genome or within the loci of a set of genes involved in a specific biological process. To address these questions, we must define statistics that describe the concentration of lesions around a specific locus, within a specific gene and within a set of genes. We must also define a statistical model to evaluate significance.
2.2 Overlap statistics
Here, we define statistics that describe the abundance of lesions that overlap one fixed set of loci 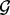 . The fixed set of loci
. The fixed set of loci 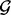 may be a single point locus, the interval locus of an individual gene or the interval loci of a set of biologically related genes that are in the same pathway or have a common ontology. Note that the fixed set of loci may be scattered on different chromosomes throughout the genome. In general,
may be a single point locus, the interval locus of an individual gene or the interval loci of a set of biologically related genes that are in the same pathway or have a common ontology. Note that the fixed set of loci may be scattered on different chromosomes throughout the genome. In general, 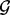 may be represented as a set of
may be represented as a set of 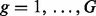 interval loci with the form
interval loci with the form 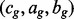 where cg is the chromosome of locus
where cg is the chromosome of locus 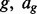 is the start position of locus g and bg is the end of locus g. We will use the acronym FLI to refer to one fixed locus of interest within the fixed set of loci
is the start position of locus g and bg is the end of locus g. We will use the acronym FLI to refer to one fixed locus of interest within the fixed set of loci 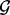 and the acronym FLIs to refer to multiple fixed loci within the set
and the acronym FLIs to refer to multiple fixed loci within the set 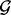 .
.
First, we define a statistic that indicates whether each lesion l overlaps each FLI g. Let 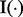 be the indicator function that equals one if the enclosed statement is true and equals zero if the enclosed statement is false. For each lesion
be the indicator function that equals one if the enclosed statement is true and equals zero if the enclosed statement is false. For each lesion 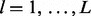 and each FLI
and each FLI 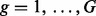 , the product
, the product
| (1) |
indicates whether lesion l overlaps FLI g because 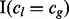 indicates whether the lesion and FLI are on the same chromosome,
indicates whether the lesion and FLI are on the same chromosome, 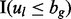 indicates that the lesion start locus is left of the FLI end locus and
indicates that the lesion start locus is left of the FLI end locus and 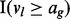 indicates that the lesion end locus is right of the FLI start locus.
indicates that the lesion end locus is right of the FLI start locus.
The abundance of lesions that overlap 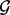 are described with statistics that are functions of the lesion-FLI overlap indicators olg in Equation (1). Each lesion l overlaps exactly
are described with statistics that are functions of the lesion-FLI overlap indicators olg in Equation (1). Each lesion l overlaps exactly
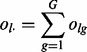 |
(2) |
FLIs. The sum
| (3) |
is the total number of overlaps. For each lesion l, let
| (4) |
indicate that the lesion overlaps at least one FLI. Then, the sum
| (5) |
is number of lesions with at least one overlap. Let 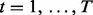 index the subjects of the study. Each subject t has
index the subjects of the study. Each subject t has
| (6) |
lesions that overlap at least one FLI because hl indicates whether lesion l overlaps at least one FLI and 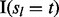 indicates whether lesion l was observed in subject t. Finally, the number of subjects with at least one overlap is
indicates whether lesion l was observed in subject t. Finally, the number of subjects with at least one overlap is
| (7) |
2.3 The GRIN model
Here, we introduce the concept of a GRIN and use it to define the null probability distributions for the descriptive statistics defined above. A GRIN with given length x base pairs on a chromosome c of length Kc base pairs may occur at each of the 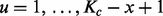 interval loci (
interval loci (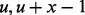 ) with uniform probability
) with uniform probability 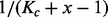 as shown in Figure 1A. A GRIN may be fully described by its chromosome c, the length Kc of chromosome c and its random start position U. Given
as shown in Figure 1A. A GRIN may be fully described by its chromosome c, the length Kc of chromosome c and its random start position U. Given 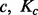 and x, the random start position U has a discrete uniform distribution over
and x, the random start position U has a discrete uniform distribution over 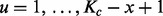 .
.
Fig. 1.

The GRIN model. (A) A GRIN of given size x may occur with uniform probability at any interval locus of equal size x along the chromosome. The overlap of a GRIN with a FLI may be represented by an indicator function of the GRIN start locus u. (B) The triangle diagram represents the overlap of any GRIN with size x and start locus u with two or more FLIs. Each horizontal line across the triangle diagram at GRIN size x defines the number of FLIs that overlap a GRIN as a function of the start locus u. For sake of illustration, a black, blue and red horizontal line are drawn across the triangle diagram, and their overlap functions are shown in the corresponding colors above the triangle diagram. (C) The probability distributions defined by the overlap functions are shown in their respective colors. (D) A probability tree that illustrates the convolution of the three probability distributions
The probability that one GRIN on chromosome c overlaps one FLI with index g (also on chromosome c) is a simple function of the FLI start position ag, the FLI end position bg, the length x of the GRIN and the length Kc of chromosome c. Figure 1A shows that a GRIN of given size x with start position u between 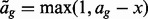 and
and 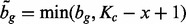 will overlap FLI g, which begins at position ag and ends at position bg. Therefore, the probability that the GRIN overlaps FLI g is simply the proportion
will overlap FLI g, which begins at position ag and ends at position bg. Therefore, the probability that the GRIN overlaps FLI g is simply the proportion
| (8) |
of the 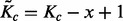 possible positions of the GRIN that overlap the FLI. Equation (8) shows that the GRIN statistical model has the intuitive property that the probability of overlap increases with the size of the GRIN and the size of the FLI.
possible positions of the GRIN that overlap the FLI. Equation (8) shows that the GRIN statistical model has the intuitive property that the probability of overlap increases with the size of the GRIN and the size of the FLI.
It is also straightforward to derive the probability that multiple independent GRINs overlap multiple FLIs on the same chromosome. The triangle diagram of Figure 1B geometrically represents the number of FLIs that overlap a GRIN as a function of the GRIN size x and GRIN start locus u. The shaded overlap regions are determined by computing 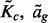 and
and 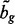 for each possible GRIN size x according to Figure 1A as described above. A horizontal line at the given GRIN size x defines the number of overlapping FLIs as a function of the GRIN start locus u (Fig. 1B). In turn, the overlap function for a given GRIN of size x defines the null probability distribution for the number of FLIs that overlap a GRIN of the given size by the proportion of possible GRIN start positions that overlap each possible number of FLIs (Fig. 1C). Finally, the null distribution for the total number of GRIN–FLI overlaps is determined by serial convolution of the distribution of the number of FLI that overlap each GRIN as illustrated by the probability tree of Figure 1D.
for each possible GRIN size x according to Figure 1A as described above. A horizontal line at the given GRIN size x defines the number of overlapping FLIs as a function of the GRIN start locus u (Fig. 1B). In turn, the overlap function for a given GRIN of size x defines the null probability distribution for the number of FLIs that overlap a GRIN of the given size by the proportion of possible GRIN start positions that overlap each possible number of FLIs (Fig. 1C). Finally, the null distribution for the total number of GRIN–FLI overlaps is determined by serial convolution of the distribution of the number of FLI that overlap each GRIN as illustrated by the probability tree of Figure 1D.
2.4 Null distribution of overlap statistics
We now derive the null probability distribution for each overlap statistic listed in Section 2.2 by representing each lesion as an independent GRIN of the same size on the same chromosome in the same subject.
The null probability 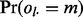 that each lesion l overlaps m FLIs is the probability that a GRIN of equal length on the same chromosome overlaps exactly m FLIs. This probability is calculated as shown in Figure 1B. The null probability
that each lesion l overlaps m FLIs is the probability that a GRIN of equal length on the same chromosome overlaps exactly m FLIs. This probability is calculated as shown in Figure 1B. The null probability 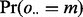 that the total number of overlaps
that the total number of overlaps 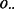 equals m is determined by serial convolution of
equals m is determined by serial convolution of 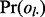 over all lesions
over all lesions 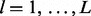 as shown in Figure 1D.
as shown in Figure 1D.
The null probability 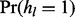 that lesion l overlaps at least one FLI is a Bernoulli distribution with success probability
that lesion l overlaps at least one FLI is a Bernoulli distribution with success probability 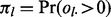 . The null probability
. The null probability 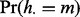 that there are m lesions with at least one overlap is determined by serial convolution of the Bernoulli(
that there are m lesions with at least one overlap is determined by serial convolution of the Bernoulli(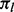 ) distributions over all
) distributions over all 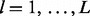 lesions.
lesions.
The null probability 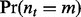 that subject t has m lesions that overlap at least one FLI is determined by serial convolution of the Bernoulli(
that subject t has m lesions that overlap at least one FLI is determined by serial convolution of the Bernoulli(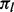 ) distributions over that subject’s lesions l, i.e. all l such that
) distributions over that subject’s lesions l, i.e. all l such that 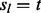 . The null probability
. The null probability 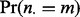 that there are m subjects with at least one overlap is determined by serial convolution of the Bernoulli distributions with success probability
that there are m subjects with at least one overlap is determined by serial convolution of the Bernoulli distributions with success probability 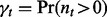 over all
over all 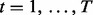 subjects.
subjects.
2.5 Questions addressed by GRIN analysis
For any particular FLI, the GRIN analysis method may be used to compute a P-value to quantify the significance of any of the overlap statistics of Section 2.2 according to the GRIN null model of Section 2.3. In particular, the GRIN model may be used to calculate a P-value for the total number 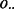 of lesion–FLI overlaps as defined in Equation (3), the total number
of lesion–FLI overlaps as defined in Equation (3), the total number 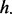 of lesions that overlap at least one FLI in Equation (4), the number nt of lesions in each subject t that overlap at least one FLI as defined in Equation (6) and the number
of lesions that overlap at least one FLI in Equation (4), the number nt of lesions in each subject t that overlap at least one FLI as defined in Equation (6) and the number 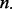 of subjects that have at least one overlap as defined in Equation (7).
of subjects that have at least one overlap as defined in Equation (7).
In practice, GRIN may be used to screen multiple sets of fixed loci by performing a separate GRIN analysis for each distinct set of loci. For example, a GRIN analysis may be performed with a particular KEGG pathway (www.kegg.jp) as the set 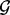 . This gene-set GRIN analysis will determine whether the lesions significantly target the particular KEGG pathway. A separate gene-set GRIN analysis can be performed for each KEGG pathway as the set
. This gene-set GRIN analysis will determine whether the lesions significantly target the particular KEGG pathway. A separate gene-set GRIN analysis can be performed for each KEGG pathway as the set 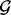 to evaluate the significance of lesion overlap with each KEGG pathway.
to evaluate the significance of lesion overlap with each KEGG pathway.
A gene-level GRIN analysis may be performed by performing a test with the locus of one individual gene as the set 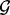 . Each overlap statistic and its corresponding P-value can be computed for this particular gene. A separate GRIN analysis can be performed using the locus of each individual gene as the set
. Each overlap statistic and its corresponding P-value can be computed for this particular gene. A separate GRIN analysis can be performed using the locus of each individual gene as the set 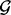 to screen every gene in the genome.
to screen every gene in the genome.
A marker-level GRIN analysis may also be used to screen the entire genome for ‘hot spot’ loci that have a significant abundance of lesions. Conceptually, one could perform a separate GRIN analysis with each microarray marker or point locus serving as the fixed set 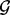 . This approach would compute overlap statistics and P-values for each point locus or microarray marker. Those loci with significant P-values would be identified as hot spots. However, screening the entire genome for hot spots by performing a separate GRIN analysis for each point locus or microarray marker in the genome is computationally prohibitive and involves a massive statistical multiplicity.
. This approach would compute overlap statistics and P-values for each point locus or microarray marker. Those loci with significant P-values would be identified as hot spots. However, screening the entire genome for hot spots by performing a separate GRIN analysis for each point locus or microarray marker in the genome is computationally prohibitive and involves a massive statistical multiplicity.
Therefore, we use a different strategy to use GRIN to screen the entire genome for hot spots that have a significant abundance of lesions. The chromosome cl and endpoints 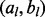 of each lesion define a set of boundaries that partition the genome into a set of
of each lesion define a set of boundaries that partition the genome into a set of 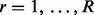 regions. Each region r is a point or interval locus that can be represented by
regions. Each region r is a point or interval locus that can be represented by 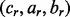 where cr is the chromosome of region
where cr is the chromosome of region 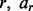 is the start locus of region r and br is the end locus of region r. A separate GRIN analysis can be applied with each region r as the set
is the start locus of region r and br is the end locus of region r. A separate GRIN analysis can be applied with each region r as the set 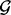 to compute overlap statistics and P-value for each region r. This strategy screens the entire genome for hot spots with one GRIN analysis per region instead of one GRIN analysis per marker. The number of regions R is on the order of the number of lesions L. The number of lesions L is typically several orders of magnitude smaller than the number of markers. Thus, the strategy to perform one analysis per region greatly reduces the computational burden and statistical multiplicity of the analysis.
to compute overlap statistics and P-value for each region r. This strategy screens the entire genome for hot spots with one GRIN analysis per region instead of one GRIN analysis per marker. The number of regions R is on the order of the number of lesions L. The number of lesions L is typically several orders of magnitude smaller than the number of markers. Thus, the strategy to perform one analysis per region greatly reduces the computational burden and statistical multiplicity of the analysis.
The test-per-region strategy to screen the entire genome for hot spots is also more conservative than the test-per-marker strategy. For each region r, the P-value from GRIN using the region r as the set 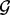 will be greater than or equal to the P-value from using any point locus within the region as the set
will be greater than or equal to the P-value from using any point locus within the region as the set 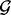 . Let a, b and y be point loci on a chromosome of size K such that y is between a and b, i.e.
. Let a, b and y be point loci on a chromosome of size K such that y is between a and b, i.e. 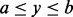 . Equation (8) clearly indicates that the probability that any GRIN overlaps the point locus y is less than or equal to the probability that the same GRIN overlaps the interval locus
. Equation (8) clearly indicates that the probability that any GRIN overlaps the point locus y is less than or equal to the probability that the same GRIN overlaps the interval locus 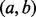 , which includes y. Thus, at every stage of the serial convolution used to compute the null distribution for an overlap statistic, the null probability of overlap will be greater for the interval locus
, which includes y. Thus, at every stage of the serial convolution used to compute the null distribution for an overlap statistic, the null probability of overlap will be greater for the interval locus 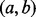 than for the point locus y. Therefore, for any of the overlap statistics, the P-value for overlap with the interval locus
than for the point locus y. Therefore, for any of the overlap statistics, the P-value for overlap with the interval locus 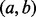 will be larger than the P-value for overlap with the point locus y within the interval locus
will be larger than the P-value for overlap with the point locus y within the interval locus 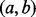 .
.
2.6 Comparison with other methods
The GRIN analysis method has several advantages over the analysis methods mentioned in the introduction. First, GRIN can address a broader spectrum of biological questions than do the other methods. Second, GRIN works with a broader diversity of genomic lesions than do the other methods. Third, the GRIN statistical model retains some biologically important properties of genomic lesions that the other methods ignore. Finally, by retaining those biological properties, the GRIN analysis method has some distinct statistical and computational advantages over the other methods. These advantages are summarized in Table 2 and described in detail below.
Table 2.
Capabilities of methods
| Capability | GRIN | GISTIC 2 | TAGCNA | SIAC |
|---|---|---|---|---|
| Marker-level tests |  |
 |
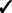 |
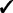 |
| Gene-level tests | 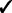 |
|||
| Gene-set tests | 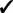 |
|||
| Analyzes gains |  |
 |
 |
 |
| Analyzes losses |  |
 |
 |
 |
| Analyzes LOH |  |
 |
 |
 |
| Analyses SB |  |
|||
| Analyzes PM |  |
|||
| No permutation |  |
|||
| Retains continuity |  |
|||
| Low memory algorithm |  |
Note: Checks indicate that the method has the indicated capability.
GRIN addresses a broader variety of biological questions than do the other methods. The other methods identify hot spot loci within the genome that have a significant abundance of lesions. GRIN can also identify hot spot loci as described in Section 2.5. Moreover, GRIN performs this analysis with much less computation because it does not resort to permutation. Section 2.5 also describes gene-level and gene-set level analyses that GRIN can perform. The other methods do not perform any analyses at the gene or gene-set level.
Additionally, GRIN works with a much broader variety of genomic lesions than do the other methods. The other methods limit consideration to copy number alterations or loss of heterozygosity (LOH). In contrast, every lesion with a well-defined interval or point locus on the reference genome coordinate system can be included in a GRIN analysis. Copy number alterations and LOH have an interval locus on the reference genome; non-silent substitutions and indels have a point locus on the reference genome; and each breakpoint of a structural rearrangement has a point locus on the reference genome. Thus, all these types of genomic lesions can be incorporated into a GRIN analysis. Furthermore, GRIN can consider each type of lesion separately just as some other methods consider amplifications and deletions separately.
The GRIN statistical model differs substantially from the POM model used by other analysis methods. Figure 2 illustrates the POM and GRIN models of chance for the genomic lesion data of one subject. The GRIN model of chance constrains lesions to retain their size, continuity and to stay located on the same chromosome (Fig. 2A). In contrast, POM assigns lesion status to markers by permutation (Fig. 2B). The GRIN model of chance is constrained to retain many of the observed properties of the lesions. Thus, the GRIN model generates data that more closely resemble these characteristics of the real data than does the POM model.
Fig. 2.

Illustration of GRIN and POM model realizations. The left column of panel A has an illustrative set of genomic lesions of one subject. The lesions are shaded in gray and chromosomes are separated by horizontal black lines. The subsequent 10 columns show 10 realizations of the GRIN model generated from those lesions. Panel B has the same set of genomic lesions and 10 realizations of the POM model generated from those lesions
The two models have different statistical properties. Under the POM model, every marker has an equal probability of being affected by a lesion; this probability is equal to the proportion of markers that are affected by a lesion in the observed data. In the example of Figure 2, 30% of the markers are affected by a lesion and thus the POM model assigns every marker a 30% null probability of being affected by a lesion by chance. In contrast, the GRIN model assigns a different probability of being affected by chance to each marker. In the example of Figure 2A, GRIN assigns a 100% probability of being affected by chance to markers on the chromosome that has a whole-chromosome event and a 0% probability to every marker on the chromosome with no lesions. These stark differences in null probabilities trickle through all calculations and may ultimately define grossly distinct null distributions.
The differences between the two statistical models becomes more profound when considering the number of lesions that affect entire genes or sets of biologically related genes that may be scattered at different loci across the genome. For instance, consider computing the null probability that a lesion overlaps any portion of a gene locus that includes x markers. For the example of Figure 2A, the probability of this event is approximately 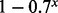 under the POM model. For x = 10, this null probability is 0.97. Such large null probabilities make it difficult for a gene-level analysis with a POM model to identify anything as statistically significant. In contrast, the null probability of overlap under the GRIN model is defined by Equation (8). Under the GRIN model, the null probability of overlap increases more gradually with the size of the gene and there is no multiplicity due to consideration of multiple markers within the gene.
under the POM model. For x = 10, this null probability is 0.97. Such large null probabilities make it difficult for a gene-level analysis with a POM model to identify anything as statistically significant. In contrast, the null probability of overlap under the GRIN model is defined by Equation (8). Under the GRIN model, the null probability of overlap increases more gradually with the size of the gene and there is no multiplicity due to consideration of multiple markers within the gene.
The GRIN model also requires less memory and computing time than do methods that rely on the POM model. Methods that use the POM model represent the genomic lesion data by a large matrix with one row per marker and one column per subject. In contrast, GRIN represents the genomic lesion data using the format shown in Table 1 and described in Section 2.1. This format requires only four items of information per lesion. The GRIN representation of the data clearly requires much less memory than the matrix representation. GRIN also requires less computing time than does POM. For each lesion and FLI, GRIN updates the overlap statistics and performs a simple convolution to update the null distribution of those overlap statistics. Thus, the total number of such operations is the product of the number of FLIs and the number of lesions. However, the POM methods update the overlap statistics for each subject and each marker within each permutation. Thus, the total number of update operations is the product of the number of markers, the number of subjects and the number of permutations. The permutation may be accurately approximated by a convolution in some settings. However, the number of markers will greatly exceed the number of lesions in most applications, and thus, GRIN will typically require much less computing time than POM methods.
3 RESULTS
3.1 Simulation study
We used simulation to evaluate GRIN, GISTIC 2.0, TAGCNA and SAIC as methods for marker-level analysis of genomic lesion data. We generated 100 independent datasets for each sample size n = 10, 50 or 100 as described below. Each method was applied to each of these 300 datasets.
For each subject, a set of random lesions and a set of targeted lesions were generated. For each subject, the number of random lesions was generated from a Poisson distribution with mean 5. Each random lesion was assigned to a chromosome with probability proportional to chromosome size. Given the assigned chromosome, the size of a random lesion was uniform. Given the assigned chromosome and lesion size, the position of the random lesion was uniform as shown in Figure 1A. Additionally, each subject could have a targeted lesion centering over RB1, TP53, CDKN2A and/or AML1. For each subject, each of these genes had a 50% probability of having a targeted lesion. The endpoints of each targeted lesion were defined by a pair of observations generated from a scaled beta distribution with mean equal to the midpoint of the targeted gene and a sum of shape parameters equal to 1000. To address multiple testing, we used the robust false discovery rate (FDR) estimation procedure (Pounds and Cheng, 2006) to compute q-values (Storey, 2002) for TAGCNA, SAIC and GRIN. For GISTIC 2.0, we used the q-values that it reports.
The average power, FDR and area under the curve (AUC) were computed for each method and sample size. The average power at the q-value threshold of 0.01 was the proportion of base pairs inside a target locus declared significant at that level averaged across simulation replications. The FDR was the average proportion of regions declared significant that did not overlap one of the four targets mentioned above. The FDR was set to zero in each simulation that a method had no significant findings. The AUC was computed by averaging the AUC of the proportion of target base pairs captured as a function of non-target base pairs captured across all replications. Good performance is indicated by keeping the FDR below 0.01 and greater power and AUC. We also recorded the average computing time for each method.
Table 3 gives the simulation results. GRIN is the only method to maintain the FDR level below 0.01 for all sample sizes. Under our simulation model, lesions affect contiguous intervals of the genome, which violate the POM model of chance (Fig. 2). Thus, there are regions that will have a number of overlapping random non-targeted lesions that is significant against the POM model of chance. The GISTIC 2.0, TAGCNA and SAIC methods all use some type of POM model and thus obtain many false-positive results.
Table 3.
Simulation results
| n | Metric | GRIN | GISTIC 2.0 | TAGCNA | SAIC |
|---|---|---|---|---|---|
| 10 | FDR | 0.0068 | 0.0200 | 0.0000 | 0.1600 |
| Power | 0.1604 | 0.0051 | 0.0000 | 0.0003 | |
| AUC | 0.7515 | 0.5314 | 0.5011 | 0.5091 | |
| Time | 0.04 | 3.9 | 8.4 | 567.2 | |
| 50 | FDR | 0.0029 | 0.0808 | 0.0320 | 0.4100 |
| Power | 0.4942 | 0.0175 | 0.0050 | 0.0000 | |
| AUC | 0.8801 | 0.5506 | 0.5919 | 0.4104 | |
| Time | 0.75 | 31.4 | 71.1 | 712.4 | |
| 100 | FDR | 0.0028 | 0.2248 | 0.1609 | 0.6880 |
| Power | 0.5853 | 0.1154 | 0.0824 | 0.0034 | |
| AUC | 0.8982 | 0.5658 | 0.6765 | 0.3835 | |
| Time | 3.7 | 152.6 | 240.5 | 747.7 |
Note: Average computing times are given in minutes.
GRIN also has the greatest statistical power in each of these simulations (Table 3). The POM model uses lesions from the entire genome to compute the probability that a lesion affects any marker by chance. Thus, large lesions increase the null probability of overlap for every marker under the POM model. Under the GRIN model, large lesions impact the null probability of overlap only for loci on the same chromosome. Thus, other regions of the genome can still have a small null probability of overlap and be assigned a significant P-value under the GRIN model.
3.2 ETP leukemia
Early T-cell precursor acute lymphoblastic leukemia (ETP-ALL) has recently been recognized as a disease entity with a poor prognosis (Coustan-Smith et al., 2009). Zhang et al. (2012) performed WGS of matched tumor and non-tumor DNA for 12 childhood ETP-ALL cases. DNA deletions, amplifications, structural rearrangements and sequence mutations were identified for each tumor by comparison of its sequence data to that of a paired control. Figure 3A shows the data.
Fig. 3.

Genomic lesion data and analysis results. Panel (A) shows the genomic data from the ETP-ALL study. Lesions are represented as shaded rectangles. The columns correspond to subjects, and rows correspond to genomic loci. The chromosome is indicated by the vertical gray and white bar to the left. The type of lesion is indicated by the color legend at the bottom. Panel (B) shows the 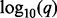 values as horizontal bars with scale indicated at the bottom. The
values as horizontal bars with scale indicated at the bottom. The 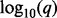 values for gains extend to the right and the
values for gains extend to the right and the 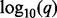 values for losses extend to the left. The methods are indicated by different colors as shown in the legend at the bottom. The vertical black and white bar in the middle indicates chromosome. Panels (C) and (D) show the analogous information for the hypodiploid leukemia dataset. The Manhattan-style plots are truncated at
values for losses extend to the left. The methods are indicated by different colors as shown in the legend at the bottom. The vertical black and white bar in the middle indicates chromosome. Panels (C) and (D) show the analogous information for the hypodiploid leukemia dataset. The Manhattan-style plots are truncated at 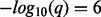
We applied GISTIC 2.0, SAIC and TAGCNA to the DNA copy number gains and losses of this dataset. We also performed separate marker-level GRIN analyses on the losses and gains using the total number of overlaps statistic defined by Equation (3). We accounted for multiple testing by using the robust FDR method developed for one-sided tests with discrete P-values (Pounds and Cheng, 2006) to compute q-values (Storey, 2002) for SAIC, TAGCNA and GRIN. We used the GISTIC 2.0 q-values for that method. The results are shown in Figure 3B and Supplementary Table S1 (the Table S1 tab of the file supplemental-tables.xlsx). For all analyses and methods, we deem results with 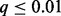 to be statistically significant. TAGCNA and SAIC failed to identify any locus as significant. GISTIC 2.0 identified a locus on chromosome 12 as having a significant number of losses (
to be statistically significant. TAGCNA and SAIC failed to identify any locus as significant. GISTIC 2.0 identified a locus on chromosome 12 as having a significant number of losses (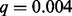 ). Zhang et al. (2012) describe the biological relevance of this loss to ETP-ALL. GRIN also determined that this locus has a significant number of losses (
). Zhang et al. (2012) describe the biological relevance of this loss to ETP-ALL. GRIN also determined that this locus has a significant number of losses (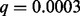 ). Moreover, GRIN identified 11 loci with a significant number of overlapping losses and 16 loci with a significant number of overlapping gains. In this example, GRIN clearly identified the greatest number of loci as significant. This is consistent with the simulation study showing that GRIN has greater statistical power than the other methods.
). Moreover, GRIN identified 11 loci with a significant number of overlapping losses and 16 loci with a significant number of overlapping gains. In this example, GRIN clearly identified the greatest number of loci as significant. This is consistent with the simulation study showing that GRIN has greater statistical power than the other methods.
We also performed marker, gene and gene-set GRIN analysis of all genomic lesions (Supplementary Table S2). The marker-level GRIN analysis identified 47 loci with a significant number of overlaps [defined by Equation (3)] and 37 loci with a significant number of subjects with at least one overlap [defined by Equation (7)]. The gene-level GRIN analysis computed overlap statistics and P-values for each of 29 176 genes (Supplementary Table S3). This analysis found that seven genes (AML1, SUZ12, ETV6, JAK3, TRG@, FBXW7 and TRDD2) have a significant number of overlaps and that three genes (AML1, JAK3 and SUZ12) have a significant number of subjects with at least one overlap. These genes are targeted by a variety of lesion types including structural rearrangements and sequence mutations as well as copy number alterations. Zhang et al. (2012) describe the biological relevance of these lesions.
Finally, we used GRIN to test the overlap of lesions with the gene loci for each of 192 KEGG pathways (Supplementary Table S4). This analysis found one pathway (dorsoventral axis formation) with a significant number of overlaps. GRIN also found three pathways (dorsoventral axis formation, melanoma and acute myeloid leukemia) with a significant number of lesions that overlap at least one FLI [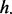 of Equation (5)]. GRIN found that the acute myeloid leukemia (AML) pathway has a significant number of subjects with at least one overlap [defined by Equation (7)]. This result and the observation that ETP-ALL has expression patterns similar to AML suggest that myeloid-directed therapies may be effective treatment for ETP-ALL (Zhang et al., 2012).
of Equation (5)]. GRIN found that the acute myeloid leukemia (AML) pathway has a significant number of subjects with at least one overlap [defined by Equation (7)]. This result and the observation that ETP-ALL has expression patterns similar to AML suggest that myeloid-directed therapies may be effective treatment for ETP-ALL (Zhang et al., 2012).
3.3 Hypodiploid acute lymphoblastic leukemia
Holmfeldt et al. (2013) performed a detailed study of hypodiploid acute lymphoblastic leukemia. Hypodiploid acute lymphoblastic leukemia is an extremely aneuploid tumor that exhibits somatic loss of at least 10 whole chromosomes. Occassionally, the tumor genome duplicates after the initial acquisition of aneuploidy. Holmfeldt et al. (2013) studied 140 cases using a variety of technologies; we use their WGS data for 20 subjects (Fig. 3C) as our example below.
We applied marker-level GRIN and the other methods to the copy number gains and losses of this dataset (Fig. 3D, Supplementary Table S5). Again, we considered results with 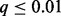 to be statistically significant. SAIC identified loss of chromosomes 2, 3, 4, 7, 12, 13 and 14 and gain of chromosome 21 as significant but did not identify any focal region as significant. TAGCNA identified the T-cell rearrangement and immunoglobin heavy (IGH) loci as regions with a significant number of losses. These loci are validated deletions that arise during normal lymphoid development rather than alterations that are specific to leukemic cells. GISTIC 2.0 found five loci with a significant number of gains and nine loci with a significant number of losses. Thirteen of these 14 loci are related to normal lymphoid development; the other locus overlaps the RB1 tumor suppressor gene. GRIN identified 55 loci with a significant number of losses and 11 loci with a significant number of gains. GRIN captured every focal locus identified as significant by GISTIC 2.0 or TAGCNA except for the number of gains in the TRA cluster (for which GRIN obtained
to be statistically significant. SAIC identified loss of chromosomes 2, 3, 4, 7, 12, 13 and 14 and gain of chromosome 21 as significant but did not identify any focal region as significant. TAGCNA identified the T-cell rearrangement and immunoglobin heavy (IGH) loci as regions with a significant number of losses. These loci are validated deletions that arise during normal lymphoid development rather than alterations that are specific to leukemic cells. GISTIC 2.0 found five loci with a significant number of gains and nine loci with a significant number of losses. Thirteen of these 14 loci are related to normal lymphoid development; the other locus overlaps the RB1 tumor suppressor gene. GRIN identified 55 loci with a significant number of losses and 11 loci with a significant number of gains. GRIN captured every focal locus identified as significant by GISTIC 2.0 or TAGCNA except for the number of gains in the TRA cluster (for which GRIN obtained 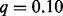 ). GRIN identified many genes of known relevance to leukemia or other cancers such as CDKN2A, CDKN2B, RB1 and CREBBP.
). GRIN identified many genes of known relevance to leukemia or other cancers such as CDKN2A, CDKN2B, RB1 and CREBBP.
We also applied GRIN to all lesions in the hypodiploid dataset. The marker level analysis determined that 62 loci have a significant total number of overlaps and that 147 loci have a significant number of subjects with at least one overlap (Supplementary Table S6). The gene-level analysis found that 1861 genes have a significant number of overlapping lesions and 2271 genes have a significant number of subjects with an overlapping lesion (Supplementary Table S7; note that many of these genes belong to gene clusters). The gene-set analysis determined that 75 gene-sets have a significant total number of overlaps and 52 gene-sets have a significant number of lesions that overlap at least one FLI (Supplementary Table S8). Many of the significant gene-sets define biological processes related to cancer (cell cycle, apoptosis, P53 signaling, etc), are involved in various forms of cancer (chronic myeloid leukemia, melanoma, prostate cancer, basal cell carcinoma, glioma, etc) or are involved in hematopoietic processes (T-cell receptor signaling, B-cell receptor signaling, hematopoietic cell lineage). No KEGG pathway had a significant number of subjects with at least one overlap according to the GRIN model because such a large portion of each subject’s genome is affected in this disease.
4 DISCUSSION
Genomic lesion data can provide useful insights regarding the development and prognosis of cancer. A thorough interpretation of genomic lesion data includes a statistical analysis that allows investigators to prioritize some findings by attributing other findings to random chance. The statistical challenge is to formally define and apply a biologically meaningful model of chance. The POM model has been used to develop analysis tools that have been useful in some studies. However, the POM does not have an explicit representation on the genome. Intuitively, it should be possible to further improve performance by developing a statistical model with an explicit genomic representation.
Therefore, we propose GRIN as a model and analysis method that explicitly represents lesions as contiguous entities with distinct loci on the reference genome. With this explicit genomic representation, the GRIN model achieves several statistical, computational and practical advantages over the widely used POM model. First, the multiplicity of the GRIN model is much less than that of the POM model. Each lesion is one random event under the GRIN model; however, there is one random event for each marker in each tumor under the POM model. In most applications, each tumor has orders of magnitude fewer lesions than markers. Thus, by reducing the multiplicity by orders of magnitude, GRIN simplifies the technical interpretation of the statistical analysis results. Additionally, GRIN defines simple null distributions for statistics that measure the abundance of lesions that overlap any fixed locus or set of loci in the genome. In this way, GRIN can simultaneously perform marker, gene and gene-set level analyses. In contrast, the POM model defines a simple null distribution only for the number of lesions that affect a point locus. The POM model conceptually defines a null distribution for the number of lesions that overlap a gene or gene-set, but this null distribution must be approximated computationally by simulation or permutation. Moreover, the statistical power of an analysis that uses the POM model to determine the significance of the number of lesions that overlap a gene or a gene-set would be extremely small due to the multiplicity of the POM model described in Section 2.6. Thus, GRIN provides a computationally efficient way to evaluate the statistical significance of patterns such as lesions affecting different loci within the same gene or gene-set.
There are a number of extensions and related problems that should be explored in future research. The model proposed here restricts the GRINs to have fixed length. The GRIN model can be generalized to allow GRINs to have random lengths. We are currently exploring ways to incorporate random length GRINs into our model. These models may further enrich our understanding of how to statistically interpret genomic lesion data. The interpretation of any statistical analysis depends on the underlying statistical model. These models will interrogate statistical significance against a more general concept of randomness.
It is also interesting to consider how to integrate other sources of genomic data to identify important loci in cancer. Some methods have been developed that perform an integrative analysis of genomic lesion data and expression data. Witten et al. (2009) and Witten and Tibshirani (2009) propose sparse canonical correlation analysis method to characterize the relationships between copy number and expression data. Fontanillo et al. (2012) also propose methods to perform an integrated analysis of expression and copy number data to identify important genomic alterations in cancer. It may be possible to use GRIN in conjunction with these methods in innovative ways to enhance our ability to expand our understanding of cancer biology. For example, GRIN may be used to identify specific pathways for a focused exploration of the association of genomic lesions with the expression of genes in those pathways.
Funding: NCI, NIGMS and NHLBI of the National Institutes of Health (CA21765, U01GM 92666, U19HL065962; in part), and the American Lebanese Syrian Associated Charities (ALSAC).
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Beroukhim R, et al. Assessing the significance of chromosomal aberration in cancer: methodology and application to glioma. Proc. Natl Acad. Sci. USA. 2007;104:20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coustan-Smith E, et al. Early T-cell precursor leukaemia: P a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10:147–156. doi: 10.1016/S1470-2045(08)70314-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanillo C, et al. Combined analysis of genome-wide expression and copy number profiles to identify key altered genomic regions in cancer. BMC Genomics. 2012;13(Suppl. 5):5. doi: 10.1186/1471-2164-13-S5-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmfeldt L, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat. Genet. 2013;45:242–252. doi: 10.1038/ng.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullighan CG, et al. Genes regulating B cell development are mutated in acute lymphoid leukaemia. Nature. 2007;446:758–764. [Google Scholar]
- Mullighan CG, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N. Eng. J. Med. 2009;360:470–480. doi: 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pounds S, Cheng C. Robust estimation of the false discovery rate. Bioinformatics. 2006;22:1979–1987. doi: 10.1093/bioinformatics/btl328. [DOI] [PubMed] [Google Scholar]
- Sanchez-Garcia F, et al. JISTIC: identification of significant targets in cancer. BMC Bioinformatics. 2010;11:189. doi: 10.1186/1471-2105-11-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD. A direct approach to false discovery rates. J. R. Stat. Soc. B. 2002;64:479–498. [Google Scholar]
- Wang J, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat. Methods. 2011;8:652–654. doi: 10.1038/nmeth.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten DM, et al. A penalized matrix decomposition, with applications to sparse principal components and canonical correlation analysis. Biostatistics. 2009;10:515–534. doi: 10.1093/biostatistics/kxp008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten DM, Tibshirani R. Extensions of sparse canonical correlation analysis, with applications to genomic data. Stat. Appl. Genet. Mol. Biol. 2009;8:Article 28. doi: 10.2202/1544-6115.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, et al. Genome-wide identification of significant aberrations in cancer genome. BMC Genomics. 2012a;13:342. doi: 10.1186/1471-2164-13-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, et al. TAGCNA: a method to identify significant consensus events of copy number alterations in cancer. PLoS One. 2012b;7:e41082. doi: 10.1371/journal.pone.0041082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, et al. Discovery of novel recurrent mutations in childhood early T-cell precursor lymphoblastic leukaemia by whole genome sequencing. Nature. 2012;481:157–63. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.