

Abstract
Although once widely anticipated to unlock how human type 1 diabetes (T1D) develops, extensive study of the nonobese diabetic (NOD) mouse has failed to yield effective treatments for patients with the disease. This has led many to question the usefulness of this animal model. While criticism about the differences between NOD and human T1D is legitimate, in many cases disease in both species results from perturbations modulated by the same genes or different genes that function within the same biological pathways. Like in humans, unusual polymorphisms within an MHC class II molecule contributes the most T1D risk in NOD mice. This insight supports the validity of this model and suggests the NOD has been improperly utilized to study how to cure or prevent disease in patients. Indeed, clinical trials are far from administering T1D therapeutics to humans at the same concentration ranges and pathological states that inhibit disease in NOD mice. Until these obstacles are overcome it is premature to label the NOD mouse a poor surrogate to test agents that cure or prevent T1D. An additional criticism of the NOD mouse is the past difficulty in identifying genes underlying T1D using conventional mapping studies. However, most of the few diabetogenic alleles identified to date appear relevant to the human disorder. This suggests that rather than abandoning genetic studies in NOD mice, future efforts should focus on improving the efficiency with which diabetes susceptibility genes are detected. The current review highlights why the NOD mouse remains a relevant and valuable tool to understand the genes and their interactions that promote autoimmune diabetes and therapeutics that inhibit this disease. It also describes a new range of technologies that will likely transform how the NOD mouse is used to uncover the genetic causes of T1D for years to come.
Keywords: type 1 diabetes, genetics, NOD mouse, HLA, MHC, mt-ND2, epistasis
Abbreviations: ADA – American Diabetes Association; ALR – alloxan-resistant; ATG – anti-thymocyte globulin; B2m – beta2-microglobulin; BC1 – backcross one; BCR – B cell receptor; BM – bone marrow; CPE - carboxypeptidase E; CTLA-4 – cytotoxic T lymphocyte antigen 4; DSB - double strand break; ESC – embryonic stem cell; FokI – Flavobacterium okeanokoites enzyme; GAD65 – glutamic acid decarboxylase 65; G-CSF – granulocyte colony-stimulating factor; GWAS – genome-wide association studies; HbA1c - glycated hemoglobin; HLA – human leukocyte antigen; HSC – hematopoetic stem cell; HSP60 – heat shock protein 60; IA-2 – islet cell antigen 512; IAPP - islet amyloid polypeptide; ICA69 – islet cell autoantigen 1; IDDM1 – insulin-dependent diabetes mellitus 1; IGRP – islet-specific glucose-6-phosphatase catalytic subunit-related protein; IL – interleukin; MHC – major histocompability complex; mt – mitochondrial ; mtDNA – mitochondrial deoxyribonucleic acid; mt-ND2 – mt encoded NADH dehydrogenase 2; NADH – nicotinamide adenine dinucleotide hydrogen; NGS – next generation sequencing; NHEJ – non-homologous end joining; NOD – nonobese diabetic; NOR – nonobese resistant; NSG – NOD scid gamma; nPOD – Network for Pancreatic Organ Donors with Diabetes; Pdx1 - pancreatic and duodenal homeobox 1; PTPRN - protein tyrosine phosphatase, receptor type, N; PTPN22 – protein tyrosine phosphatase, non-receptor type 22; ROS – reactive oxygen species; RNA – ribonucleic acid; RNAi – RNA interference; shRNA – short hairpin RNA; siRNA - small interfering RNA; SNP – single nucleotide polymorphism; SSRP – simple sequence repeat polymorphic; T1D – type 1 diabetes; TALEN – transcription activator-like effector nuclease; TCR – T cell receptor; VNTR – variable number of tandem repeat; WGS – whole genome sequencing; ZFN – zinc finger nuclease; ZnT8 – zinc transporter 8
1. Introduction
The fortuitous development of the NOD mouse [1] resulted in the generation of the most widely used animal model for the study of autoimmune diabetes. There are many similarities between autoimmune diabetes in the NOD mouse and in humans (Table 1). Due to the high level of use, this inbred mouse strain has been the subject of a multitude of reviews, books, and commentaries since its introduction over thirty years ago. These works have detailed the strengths of the NOD mouse model and have also pointed to significant weaknesses. Our purpose here is to provide some considerations for those working with the NOD to promote use of this model in a way that will benefit the understanding of how genetics impact human autoimmune disease.
Table 1. Characteristics of spontaneous type 1 diabetes diagnosed in man and the NOD mouse.
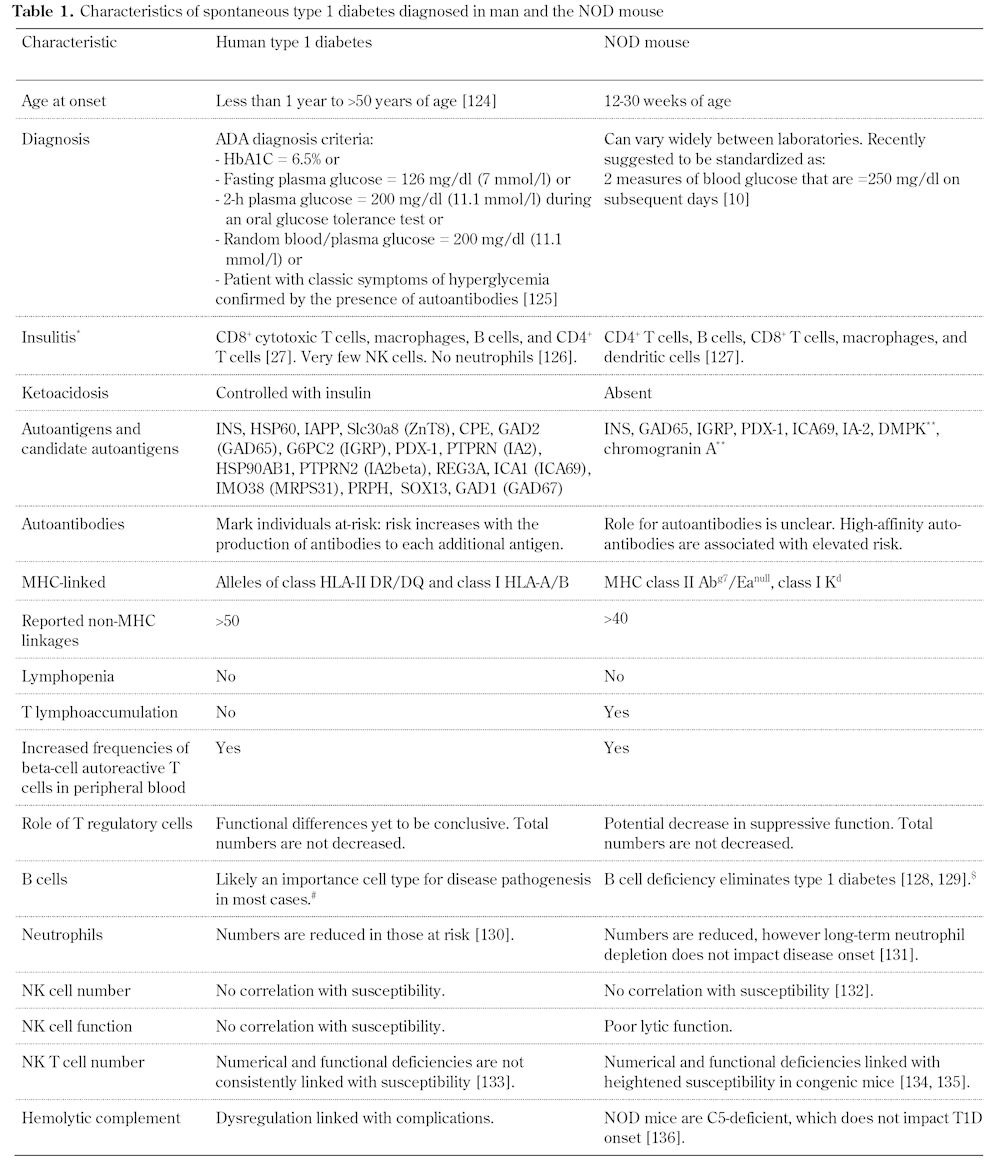
Legend: * Listing order by frequency of cell type. ** Not yet identified as autoantigens in humans. # A single individual with X-linked agammaglobulinemia (severe hereditary B cell deficiency) developed diabetes with T cell reactivity to GAD65 and IA-2 [137]. $ B cell-deficient NOD mice (NOD.129S2-Ighmtm1Cgn/Dvs) are strongly resistant to spontaneous autoimmune diabetes. These mice are susceptible to mild insulitis and, on treatment with cyclophosphamide, develop diabetes. References [124-136].
2. Use of NOD mice in preclinical trials
We have recently published an extensive review on the similarities of diabetes development in humans and NOD mice [2], including comparisons of the immunologic and genetic contributions (Table 1). However, emerging evidence from translational efforts forms a unique starting point for a comparison of human and NOD mouse autoimmunity. The NOD mouse was used early and has since been employed very often to test agents to prevent and/or reverse the disease. In general, there are three types of models, namely:
1. Early prevention where a therapy is initiated at 4-8 weeks of age, which would be before establishment of overt islet destruction.
2. Late prevention, 8-12 weeks, with the intent to arrest active pathogenesis.
3. Reversal of disease in mice with clinical symptoms that are treated with the intent of restoring glycemic control.
While many agents can prevent T1D when used early in NOD mice, the number of agents that are effective in late prevention declines, and only a handful have been demonstrated to reverse established disease.
In 1982, the first study demonstrating the therapeutic potential of a compound on T1D was published [3]. This study demonstrated that nicotinamide had the ability to both prevent T1D in prediabetic NOD mice and to reverse it in mice that exhibited hyperglycemia. However, the translation of this preclinical protocol to humans failed to have any preventative effect [4]. A pattern has followed with success in the NOD followed by failure in translating these "cures" to man in trials utilizing a number of agents; a partial list would include the agents daclizumab, anakinra, or teplizumab [5].
Several factors could have contributed to the lack of translation, including clear differences in dosing when moving from mouse to man, a lack of assessment of beta-cell secretory function in mouse, the absence of immunologic or metabolic biomarkers, and an extreme difference in the timeline for treatment initiation [5-7]. The timing of the treatment may be essential for disease remission. Preclinical trials using anti-CD3 monoclonal antibodies in the NOD mouse had a 10% success when the therapy was initiated in mice with a blood sugar greater than 350 mg/dl, compared with reversal in 23 of 35 (66%) mice with circulating glucose levels less than 350 mg/dl [8]. This has prompted investigators to screen NOD mice intensively for onset so that treatments could be initiated as close to that point as possible [8, 9], resulting in inflated efficacy percentages. In contrast, an inclusion criterion for clinical trials is less than 100 days post onset. Therefore, targeting patients immediately after diagnosis of diabetes may result in increased efficacy [6, 7]. Furthermore, a lack of consistency in methodologies has marked studies employing NOD mice for reversal studies. An exceptional commentary appearing in Science Translation Medicine has proposed a detailed plan for improving use of the NOD for preclinical studies [10].
While efforts to move from bench to bedside have met with difficulties, some success has resulted from the bedside-to-bench approach. The only pharmacologic therapy known to reverse T1D in humans and to provide for increased C-peptide production (alongside of gaining exogenous insulin independence) is a method that is commonly termed the "Brazilian protocol" [11-17]. This method utilized a combination therapy of anti-thymocyte globulin (ATG), granulocyte colony-stimulating factor (G-CSF), cytoxan, and autologous stem cell infusion; it has reported remarkably promising results. Yet, the mechanisms underlying the effectiveness of this treatment regimen remain unclear. Recent work has demonstrated ATG as effective in reversing NOD mice with recent onset disease [18]. Furthermore, enhanced efficacy in this reversal process can be obtained by adding G-CSF to this therapy [9]. The combination of ATG and G-CSF was able to reverse the majority of recent onset diabetic NOD mice with blood glucose levels ≤450 mg/dl (~65%; n = 73), yet mice with blood glucose levels >450 mg/dl also exhibited a significant reduction in reversal rate (36%, n = 39). While we conclude, similar to the summary of the anti-CD3 preclinical trials [8], that these blood glucose levels represent the level of beta-cell mass at onset, these data convey our inadequate knowledge of the natural history of beta-cell failure in the NOD mouse. They also highlight, that even under conditions of genetic homogeneity the onset of disease is dissimilar; not unlike that observed in monozygotic twins where the concordance is 65% [19]. This suggests, as discussed elsewhere in this edition, that environmental factors in combination with susceptibility alleles act to control timing and perhaps severity of T1D onset.
Further, efforts such as the Network for Pancreatic Organ Donors with Diabetes (nPOD) [20, 21] and those in Europe [22-27] are highlighting differences between the insulitic lesion comparing individuals with T1D and autoantibody-positive organ donors. This high level of heterogeneity in disease, even amongst twins or the NOD mouse, should refocus the attention on the genetic causes. It is the genetic similarities where the NOD mouse likely gains the greatest amount of traction for assisting in the understanding of human T1D.
3. Similarities in genetic causation
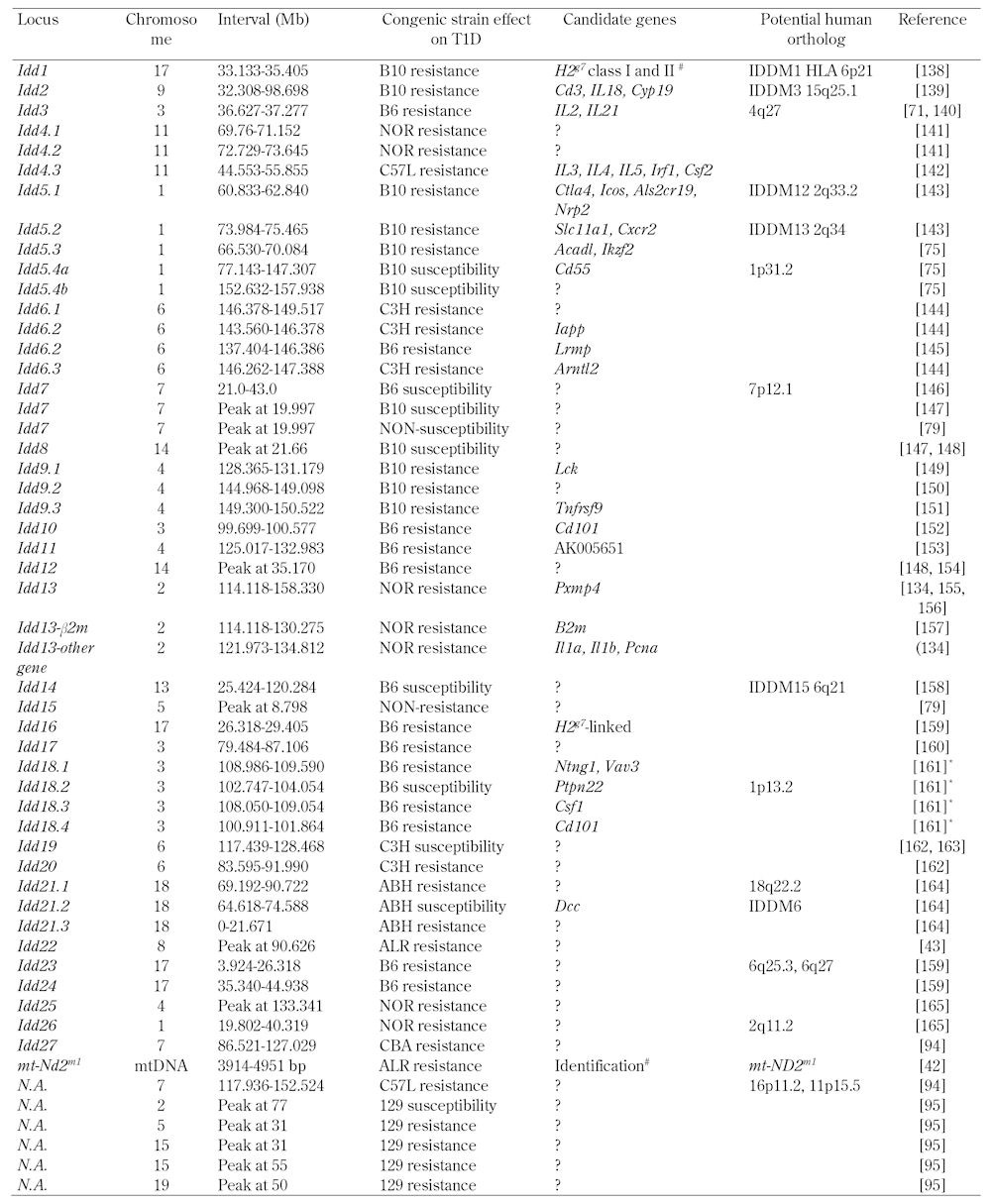
T1D is a polygenic disease, with over 50 genetic linkages identified in both human and mouse that are associated with this autoimmune disease (recently reviewed in [2]). The identification of loci contributing to T1D has been accomplished through arranged marriages of NOD with more than 10 inbred mouse strains (Table 2). Similar to the genome wide studies in human populations, some loci are unique to a specific outcross partner, while others, such as genes within the major histocompatibility complex (MHC) locus, appear to contribute in most of these studies. A major reason that the NOD mouse has been thought to serve well as a model for the human form of T1D are the similarities in genes/loci that impact disease, comparing those identified in human genome-wide scans or genome-wide association studies (GWAS) to the regions of the genome associated with causation in NOD. As there is not enough space here for a comprehensive review of all linkages, we will highlight some specific linkages as they provide useful examples. These examples will go from almost identical disease-associated allotypes, to gene systems that are similar in function, and genetic changes that impact similar pathways in human and mouse.
Table 2. Type 1 diabetes susceptibility loci mapped in the NOD mouse.
4. HLA and MHC: master genes for susceptibility
A preponderance of evidence implicated acquired immune cells, specifically both CD4+ and CD8+ T lymphocytes as final effectors of beta-cell death. Autoreactive responses of CD4+ and CD8+ T cells to over 15 different antigens have been measured in T1D patients and at risk individuals. In the NOD mouse, immune responses have been measured against 8 antigens [2]. The T cell receptor (TCR) of a T lymphocyte recognizes a particular peptide antigen in the context of a specific MHC molecule, also known as human leukocyte antigen (HLA). This TCR-peptide-MHC interaction is therefore essential for T1D pathogenesis [28]. Likewise, polymorphisms in MHC/HLA are very highly associated with disease susceptibility in both human and mouse.
The linkage to HLA class II, termed insulin-dependent diabetes mellitus 1 (IDDM1) [29], and the class II MHC allele in NOD [30] are by far the most significant susceptibility loci [31]. DQB alleles with Ser, Ala, or Val at amino acid residue 57 are associated with T1D susceptibility, while those alleles containing an Asp residue are considered protective. These risk HLA molecules are included within HLA haplotypes that are significantly enriched in the human form of the disease. Likewise, the only class II MHC allele expressed on the surface of cells from NOD mice, H2-Ag7, is a non-asp 57-containing allele [32]. The lack of a full complement of MHC class II molecules is an important difference when compared to the human population, including individuals with T1D, which in general express HLA allotypes from all class II loci.
The association of T1D with the MHC class II region was confirmed through the generation of NOD mice congenic for protective MHC haplotypes. Several congenic NOD strains have been generated by selective breeding using simple sequence repeat polymorphic (SSRP) or microsatellite markers to introgress the MHC region from diabetes resistant mouse strains into the NOD genetic background. In cases where the NOD allele at H2-A was replaced, the resulting mice were resistant to T1D. These congenic mice confirmed the importance of the contributions of this region of chromosome 17 to T1D pathogenesis. However, due to the presence of hundreds of genes in addition to the MHC class II loci, these congenic systems have not yet been able to directly implicate the MHC class II Ag7 allele. Use of transgenic NOD mice expressing a genetically engineered H2-Ag7 where serine at position 57 was replaced with an aspartic acid, have helped to address the importance of this single amino acid in NOD mice [33]. These NOD-Ag7Asp mice exhibited a significant reduction in T1D incidence, from ~55% to ~15% by 40 weeks of age. While the protection was present, the lack of complete penetrance of the protection provided by Asp-57 suggests that there is more to the contribution of MHC/HLA class II alleles than this amino acid residue. Furthermore, as these studies were performed using a "low-incidence" colony, it is not clear how protective this amino acid substitution would be in a colony with a T1D incidence of up to 90% in the control NOD mice [34]. This underscores the importance of the interplay between genes and environment for T1D onset [35].
The homology of the NOD allele to the predisposing alleles in humans is a major strength of the NOD model. It is believed that the non-asp-containing alleles cause a local rearrangement within the peptide-binding site that alters the peptide-binding specificity [36]. Yet, data demonstrating a clear mechanism for how Ag7 or the non-asp-containing HLA alleles cause T1D remains elusive. It should be noted that mechanistic studies using human cells are complicated by high genetic diversity at the HLA loci and by differences in allele frequencies [37]. The ability to limit these variables and to perform invasive studies indicates that the NOD is an excellent system to study the impact of class II MHC on T1D pathogenesis. As discussed below and elsewhere in this volume, the emergence of NOD embryonic stem cells and knock-in technologies such as zinc finger nucleases (ZFN) and transcription activator-like effector nucleases (TALEN) will provide ample opportunities for investigators to directly probe the MHC contributions to T1D.
4.1 mt-ND2 and mt-Nd2
A hypothesis-driven approach to a biologic problem can create self-fulfilling prophecies as experiments can be designed or results interpreted towards supporting rather than testing a postulate. T1D is an autoimmune disease studied by immunologists. When loci are mapped immunologists likely point to genes influencing immune function as candidates rather than taking a non-biased approach. Certainly, there is excellent support for genes involved in immunological processes in T1D as many of the genetic determinants are expressed in cells from the hematopoietic compartment, including immune cells [38]. On the contrary, genes may also influence the susceptibility of the beta-cell to immune-mediated destruction. For this to occur, it may take a unique outcross partner. To better understand the role of beta-cell-expressed T1D genes, NOD mice were outcrossed to the alloxan-resistant (ALR) mouse, which was derived to have beta-cells that are extremely resistant to destruction [39-49].
By outcrossing ALR to NOD mice, following these progeny for onset of clinical and pathological features of T1D, and then performing a genome-wide screen, three loci in the nuclear genome and an association with the mitochondrial genome (mtDNA) were identified. Sequencing of the entire mtDNA from several strains identified a single nucleotide polymorphism (SNP) as the only difference between NOD and ALR [42]. This C to A SNP results in a leucine to methionine amino acid substitution. The affected gene, mt-Nd2, is a subunit of complex I of the mitochondrial electron transport chain. To determine the role of the ALR-derived mt-Nd2a allele, NOD mice with the ALR mtDNA were generated. Using adoptive transfer models, it was determined that, while the gene is expressed systemically, the protective allele, mt-Nd2a, has an impact on T1D onset only at the beta-cell level. Study of the mitochondria of these NOD.mtALR conplastic mice (i.e. mice where the cytoplasmic genomes have been exchanged) demonstrated a significant difference in basal and stimulated mitochondrial reactive oxygen species (ROS) production from complex I of the mitochondrial electron transport chain [50-53]. As mitochondrial ROS production has been tied to the induction of apoptosis, the reduction in mitochondrial ROS production was directly linked to protection against beta-cell apoptosis [39, 50, 51, 54]. After exposure to autoreactive CD8+ T cells, Fas-agonistic antibody, or proinflammatory cytokines, NOD.mtALR beta-cells failed to increase mitochondrial ROS, exhibited an absence of caspase 8 activation, and remained viable [39, 55]. The importance of ROS in this system was confirmed by adding hydrogen peroxide (H2O2) in combination with cytokines or FAS-agonistic antibody. Addition of H2O2 with the pro-apoptotic agents results in caspase 8 activation in NOD.mtALR beta-cells and decreased viability. This effect was caspase-dependent as inhibition of caspase 8 after incubating NOD.mtALR beta-cells with H2O2 and FAS-agonistic antibody blocked cell death.
Similar to these findings in the NOD mouse, a C to A transversion at mtDNA nucleotide position 5178 within the nicotinamide adenine dinucleotide hydrogen (NADH) dehydrogenase subunit 2 gene (mt-ND2), resulting in a leucine to methionine amino acid substitution at amino acid residue 237, had been reported as being protective against T1D onset in humans [56]. In a population-based study of 385 Japanese patients (154 male and 231 female) with T1D and 469 age- and sex-matched controls, the methionine-containing allele was associated with protection from T1D (p = 0.017) and a significantly reduced frequency (p = 0.0046) of the T1D-associated auto-antibodies (GAD, IA-2, and IAA). To study the impact of mt-ND2a on human beta-cell destruction, cytoplasmic hybrid (or cybrid) human beta-cells were generated by fusing mitochondrial DNA-depleted human beta-cells with platelets [57, 58], because they are anuclear. Platelet donors were chosen based on their mtDNA haplotype [59-61]. Individuals from haplogroup D served as donors for the mt-ND2a-containing human beta-cell lines, while platelets from donors with haplogroup M9 were fused to produce the mt-ND2c-containing human beta-cells. For these cells, M9 was chosen as it is the most genetically related haplogroup to D [62]. These novel human beta-cell lines were then tested for resistance in assays similar to those described above for the mouse beta-cells. When the protective mt-ND2a is present, human beta-cells exhibit reduced mitochondrial ROS production and resistance to apoptosis induced by autoimmune mediated stress, including autoreactive CD8+ cytotoxic T lymphocytes [55]. This is in accord with the findings using NOD.mtALR beta-cells. These studies highlight the importance of crosstalk between research involving samples from the NOD mouse with those from human subjects, and emphasize the important fact that genes influencing T1D may modulate the disease by impacting beta-cell function and/or viability.
4.2 Homology in gene systems and pathways in genetic pathogenesis
MHC class II is perhaps an extreme example where an orthologous amino acid change contributes to T1D susceptibility in both humans and mice. While the majority of loci linked to T1D in NOD mice remain undiscovered, of those identified to date not all have been associated with T1D in humans (Table 2). For example, beta2-microglobulin (B2m) has been confirmed as an Idd gene in the mouse [63], but has not been confirmed as a T1D gene in humans. Similarly, genes have been identified in humans as causative, but the syntenic region in mouse has not been associated with disease. An example is the insulin gene, INS, in the IDDM2 interval. It is widely believed that a variable number of tandem repeats (VNTR) in the INS promoter plays a significant role in human T1D [64]. A low number of repeats (30-44; class I) is associated with risk of T1D compared to alleles with more than 110 repeats (class III). An intermediate length VNTR (class II) is rare. The VNTR regulates insulin expression in a tissue-specific fashion. A class I VNTR results in high pancreatic islet insulin expression and low expression in the thymus, while class III VNTRs are associated with the opposite (low pancreatic expression and a two-fold increase in thymic expression). The elevated expression in the thymus associated with the class III VNTR has been associated with elevated immune regulation to insulin [65].
In the mouse, there are two genes that encode insulin, these are insulin I (Ins1; chromosome 19) and insulin II (Ins2; chromosome 7). At the time of writing, neither insulin gene had been linked to T1D, as the biological phenomenon associated with expression of these two isoforms exists in all mouse strains tested. In the mouse, both Ins1 and Ins2 are expressed in pancreatic islets. However, only Ins2 is expressed in the thymus [66]. Genetic ablation of Ins2 in the NOD mouse results in an accelerated onset of diabetes with an observed failure of tolerance to insulin [67]. These Ins2-deficient NOD mice could be employed for the development of therapies that produce tolerance, which could be translated for induction of tolerance in individuals with a class I VNTR.
In other cases, while the gene linked to T1D may differ between these species, an identical pathway is affected. CD25, a subunit of the high-affinity trimeric IL-2 receptor, has been linked in humans, while the gene encoding IL-2 itself, Il2, is likely the gene contributing disease susceptibility within the Idd3 region. In both human and mouse, investigators portend that the disease-associated alleles create a reduction in IL-2 signaling [68, 69]. Deficiency of IL-2 or IL-2 signaling in human or mouse results in lymphoproliferative disease [68, 70], highlighting the importance of this signaling pathway in immune homeostasis. In the mouse, Il2NOD is associated with a two-fold reduction in IL-2 production, a level that was confirmed as contributory for disease using IL-2 haploinsufficient NOD-mice [71]. The human risk allele of CD25 is associated with reduced expression. In both cases, the lack of IL-2 signaling has been proposed to contribute to reduced Treg activity and a reduction in activation-induced cell death of effector T cells [68]. In either case, the exact genetic mechanism for reduced expression remains to be elucidated.
The identification of loci and genes that impact T1D in the NOD mouse has allowed for disease modulation using congenic and transgenic approaches. Certainly, the NOD MHC haplotype (H2g7) is necessary, but not alone sufficient, to bring on T1D when congenically introgressed into a different genetic background. In fact, a combination of genes necessary for disease induction on a different background has not yet been identified. This strongly suggests that there are essential, but unidentified, genetic interactions present that greatly modulate susceptibility and resistance.
5. Gene-gene interactions and epistatic effects
T1D genetic research is largely focused on uncovering individual genes that impact the disease. However, it is the combined effect of multiple T1D susceptibility and resistance alleles, which often interact with each other, that ultimately determine an individual's risk [72]. The difficult task of unraveling this complexity has been carried forward mainly in animal models, including NOD strains congenically expressing different combinations of Idd loci from T1D resistant strains (Table 2 and [73]). These animals have proven a useful tool for understanding how, when expressed on an autoimmune-permissive background, different disease resistance loci modulate T1D or various subphenotypes of the disease. Congenic mice have also uncovered numerous gene-gene interactions and gene-masking effects between Idd loci that impact T1D [69]. These effects are normally hidden in a segregating population such as backcross one (BC1) or F2 mouse generations or humans, and are therefore very difficult to detect using conventional genetic association studies [69].
One of the most striking examples of how genetic interactions affect T1D development is the synergism between the partially disease-protective Idd3 and Idd5 congenic intervals. Combining these two loci on the NOD background results in near complete elimination of diabetes and insulitis [74]. However, combining Idd3 with various Idd5 subregions (Idd5.1, 5.2, 5.3, and 5.4) results in a range of disease-protective effects (reviewed in [69]). These include Idd3/Idd5.1 mice that were not more protected than Idd3 mice and Idd3/Idd5.1/Idd5.3 mice that were equally resistant to T1D compared to the Idd3/Idd5 strain, but develop more insulitis.
Other studies have detected strong gene-gene interactions within the Idd5 locus. One is between Idd5.1 that is suggested to encode a protective allele of Ctla4 and Idd5.4 that encodes a B10-derived susceptibility allele without known causative gene product. Hunter et al. found that the Idd5.4 allele has a dramatic effect on T1D susceptibility in the presence of Idd5.2 and Idd5.3, but no effect when protective alleles at Idd5.1 are also present [75]. This suggests that Idd5.4 can mask the effects of protective alleles at Idd5.2 and Idd5.3, but that the Idd5.4 susceptibility allele in turn is masked by the protective effect of Idd5.1/Ctla4. One interpretation of these findings is that the B10-derived Idd5.4 susceptibility allele may modulate an immune event that is counter-regulated by Ctla4.
Congenic mice are also useful for cell transfer experiments to determine how epistatic interactions affect diabetogenic immune responses within specific cell types. This strategy was used to reveal complex genetic interactions within Idd9 and Idd11 on distal chromosome 4 that control how B cells contribute to disease [76, 77]. For these studies, lethally irradiated B cell-deficient and T1D-resistant NOD.IgHnull mice were transplanted with syngeneic bone marrow (BM) and B cells from various chromosome 4 subcongenic donors. T1D incidence was subsequently monitored to establish whether B cells expressing different subcongenic intervals from the nonobese resistant (NOR) strain protected recipient mice from disease compared with standard NOD B cells. These studies identified four adjacent regions (designated R1, R2, R3, and R4) that interactively control the efficiency with which autoreactive B cells become tolerized or induce disease [77]. Several of these interactions appear to alter immunological responses involved in various tolerance induction mechanisms. For instance, NOR-derived alleles at R1 and R4 combine to increase the efficiency of B cell anergy, with counterbalancing negative regulation by R2. The NOR-derived congenic regions also contain genes that increase the diabetogenic capacity of B cells. These are encoded within R2 and R4 that interact to promote B cell hyperresponsiveness to B cell receptor (BCR) stimulation. However, the pathogenic effect of this interaction must normally be masked by the large number of other diabetes resistance alleles within the chromosome 4 interval.
Similar complex interactions appear to contribute to many other T1D subclinical phenotypes. Fox et al. described how the T cell-dependent progression of insulitis from a benign to a destructive state requires NOD-derived alleles at both the Idd5 and Idd13 loci [78]. Indeed, (NOD x NOR)F2 intercross mice that inherit at least one NOD-derived allele at Idd5 and Idd13 display invasive insulitis. In contrast, animals homozygous for NOR-derived C57BLKS/J alleles at either Idd5 or Idd13 develop mild peri-insulitis. Syntenic intervals that map to regions of human chromosomes 2q [79-82] and 15q [83, 84] may interact to control the same processes, which could explain why both are considered central regulators of disease pathogenesis.
There is also strong evidence that genetic loci that independently influence the risk for T1D interact in important ways to modulate disease progression in humans. One recent study tested interactions between 38 T1D-associated non-HLA regions with various HLA class II genotypes in a large collection of T1D samples [85]. HLA*non-HLA gene interactions were selected for testing because HLA class II genes have the largest effects on T1D, and therefore have a higher probability of showing interactions with a non-HLA locus. The results identified SNPs within two T1D-associated genes, PTPN22 and CTLA4, that alter the predicted disease risk of various HLA haplotypes. The interaction between PTPN22 and HLA class II genotypes confirms earlier work showing that the effect of a susceptibility allele at PTPN22 is greater in low-risk compared to high-risk HLA class II genotypes [86-88].
In another study, Winckler et al. determined how different combinations of non-HLA susceptibility genes stratify islet autoimmunity and/or T1D risk [89]. Children of T1D patients were genotyped for 12 T1D-associated genes (ERBB3, PTPN2, IFIH1, PTPN22, KIAA0350, CD25, CTLA4, SH2B3, IL2, IL18RAP, IL10, and COBL) and followed prospectively from birth for the development of islet autoantibodies and disease development. This strategy predicted with surprising accuracy the risk for developing islet autoantibodies and T1D, and the progression from islet autoimmunity to T1D especially in children carrying high-risk HLA genes. The authors anticipated that some gene combinations would be more useful than others. Therefore, the prevalence of individual gene SNPs in different combinations was examined to determine which subpopulations were more predictive of disease. SNPs of four genes (CTLA4, PTPN22, IL18RAP, and IFIH1) were present in almost all combinations, while the IL-2 SNP was infrequently detected. It was also determined that a collection of 8 genes (IFIH1, CTLA4, PTPN22, IL18RAP, SH2B3, KIAA0350, COBL, and ERBB3) predicted with greatest accuracy the chance of high-risk HLA carriers developing disease. The worse predictive power of all 12 SNPs may reflect a higher frequency of gene-gene interactions that mask the influence of individual T1D susceptibility alleles.
These and other GWAS studies make clear how some genes are strongly modulated by masking effects and gene-gene interactions, while others are not. Interpreting how statistically significant effects and interactions for T1D genes translate into biological actions, presents a challenging problem that experimentation with human samples alone are unlikely to solve. Therefore, future strategies to identify how crosstalk between genes underlies the development of T1D should include the continued use of congenic mice, with a particular focus on genes already known to impact both the mouse and human disease. These efforts should be greatly enhanced by the availability of new genetic tools (discussed below) that will allow the modification of two or more genes within congenic intervals, including the replacement of mouse genes with human variants.
6. Improving the efficiency of candidate gene identification
Enthusiasm for the use of NOD mice to identify genes that contribute to T1D has been tempered by the many difficulties this approach entails. The first limitation is the large number of mice needed to detect Idd loci when hyperglycemia or insulitis levels are used as an indicator of disease [73]. This is because very few F2 or first backcross (BC1) progeny generated from outcrossing NOD mice with T1D-resistant strains develop spontaneous autoimmune diabetes even when the diabetogenic H2g7 MHC is fixed in all segregants [90]. A second limitation has been that even when Idd loci are detected and delineated, the T1D resistance or susceptibility genes they contain are difficult to validate with available technologies. This is particularly true for Idd loci containing multiple tightly linked genes not easily separated through genetic recombination. For these reasons relatively few mouse Idd genes have been identified to date [73]. As a consequence, there has been a significant decline over the last decade in the number of large-scale mouse studies to identify genes contributing to T1D.
Nonetheless, there remain compelling reasons for more investment in mouse genetics. They include the fact that of the small number of mouse Idd genes identified most also contribute to human T1D or function within the same biological pathways (see section entitled "Similarities in genetic causation" above). There is also good evidence that, just like in individual patients, differing genetic subset combinations drawn from an overall larger pool of possible susceptibility variants can contribute to disease pathogenesis in a threshold fashion in mice [72]. These subsets of genes likely control T1D susceptibility by eliciting perturbations at differing nodes within a common set of immunoregulatory pathways. Evidence for this possibility includes the previous observation that nominally resistant mouse strains can carry gene variants, which when expressed in the proper combinatorial context, contribute more strongly to T1D development than the corresponding allele from disease susceptible NOD mice (Table 2 and reviewed in [73]). Although genetic studies using inbred mice are costly because of the large number of mice required, they remain a powerful method of detecting rare T1D susceptibility alleles that are impractical to identify through GWAS analyses, which require tens or hundreds of thousand of human subjects [31, 72]. Thus, for the reasons outlined above the question is not whether pursuing the identity of T1D susceptibility and resistance alleles is worthwhile, but rather how to make this process more efficient. Considerable encouragement comes from a new generation of genetic tools that may circumvent many of the most intractable obstacles that traditionally limited the identification of Idd candidate genes. Some of these are described in the following sections.
6.1 NOD embryonic stem cells
Until recently, no robust embryonic stem cell (ESC) lines capable of germ-line transmission could be generated from NOD mice due to difficulties in controlling the differentiation of these cells in culture [91-94]. Without this tool it is necessary to validate T1D gene candidates using fully germ-line competent ESC lines derived from other mouse strains. This practice introduces into the genetic background of the NOD mouse variable numbers of linked genes, some of which impact T1D [95]. In 2009, new culture methods were described that enable the derivation of NOD ESC capable of germ-line transmission [96]. Non-genetically manipulated mice generated from these cells were susceptible to T1D onset. The cells were grown based on a previous discovery that inclusion of the small molecule inhibitors PD0325901 and Chir99021 to a defined serum-free culture medium could facilitate ESC isolation from a wide number of strains [97]. This development should allow for much more efficient validation of genes within Idd loci because these genes permit the replacement of candidate alleles to be carried out individually and selectively. Indeed, these cells have been used to begin testing the importance of candidate genes within the MHC locus [98]. The same culture techniques were used to generate germ-line-competent ESC from immune-deficient NOD scid gamma (NSG) mice [99]. This resource should enable the future modification of the NSG strain to better accept engraftment of human tissues and cells.
Additionally, an ESC line from T1D susceptible NOD.CBALs-Tyr+/LtJ (Agouti NOD) mice has already been used to generate a genetically modified NOD stock at The Jackson Laboratory (Dr. Lenny Shultz, personal communication). This strain's background carries a wild type Tyr (tyrosinase) allele from CBA mice, which allows for expression of the Agouti allele [94]. This dominant coat color marker provides an alternative for tracking germ-line transmissions by laboratories that routinely use albino blastocysts. After delivery of the DNA construct into the Agouti NOD-ES cells, these cells can be injected into a blastocyst derived from an albino mouse, and chimeric progeny bred with NOD.
6.2 Zinc finger nucleases (ZFN)
While germ-line-competent NOD ESC lines amenable to genetic modification have become available, recent development of other genetic tools will further facilitate gene-mapping studies in NOD mice. Among them, the ZFN technology has emerged as a powerful means to specifically target genes in a variety of cells and organisms [100]. The benefits of using the ZFN mutagenesis technology over the ESC approach include its applicability to various genetic backgrounds, its high targeting efficiency, and a short period of time required to generate a knockout animal [100].
ZFNs are fusion proteins containing a sequence-specific DNA-binding zinc finger domain and a nuclease domain [100, 101]. Engineered ZFNs specifically recognize and bind a defined target gene sequence within the nucleus of a cell and introduce a double strand break (DSB) [102, 103]. The cellular DNA repair machinery then fixes these breaks, most frequently via the non-homologous end joining (NHEJ) mechanism, resulting in small deletions of the gene sequence (few to hundreds of base pairs) and disruption (knockout) of the target gene [102, 103]. Injected as synthetic mRNAs, ZFNs typically work at the one-cell embryo stage, resulting in single-step, whole animal gene disruption, and infrequent mosaics [104]. More precise genetic engineering can be achieved as well because a DSB also stimulates repair via a homology-directed repair (HDR) mechanism if a homologous DNA template is cointroduced into the cell [105]. It is expected that the ZFN technology will provide an unprecedented approach to identify causal variants within previously known Idd regions or to study the function of a particular gene in the NOD model.
6.3 Transcription activator-like effector nucleases (TALEN)
The recently developed transcription activator-like effector nucleases (TALEN) technology allows for the direct targeting of any gene in zygotes of the inbred mouse strain of choice [106-109]. This rapidly evolving genetic modification method is more efficient than conventional gene targeting technologies. It employs DNA-binding TALEN repeat modules designed to have precise specificity for particular nucleotide sequences that are coupled with a modified Flavobacterium okeanokoites enzyme, FokI, nuclease domain. When combined, this creates site-specific double-stranded breaks with remarkable efficiency (~15% in mouse zygotes). TALEN technology has been employed to genetically alter mouse strains previously recalcitrant to the development of stable ESC, including NOD (personal communication with Dr. Dave Serreze). TALENs, like ZFN, represent another new and versatile technology for future manipulation of gene expression in NOD mice. It is reasonable to expect that, because of their efficiency and ease of use, engineered nucleases will significantly replace ESC as a method to edit the NOD genome.
6.4 RNA interference
While NOD ESC, ZFN, and TALEN technology will likely transform future efforts to test T1D candidate genes, RNA interference (RNAi) has already proven useful for manipulating gene expression in NOD mice without introducing genetic contamination from other strains. This approach is based on a well-established transgenesis methodology that entails the direct introduction of short hairpin RNA (shRNA) containing constructs into NOD zygotes by viral transduction [96, 110, 111]. The shRNA-containing constructs are designed to silence genes that impact T1D. shRNA is a sequence of RNA that contains a tight hairpin turn. This structure is cleaved by intracellular machinery into small interfering RNA (siRNA) that knocks down any mRNA bearing a complementary sequence [112]. Several companies are developing viral libraries that produce shRNA that integrate into the host genome and ensure stable gene silencing after integration. The silencing cassette can be incorporated into many different types of vectors, including lentiviral, adenoviral, or retroviral vectors. RNAi has already provided valuable insight into how expression of the T1D candidate genes IL-17 [113], PTPN22 [114], soluble CTLA4 [115], and Slc11a1 (Nramp1) [116] contribute to disease.
Although the effectiveness of RNAi is well established, there are some important limitations of this technology. One is the so-called "off targeting" effect that lowers the specificity of RNAi and produces false positive results [112]. This occurs when genes with incomplete complementarity with the transgenically expressed shRNA are inadvertently downregulated, which may result in data misinterpretation and even toxicity [112]. Another drawback is that many of the shRNA molecules capable of triggering RNAi do not shut down gene activity completely. Because of these problems RNAi experiments require stringent controls to minimize the risk of data misinterpretation, including transducing NOD zygotes with scrambled shRNA designed not to target any transcripts. It is also recommended that results obtained using shRNA-expressing NOD mice be confirmed by additional non-RNAi experiments.
6.5 Retrogenic mice
Retrogenics refers to a method of transgenically expressing genes in hematopoetic stem cell (HSC)-derived cell types by transducing bone marrow using retroviral vectors before adoptive transfer into lethally irradiated recipient mice [117]. This technology has helped to uncover the contribution of diabetogenic TCRs of different specificities to insulitis development in the NOD mouse [118]. This discovery entailed the transduction of bone marrow cells with retroviral vectors, containing TCRα and TCRβ chains of interest, linked with a 2A viral peptide cleavage sequence that allows for the stoichiometric translation of both TCR peptides within a single vector [119]. The advantages of retrogenic technology are manifold, including the low cost and short time interval required to generate retrogenic mice compared with transgenic animals [117]. This approach also avoids the possibility of founder effects that often complicate results obtained using transgenic mice, as each retrogenic animal is a founder [117]. Because of the flexibility of this technique, it is likely that retrogenics will be adapted by T1D researchers for a variety of additional applications to speed the identification of the genetic causes of T1D, including overexpressing in HSC-derived cell types, human TCRs, other Idd gene candidates, or the shRNA that silence them.
6.6 Next generation sequencing
Applications for next generation sequencing (NGS) hold enormous potential for accelerating the discovery of T1D causative genes. One of this technology's most important contributions has been for whole genome sequencing (WGS) of the NOD mouse and some of the different strains this stock has been outcrossed to for detection of Idd loci. These sequence data have provided a means of accurately identifying whether T1D candidate genes contain SNPs that affect protein expression levels, structure, and/or function. DNA sequences for NOD mice and 16 additional strains are publically available through the Sanger Institute’s database. However, some mouse strains, including NOR, B10, and ALR, used to generate many of the stocks congenic for T1D resistance loci on the NOD background have yet to be sequenced. When the sequences of these stains are available, their DNA code should provide much more accurate predictions of which alleles within Idd loci contribute to disease than current SNP-based methods.
Although the efficiency of WGS continues to rapidly improve, this technology is likely to remain too expensive and time-consuming to replace conventional linkage studies. At the time of writing, the cost for sequencing the genome of a single mouse (or mouse strain) including the bioinformatics time for assembly is less than $10,000 (by several institutes, including Centrillion Biosciences Inc., Palo Alto, CA, USA). In future, a more cost-effective alternative will be to sequence whole mouse exomes using NGS. Exome sequencing captures allelic variants within the ~1% of the genome where most disease variations are located [120, 121] at a current cost of ~$1300 at 50X coverage for a 50Mb array. While this approach is efficient for identifying protein-coding sequences that underlie Mendelian disorders and de novo mutations, it is also useful for detecting polymorphisms that alter risk for, rather than cause, complex genetic disease including T1D [121]. The principal disadvantage of exome sequencing is that much of the 99% of the genome that remains unsequenced contains genetic material that contributes in many critical ways to gene regulation and no doubt T1D development. Whole-exome sequencing also requires additional steps of exome enrichment compared to WGS [120, 121]. This significantly adds to the cost of each sample.
For this reason, it is often favorable to sequence the transcriptome instead. This process, also known as RNA sequencing (RNA-seq) refers to the NGS of cDNA transcribed from all RNAs in a cell or tissue [122]. RNA-seq provides a method of detecting transcribed genes and other non-coding RNA without the need for any additional enrichment steps [122]. Transcripts may also be mapped against reference genomic DNA to obtain additional information, including transcription localization and the relative frequency of different splice variants [122]. A significant advantage of transcriptome sequencing is that this technology directly detects all cDNA sequence, and therefore has very low if any background and no upper limit for quantification. In contrast, because of intrinsic experimental limitations, microarray analyses cannot detect all transcription products and lacks sensitivity for genes expressed at high and low levels [122]. While RNA-seq represents a sizable advance over microarray analysis for detection of how polymorphisms within Idd candidates affect gene expression, widespread use of this technology has been limited by cost, bioinformatics challenges, and a prerequisite for sequencing information to detect and evaluate transcripts. However, it is likely that most of these obstacles will be overcome in time.
7. Conclusions
The genetic pathogenesis of T1D is complex. While sub-phenotypes of T1D (i.e. autoantibodies) may appear in humans and mice not at genetic risk, full pathogenesis requires complex interactions of susceptibility alleles along with a network of synergizing factors, including the environment. Shared between NOD mice and humans is the paramount role of the MHC class II alleles (Tables 1 and 2). Yet, the MHC alone is not sufficient. There are substantial contributions of non-MHC genes in both species. NOD mice also have lost tolerance to an array of self-proteins that is similar to the responses observed in humans, including insulin, glutamic acid decarboxylase 65 (GAD65), IA2 (ICA512 or protein tyrosine phosphatase, receptor type, N), and ICA69 (islet cell autoantigen 1) and IGRP (glucose-6-phosphatase, catalytic subunit, 2 (G6pc2)) protein (Table 1). Furthermore, a role for environmental and dietary factors is linked. Early gluten exposure has been associated with T1D onset in humans and is essential for T1D to initiate in the NOD mouse [123].
As has been pointed out, there are key differences between disease development in NOD and that in humans. Most notable, NOD mice exhibit an extremely aggressive form of T1D. Insulitis is present at weaning and the insulitic infiltrates are heavy. In comparison, islet inflammation appears to be a rare event in man [20-27]. However, based on genetic homogeneity throughout the NOD colonies held worldwide, the mouse strain represents a single individual with diabetes or a family with a given genetic susceptibility. Without this aggressive disease, NOD mice would be significantly more difficult to study and the interpretation of results from genetic studies would present unique challenges. Understanding the differences and focusing on similarities, specifically those that modulate genetic pathogenesis, should allow for those in this field to continue to make progress with this model to better understand the human condition.
Disclosure: The authors report no conflict of interests.
Acknowledgments
Support from the Juvenile Diabetes Foundation International, the American Diabetes Association, and National Institutes of Health is gratefully acknowledged.
References
- 1.Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a non-obese, diabetic strain of mice. Exp Anim. 1980;29:1–8. doi: 10.1538/expanim1978.29.1_1. [DOI] [PubMed] [Google Scholar]
- 2.Thayer TC, Wilson SB, Mathews CE. Use of nonobese diabetic mice to understand human type 1 diabetes. Endocrinol Metab Clin North Am. 2010;39(3):541–561. doi: 10.1016/j.ecl.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamada K, Nonaka K, Hanafusa T, Miyazaki A, Toyoshima H, Tarui S. Preventive and therapeutic effects of large-dose nicotinamide injections on diabetes associated with insulitis. An observation in nonobese diabetic (NOD) mice. Diabetes. 1982;31(9):749–753. doi: 10.2337/diab.31.9.749. [DOI] [PubMed] [Google Scholar]
- 4.Gale EA, Bingley PJ, Emmett CL, Collier T. European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet. 2004;363(9413):925–931. doi: 10.1016/S0140-6736(04)15786-3. [DOI] [PubMed] [Google Scholar]
- 5.Staeva TP, Chatenoud L, Insel R, Atkinson MA. Recent lessons learned from prevention and recent-onset type 1 diabetes immunotherapy trials. Diabetes. 2013;62(1):9–17. doi: 10.2337/db12-0562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sherry N, Hagopian W, Ludvigsson J, Jain SM, Wahlen J, Ferry RJ Jr, Bode B, Aronoff S, Holland C, Carlin D. et al. Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial. Lancet. 2011;378(9790):487–497. doi: 10.1016/S0140-6736(11)60931-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bach JF. Anti-CD3 antibodies for type 1 diabetes: beyond expectations. Lancet. 2011;378(9790):459–460. doi: 10.1016/S0140-6736(11)60980-X. [DOI] [PubMed] [Google Scholar]
- 8.Sherry NA, Chen W, Kushner JA, Glandt M, Tang Q, Tsai S, Santamaria P, Bluestone JA, Brillantes AM, Herold KC. Exendin-4 improves reversal of diabetes in NOD mice treated with anti-CD3 monoclonal antibody by enhancing recovery of beta-cells. Endocrinology. 2007;148(11):5136–5144. doi: 10.1210/en.2007-0358. [DOI] [PubMed] [Google Scholar]
- 9.Parker MJ, Xue S, Alexander JJ, Wasserfall CH, Campbell-Thompson ML, Battaglia M, Gregori S, Mathews CE, Song S, Troutt M, Eisenbeis S, Williams J, Schatz DA, Haller MJ, Atkinson MA. Immune depletion with cellular mobilization imparts immunoregulation and reverses autoimmune diabetes in nonobese diabetic mice. Diabetes. 2009;58(10):2277–2284. doi: 10.2337/db09-0557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Atkinson MA. Evaluating preclinical efficacy. Sci Transl Med. 2011;3(96):96cm22. doi: 10.1126/scitranslmed.3002757. [DOI] [PubMed] [Google Scholar]
- 11.Couri CE, Foss MC, Voltarelli JC. Secondary prevention of type 1 diabetes mellitus: stopping immune destruction and promoting beta-cell regeneration. Braz J Med Biol Res. 2006;39(10):1271–1280. doi: 10.1590/s0100-879x2006001000002. [DOI] [PubMed] [Google Scholar]
- 12.Couri CE, Oliveira MC, Stracieri AB, Moraes DA, Pieroni F, Barros GM, Madeira MI, Malmegrim KC, Foss-Freitas MC, Simoes BP, Martinez EZ, Foss MC, Burt RK, Voltarelli JC. C-peptide levels and insulin independence following autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus. JAMA. 2009;301(15):1573–1579. doi: 10.1001/jama.2009.470. [DOI] [PubMed] [Google Scholar]
- 13.Couri CE, Voltarelli JC. Autologous stem cell transplantation for early type 1 diabetes mellitus. Autoimmunity. 2008;41(8):666–672. doi: 10.1080/08916930802200208. [DOI] [PubMed] [Google Scholar]
- 14.de Oliveira GL, Malmegrim KC, Ferreira AF, Tognon R, Kashima S, Couri CE, Covas DT, Voltarelli JC, de Castro FA. Up-regulation of fas and fasL pro-apoptotic genes expression in type 1 diabetes patients after autologous haematopoietic stem cell transplantation. Clin Exp Immunol. 2012;168(3):291–302. doi: 10.1111/j.1365-2249.2012.04583.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leal AM, Oliveira MC, Couri CE, Moraes DA, Stracieri AB, Pieroni F, Soares RD, Barros GN, Simoes BP, Foss MC, Voltarelli JC. Testicular function in patients with type 1 diabetes treated with high-dose CY and autologous hematopoietic SCT. Bone Marrow Transplant. 2012;47(3):467–468. doi: 10.1038/bmt.2011.113. [DOI] [PubMed] [Google Scholar]
- 16.Voltarelli JC, Couri CE, Stracieri AB, Oliveira MC, Moraes DA, Pieroni F, Barros GM, Madeira MI, Malmegrim KC, Foss-Freitas MC, Simoes BP, Foss MC, Squiers E, Burt RK. Autologous hematopoietic stem cell transplantation for type 1 diabetes. Ann N Y Acad Sci. 2008;1150:220–229. doi: 10.1196/annals.1447.048. [DOI] [PubMed] [Google Scholar]
- 17.Voltarelli JC, Couri CE, Stracieri AB, Oliveira MC, Moraes DA, Pieroni F, Coutinho M, Malmegrim KC, Foss-Freitas MC, Simoes BP, Foss MC, Squiers E, Burt RK. Autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus. JAMA. 2007;297(14):1568–1576. doi: 10.1001/jama.297.14.1568. [DOI] [PubMed] [Google Scholar]
- 18.Simon G, Parker M, Ramiya V, Wasserfall C, Huang Y, Bresson D, Schwartz RF, Campbell-Thompson M, Tenace L, Brusko T. et al. Murine antithymocyte globulin therapy alters disease progression in NOD mice by a time-dependent induction of immunoregulation. Diabetes. 2008;57(2):405–414. doi: 10.2337/db06-1384. [DOI] [PubMed] [Google Scholar]
- 19.Redondo MJ, Jeffrey J, Fain PR, Eisenbarth GS, Orban T. Concordance for islet autoimmunity among monozygotic twins. N Engl J Med. 2008;359(26):2849–2850. doi: 10.1056/NEJMc0805398. [DOI] [PubMed] [Google Scholar]
- 20.Campbell-Thompson M, Wasserfall C, Kaddis J, Albanese-O'Neill A, Staeva T, Nierras C, Moraski J, Rowe P, Gianani R, Eisenbarth G, Crawford J, Schatz D, Pugliese A, Atkinson M. Network for Pancreatic Organ Donors with Diabetes (nPOD): developing a tissue biobank for type 1 diabetes. Diabetes Metab Res Rev. 2012;28(7):608–617. doi: 10.1002/dmrr.2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Atkinson MA, Gianani R. The pancreas in human type 1 diabetes: providing new answers to age-old questions. Curr Opin Endocrinol Diabetes Obes. 2009;16(4):279–285. doi: 10.1097/MED.0b013e32832e06ba. [DOI] [PubMed] [Google Scholar]
- 22.In't Veld P. Insulitis in human type 1 diabetes: The quest for an elusive lesion. Islets. 2011;3(4):131–138. doi: 10.4161/isl.3.4.15728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.In't Veld P. Insulitis in type 1 diabetes: a sticky problem. Diabetes. 2009;58(6):1257–1258. doi: 10.2337/db09-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.In't Veld P, Lievens D, De Grijse J, Ling Z, Van der Auwera B, Pipeleers-Marichal M, Gorus F, Pipeleers D. Screening for insulitis in adult autoantibody-positive organ donors. Diabetes. 2007;56(9):2400–2404. doi: 10.2337/db07-0416. [DOI] [PubMed] [Google Scholar]
- 25.Foulis AK, Stewart JA. The pancreas in recent-onset type 1 (insulin-dependent) diabetes mellitus: insulin content of islets, insulitis and associated changes in the exocrine acinar tissue. Diabetologia. 1984;26(6):456–461. doi: 10.1007/BF00262221. [DOI] [PubMed] [Google Scholar]
- 26.Foulis AK, Liddle CN, Farquharson MA, Richmond JA, Weir RS. The histopathology of the pancreas in type 1 (insulin-dependent) diabetes mellitus: a 25-year review of deaths in patients under 20 years of age in the United Kingdom. Diabetologia. 1986;29(5):267–274. doi: 10.1007/BF00452061. [DOI] [PubMed] [Google Scholar]
- 27.Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol. 2009;155(2):173–181. doi: 10.1111/j.1365-2249.2008.03860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sosinowski T, Eisenbarth GS. Type 1 diabetes: primary antigen/peptide/register/trimolecular complex. Immunol Res. 2013;55(1-3):270–276. doi: 10.1007/s12026-012-8367-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Concannon P, Gogolin-Ewens KJ, Hinds DA, Wapelhorst B, Morrison VA, Stirling B, Mitra M, Farmer J, Williams SR, Cox NJ, Bell GI, Risch N, Spielman RS. A second-generation screen of the human genome for susceptibility to insulin-dependent diabetes mellitus. Nat Genet. 1998;19(3):292–296. doi: 10.1038/985. [DOI] [PubMed] [Google Scholar]
- 30.Hattori M, Buse JB, Jackson RA, Glimcher L, Dorf ME, Minami M, Makino S, Moriwaki K, Kuzuya H, Imura H. et al. The NOD mouse: recessive diabetogenic gene in the major histocompatibility complex. Science. 1986;231(4739):733–735. doi: 10.1126/science.3003909. [DOI] [PubMed] [Google Scholar]
- 31.Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C. et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41(6):703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singer SM, Tisch R, Yang XD, Sytwu HK, Liblau R, McDevitt HO. Prevention of diabetes in NOD mice by a mutated I-Ab transgene. Diabetes. 1998;47(10):1570–1577. doi: 10.2337/diabetes.47.10.1570. [DOI] [PubMed] [Google Scholar]
- 33.Quartey-Papafio R, Lund T, Chandler P, Picard J, Ozegbe P, Day S, Hutchings PR, O'Reilly L, Kioussis D, Simpson E. et al. Aspartate at position 57 of nonobese diabetic I-Ag7 beta-chain diminishes the spontaneous incidence of insulin-dependent diabetes mellitus. J Immunol. 1995;154(10):5567–5575. [PubMed] [Google Scholar]
- 34.Hanson MS, Cetkovic-Cvrlje M, Ramiya VK, Atkinson MA, Maclaren NK, Singh B, Elliott JF, Serreze DV, Leiter EH. Quantitative thresholds of MHC class II I-E expressed on hemopoietically derived antigen-presenting cells in transgenic NOD/Lt mice determine level of diabetes resistance and indicate mechanism of protection. J Immunol. 1996;157(3):1279–1287. [PubMed] [Google Scholar]
- 35.Wasserfall C, Nead K, Mathews C, Atkinson MA. The threshold hypothesis: solving the equation of nurture vs nature in type 1 diabetes. Diabetologia. 2011;54(9):2232–2236. doi: 10.1007/s00125-011-2244-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sato AK, Sturniolo T, Sinigaglia F, Stern LJ. Substitution of aspartic acid at beta57 with alanine alters MHC class II peptide binding activity but not protein stability: HLA-DQ (alpha1*0201, beta1*0302) and (alpha1*0201, beta1*0303) Hum Immunol. 1999;60(12):1227–1236. doi: 10.1016/s0198-8859(99)00120-2. [DOI] [PubMed] [Google Scholar]
- 37.Noble JA, Erlich HA. Genetics of type 1 diabetes. Cold Spring Harb Perspect Med. 2012;2(1):a007732. doi: 10.1101/cshperspect.a007732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wicker LS, Miller BJ, Chai A, Terada M, Mullen Y. Expression of genetically determined diabetes and insulitis in the nonobese diabetic (NOD) mouse at the level of bone marrow-derived cells. Transfer of diabetes and insulitis to nondiabetic (NOD X B10) F1 mice with bone marrow cells from NOD mice. J Exp Med. 1988;167(6):1801–1810. doi: 10.1084/jem.167.6.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen J, Gusdon AM, Piganelli J, Leiter EH, Mathews CE. mt-Nd2(a) Modifies resistance against autoimmune type 1 diabetes in NOD mice at the level of the pancreatic beta-cell. Diabetes. 2011;60(1):355–359. doi: 10.2337/db10-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pomerleau DP, Bagley RJ, Serreze DV, Mathews CE, Leiter EH. Major histocompatibility complex-linked diabetes susceptibility in NOD/Lt mice: subcongenic analysis localizes a component of Idd16 at the H2-D end of the diabetogenic H2(g7) complex. Diabetes. 2005;54(5):1603–1606. doi: 10.2337/diabetes.54.5.1603. [DOI] [PubMed] [Google Scholar]
- 41.Mathews CE, Suarez-Pinzon WL, Baust JJ, Strynadka K, Leiter EH, Rabinovitch A. Mechanisms underlying resistance of pancreatic islets from ALR/Lt mice to cytokine-induced destruction. J Immunol. 2005;175(2):1248–1256. doi: 10.4049/jimmunol.175.2.1248. [DOI] [PubMed] [Google Scholar]
- 42.Mathews CE, Leiter EH, Spirina O, Bykhovskaya Y, Gusdon AM, Ringquist S, Fischel-Ghodsian N. mt-Nd2 Allele of the ALR/Lt mouse confers resistance against both chemically induced and autoimmune diabetes. Diabetologia. 2005;48(2):261–267. doi: 10.1007/s00125-004-1644-8. [DOI] [PubMed] [Google Scholar]
- 43.Mathews CE, Graser RT, Bagley RJ, Caldwell JW, Li R, Churchill GA, Serreze DV, Leiter EH. Genetic analysis of resistance to Type-1 Diabetes in ALR/Lt mice, a NOD-related strain with defenses against autoimmune-mediated diabetogenic stress. Immunogenetics. 2003;55(7):491–496. doi: 10.1007/s00251-003-0603-8. [DOI] [PubMed] [Google Scholar]
- 44.Mathews CE, Dunn BD, Hannigan MO, Huang CK, Leiter EH. Genetic control of neutrophil superoxide production in diabetes-resistant ALR/Lt mice. Free Radic Biol Med. 2002;32(8):744–751. doi: 10.1016/s0891-5849(02)00747-5. [DOI] [PubMed] [Google Scholar]
- 45.Mathews CE, Graser RT, Savinov A, Serreze DV, Leiter EH. Unusual resistance of ALR/Lt mouse beta cells to autoimmune destruction: role for beta cell-expressed resistance determinants. Proc Natl Acad Sci U S A. 2001;98(1):235–240. doi: 10.1073/pnas.98.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mathews CE, Graser RT, Serreze DV, Leiter EH. Reevaluation of the major histocompatibility complex genes of the NOD-progenitor CTS/Shi strain. Diabetes. 2000;49:131–134. doi: 10.2337/diabetes.49.1.131. [DOI] [PubMed] [Google Scholar]
- 47.Mathews CE, Leiter EH. Constitutive differences in antioxidant defense status distinguish alloxan-resistant and alloxan-susceptible mice. Free Radic Biol Med. 1999;27(3-4):449–455. doi: 10.1016/s0891-5849(99)00084-2. [DOI] [PubMed] [Google Scholar]
- 48.Mathews CE, Leiter EH. Resistance of ALR/Lt islets to free radical-mediated diabetogenic stress is inherited as a dominant trait. Diabetes. 1999;48(11):2189–2196. doi: 10.2337/diabetes.48.11.2189. [DOI] [PubMed] [Google Scholar]
- 49.Graser RT, Mathews CE, Leiter EH, Serreze DV. MHC characterization of ALR and ALS mice: respective similarities to the NOD and NON strains. Immunogenetics. 1999;49(7-8):722–726. doi: 10.1007/s002510050673. [DOI] [PubMed] [Google Scholar]
- 50.Chen J, Gusdon AM, Mathews CE. Role of genetics in resistance to type 1 diabetes. Diabetes Metab Res Rev. 2011;27(8):849–853. doi: 10.1002/dmrr.1260. [DOI] [PubMed] [Google Scholar]
- 51.Chen J, Gusdon AM, Thayer TC, Mathews CE. Role of increased ROS dissipation in prevention of T1D. Ann N Y Acad Sci. 2008;1150:157–166. doi: 10.1196/annals.1447.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gusdon AM, Votyakova TV, Mathews CE. mt-Nd2a suppresses reactive oxygen species production by mitochondrial complexes I and III. J Biol Chem. 2008;283(16):10690–10697. doi: 10.1074/jbc.M708801200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gusdon AM, Votyakova TV, Reynolds IJ, Mathews CE. Nuclear and mitochondrial interaction involving mt-Nd2 leads to increased mitochondrial reactive oxygen species production. J Biol Chem. 2007;282(8):5171–5179. doi: 10.1074/jbc.M609367200. [DOI] [PubMed] [Google Scholar]
- 54.Chen J, Lu Y, Lee CH, Li R, Leiter EH, Mathews CE. Commonalities of genetic resistance to spontaneous autoimmune and free radical - mediated diabetes. Free Radic Biol Med. 2008;45(9):1263–1270. doi: 10.1016/j.freeradbiomed.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lightfoot YL, Chen J, Mathews CE. mt-ND2 protects human beta-cells from immune-mediated destruction. Diabetes. 2012;62:A67. [Google Scholar]
- 56.Uchigata Y, Okada T, Gong JS, Yamada Y, Iwamoto Y, Tanaka M. A mitochondrial genotype associated with the development of autoimmune-related type 1 diabetes. Diabetes Care. 2002;25(11):2106. doi: 10.2337/diacare.25.11.2106. [DOI] [PubMed] [Google Scholar]
- 57.Lightfoot YL, Chen J, Mathews CE. Role of the mitochondria in immune-mediated apoptotic death of the human pancreatic beta cell line betaLox5. PLoS One. 2011;6(6):e20617. doi: 10.1371/journal.pone.0020617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lightfoot YL, Chen J, Mathews CE. Immune-mediated beta-cell death in type 1 diabetes: lessons from human beta-cell lines. Eur J Clin Invest. 2012;42(11):1244–1251. doi: 10.1111/j.1365-2362.2012.02711.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen J, Hattori Y, Nakajima K, Eizawa T, Ehara T, Koyama M, Hirai T, Fukuda Y, Kinoshita M, Sugiyama A, Hayashi J, Onaya T, Kobayashi T, Tawata M. Mitochondrial complex I activity is significantly decreased in a patient with maternally inherited type 2 diabetes mellitus and hypertrophic cardiomyopathy associated with mitochondrial DNA C3310T mutation: a cybrid study. Diabetes Res Clin Pract. 2006;74(2):148–153. doi: 10.1016/j.diabres.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 60.Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- 61.Lightfoot YL, Chen J, Mathews CE. Role of the mitochondria in immune-mediated apoptotic death of the human pancreatic cell line beta Lox5. PLoS One. 2011;6(6):e20617. doi: 10.1371/journal.pone.0020617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tanaka M, Cabrera VM, Gonzalez AM, Larruga JM, Takeyasu T, Fuku N, Guo LJ, Hirose R, Fujita Y, Kurata M. et al. Mitochondrial genome variation in eastern Asia and the peopling of Japan. Genome Res. 2004;14(10A):1832–1850. doi: 10.1101/gr.2286304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hamilton-Williams EE, Serreze DV, Charlton B, Johnson EA, Marron MP, Mullbacher A, Slattery RM. Transgenic rescue implicates beta2-microglobulin as a diabetes susceptibility gene in nonobese diabetic (NOD) mice. Proc Natl Acad Sci U S A. 2001;98(20):11533–11538. doi: 10.1073/pnas.191383798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vafiadis P, Ounissi-Benkalha H, Palumbo M, Grabs R, Rousseau M, Goodyer CG, Polychronakos C. Class III alleles of the variable number of tandem repeat insulin polymorphism associated with silencing of thymic insulin predispose to type 1 diabetes. J Clin Endocrinol Metab. 2001;86(8):3705–3710. doi: 10.1210/jcem.86.8.7733. [DOI] [PubMed] [Google Scholar]
- 65.Durinovic-Bello I, Wu RP, Gersuk VH, Sanda S, Shilling HG, Nepom GT. Insulin gene VNTR genotype associates with frequency and phenotype of the autoimmune response to proinsulin. Genes Immun. 2010;11(2):188–193. doi: 10.1038/gene.2009.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mathews CE, Pietropaolo SL, Pietropaolo M. Reduced thymic expression of islet antigen contributes to loss of self-tolerance. Ann N Y Acad Sci. 2003;1005:412–417. doi: 10.1196/annals.1288.070. [DOI] [PubMed] [Google Scholar]
- 67.Babaya N, Nakayama M, Moriyama H, Gianani R, Still T, Miao D, Yu L, Hutton JC, Eisenbarth GS. A new model of insulin-deficient diabetes: male NOD mice with a single copy of Ins1 and no Ins2. Diabetologia. 2006;49(6):1222–1228. doi: 10.1007/s00125-006-0241-4. [DOI] [PubMed] [Google Scholar]
- 68.Hulme MA, Wasserfall CH, Atkinson MA, Brusko TM. Central role for interleukin-2 in type 1 diabetes. Diabetes. 2012;61(1):14–22. doi: 10.2337/db11-1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ridgway WM, Peterson LB, Todd JA, Rainbow DB, Healy B, Burren OS, Wicker LS. Gene-gene interactions in the NOD mouse model of type 1 diabetes. Adv Immunol. 2008;100:151–175. doi: 10.1016/S0065-2776(08)00806-7. [DOI] [PubMed] [Google Scholar]
- 70.Caudy AA, Reddy ST, Chatila T, Atkinson JP, Verbsky JW. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J Allergy Clin Immunol. 2007;119(2):482–487. doi: 10.1016/j.jaci.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 71.Yamanouchi J, Rainbow D, Serra P, Howlett S, Hunter K, Garner VE, Gonzalez-Munoz A, Clark J, Veijola R, Cubbon R. et al. Interleukin-2 gene variation impairs regulatory T cell function and causes autoimmunity. Nat Genet. 2007;39(3):329–337. doi: 10.1038/ng1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morahan G, Mehta M, James I, Chen WM, Akolkar B, Erlich HA, Hilner JE, Julier C, Nerup J, Nierras C, Pociot F, Todd JA, Rich SS, Type 1 Diabetes Genetics Consortium. Tests for genetic interactions in type 1 diabetes: linkage and stratification analyses of 4,422 affected sib-pairs. Diabetes. 2011;60(3):1030–1040. doi: 10.2337/db10-1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Driver JP, Serreze DV, Chen YG. Mouse models for the study of autoimmune type-1 diabetes: a NOD to similarities and differences to human disease. Sem Immunopathol. 2011;33(1):67–87. doi: 10.1007/s00281-010-0204-1. [DOI] [PubMed] [Google Scholar]
- 74.Hill NJ, Lyons PA, Armitage N, Todd JA, Wicker LS, Peterson LB. The NOD Idd5 locus controls insulitis and diabetes and overlaps the orthologous CTLA4/IDDM12 and NRAMP1 loci in humans. Diabetes. 2000;49:1744–1747. doi: 10.2337/diabetes.49.10.1744. [DOI] [PubMed] [Google Scholar]
- 75.Hunter K, Rainbow D, Plagnol V, Todd JA, Peterson LB, Wicker LS. Interactions between Idd5.1/Ctla4 and other type 1 diabetes genes. J Immunol. 2007;179(12):8341–8349. doi: 10.4049/jimmunol.179.12.8341. [DOI] [PubMed] [Google Scholar]
- 76.Silveira PA, Chapman HD, Stolp J, Johnson E, Cox SL, Hunter K, Wicker LS, Serreze DV. Genes within the Idd5 and Idd9/11 diabetes susceptibility loci affect the pathogenic activity of B cells in nonobese diabetic mice. J Immunol. 2006;177(10):7033–7041. doi: 10.4049/jimmunol.177.10.7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stolp J, Chen YG, Cox SL, Henck V, Zhang W, Tsaih SW, Chapman H, Stearns T, Serreze DV, Silveira PA. Subcongenic analyses reveal complex interactions between distal chromosome 4 genes controlling diabetogenic B cells and CD4 T cells in nonobese diabetic mice. J Immunol. 2012;189(3):1406–1417. doi: 10.4049/jimmunol.1200120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fox CJ, Paterson AD, Mortin-Toth SM, Danska JS. Two genetic loci regulate T cell-dependent islet inflammation and drive autoimmune diabetes pathogenesis. Am J Hum Genet. 2000;67:67–81. doi: 10.1086/302995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McAleer MA, Reifsnyder P, Palmer SM, Prochazka M, Love JM, Copeman JB, Powell EE, Rodrigues NR, Prins JB, Serreze DV. et al. Crosses of NOD mice with the related NON strain. A polygenic model for IDDM. Diabetes. 1995;44(10):1186–1195. doi: 10.2337/diab.44.10.1186. [DOI] [PubMed] [Google Scholar]
- 80.Esposito L, Hill NJ, Pritchard LE, Cucca F, Muxworthy C, Merriman ME, Wilson A, Julier C, Delepine M, Tuomilehto J. et al. Genetic analysis of chromosome 2 in type 1 diabetes: analysis of putative loci IDDM7, IDDM12, and IDDM13 and candidate genes NRAMP1 and IA-2 and the interleukin-1 gene cluster. IMDIAB Group. Diabetes. 1998;47(11):1797–1799. doi: 10.2337/diabetes.47.11.1797. [DOI] [PubMed] [Google Scholar]
- 81.Owerbach D, Gabbay KH. The HOXD8 locus (2q31) is linked to type I diabetes. Interaction with chromosome 6 and 11 disease susceptibility genes. Diabetes. 1995;44(1):132–136. doi: 10.2337/diab.44.1.132. [DOI] [PubMed] [Google Scholar]
- 82.Morahan G, Huang D, Tait BD, Colman PG, Harrison LC. Markers on distal chromosome 2q linked to insulin-dependent diabetes mellitus. Science. 1996;272(5269):1811–1813. doi: 10.1126/science.272.5269.1811. [DOI] [PubMed] [Google Scholar]
- 83.Field LL, Tobias R, Magnus T. A locus on chromosome 15q26 (IDDM3) produces susceptibility to insulin dependent diabetes mellitus. Nat Genet. 1994;8:189–194. doi: 10.1038/ng1094-189. [DOI] [PubMed] [Google Scholar]
- 84.Zamani M, Pociot F, Raeymaekers P, Nerup J, Cassiman JJ. Linkage of type I diabetes to 15q26 (IDDM3) in the Danish population. Hum Genet. 1996;98(4):491–496. doi: 10.1007/s004390050245. [DOI] [PubMed] [Google Scholar]
- 85.Howson JM, Cooper JD, Smyth DJ, Walker NM, Stevens H, She JX, Eisenbarth GS, Rewers M, Todd JA, Akolkar B. et al. Evidence of gene-gene interaction and age-at-diagnosis effects in type 1 diabetes. Diabetes. 2012;61(11):3012–3017. doi: 10.2337/db11-1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hermann R, Lipponen K, Kiviniemi M, Kakko T, Veijola R, Simell O, Knip M, Ilonen J. Lymphoid tyrosine phosphatase (LYP/PTPN22) Arg620Trp variant regulates insulin autoimmunity and progression to type 1 diabetes. Diabetologia. 2006;49(6):1198–1208. doi: 10.1007/s00125-006-0225-4. [DOI] [PubMed] [Google Scholar]
- 87.Smyth DJ, Cooper JD, Howson JM, Walker NM, Plagnol V, Stevens H, Clayton DG, Todd JA. PTPN22 Trp620 explains the association of chromosome 1p13 with type 1 diabetes and shows a statistical interaction with HLA class II genotypes. Diabetes. 2008;57(6):1730–1737. doi: 10.2337/db07-1131. [DOI] [PubMed] [Google Scholar]
- 88.Steck AK, Liu SY, McFann K, Barriga KJ, Babu SR, Eisenbarth GS, Rewers MJ, She JX. Association of the PTPN22/LYP gene with type 1 diabetes. Pediatr Diabetes. 2006;7(5):274–278. doi: 10.1111/j.1399-5448.2006.00202.x. [DOI] [PubMed] [Google Scholar]
- 89.Winkler C, Krumsiek J, Lempainen J, Achenbach P, Grallert H, Giannopoulou E, Bunk M, Theis FJ, Bonifacio E, Ziegler AG. A strategy for combining minor genetic susceptibility genes to improve prediction of disease in type 1 diabetes. Genes Immun. 2012;13(7):549–555. doi: 10.1038/gene.2012.36. [DOI] [PubMed] [Google Scholar]
- 90.Wicker LS, Miller BJ, Coker LZ, McNally SE, Scott S, Mullen Y, Appel MC. Genetic control of diabetes and insulitis in the non-obese diabetic (NOD) mouse. J Exp Med. 1987;165:1639–1654. doi: 10.1084/jem.165.6.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Arai S, Minjares C, Nagafuchi S, Miyazaki T. Improved experimental procedures for achieving efficient germ line transmission of nonobese diabetic (NOD)-derived embryonic stem cells. Exp Diabesity Res. 2004;5(3):219–226. doi: 10.1080/15438600490486877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brook FA, Evans EP, Lord CJ, Lyons PA, Rainbow DB, Howlett SK, Wicker LS, Todd JA, Gardner RL. The derivation of highly germline-competent embryonic stem cells containing NOD-derived genome. Diabetes. 2003;52(1):205–208. doi: 10.2337/diabetes.52.1.205. [DOI] [PubMed] [Google Scholar]
- 93.Nagafuchi S, Katsuta H, Kogawa K, Akashi T, Kondo S, Sakai Y, Tsukiyama T, Kitamura D, Niho Y, Watanabe T. Establishment of an embryonic stem (ES) cell line derived from a non-obese diabetic (NOD) mouse: in vivo differentiation into lymphocytes and potential for germ line transmission. FEBS Lett. 1999;455(1-2):101–104. doi: 10.1016/s0014-5793(99)00801-7. [DOI] [PubMed] [Google Scholar]
- 94.Chen J, Reifsnyder PC, Scheuplein F, Schott WH, Mileikovsky M, Soodeen-Karamath S, Nagy A, Dosch MH, Ellis J, Koch-Nolte F, Leiter EH. "Agouti NOD": identification of a CBA-derived Idd locus on Chromosome 7 and its use for chimera production with NOD embryonic stem cells. Mamm Genome. 2005;16(10):775–783. doi: 10.1007/s00335-005-0007-1. [DOI] [PubMed] [Google Scholar]
- 95.Leiter EH, Reifsnyder PC, Wallace R, Li R, King B, Churchill GC. NOD x 129.H2(g7) backcross delineates 129S1/SvImJ-derived genomic regions modulating type 1 diabetes development in mice. Diabetes. 2009;58(7):1700–1703. doi: 10.2337/db09-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nichols J, Jones K, Phillips JM, Newland SA, Roode M, Mansfield W, Smith A, Cooke A. Validated germline-competent embryonic stem cell lines from nonobese diabetic mice. Nat Med. 2009;15(7):814–818. doi: 10.1038/nm.1996. [DOI] [PubMed] [Google Scholar]
- 97.Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, Smith A. The ground state of embryonic stem cell self-renewal. Nature. 2008;453(7194):519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Morgan MA, Muller PS, Mould A, Newland SA, Nichols J, Robertson EJ, Cooke A, Bikoff EK. The nonconventional MHC class II molecule DM governs diabetes susceptibility in NOD mice. PLoS One. 2013;8(2):e56738. doi: 10.1371/journal.pone.0056738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Landel CP, Dunlap J, Patton JB, Manser T. A germline-competent embryonic stem cell line from NOD.Cg-Prkdc (scid) Il2rg (tm1Wjl)/SzJ (NSG) mice. Transgenic Res. 2013;22(1):179–185. doi: 10.1007/s11248-012-9629-8. [DOI] [PubMed] [Google Scholar]
- 100.Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11(9):636–646. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- 101.Jacob HJ, Lazar J, Dwinell MR, Moreno C, Geurts AM. Gene targeting in the rat: advances and opportunities. Trends Genet. 2010;26(12):510–518. doi: 10.1016/j.tig.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Geurts AM, Moreno C. Zinc-finger nucleases: new strategies to target the rat genome. Clin Sci (Lond) 2010;119(8):303–311. doi: 10.1042/CS20100201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Geurts AM, Cost GJ, Remy S, Cui X, Tesson L, Usal C, Menoret S, Jacob HJ, Anegon I, Buelow R. Generation of gene-specific mutated rats using zinc-finger nucleases. Methods Mol Biol. 2010;597:211–225. doi: 10.1007/978-1-60327-389-3_15. [DOI] [PubMed] [Google Scholar]
- 104.Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM, Jenkins SS, Wood A, Cui X, Meng X. et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science. 2009;325(5939):433. doi: 10.1126/science.1172447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cui X, Ji D, Fisher DA, Wu Y, Briner DM, Weinstein EJ. Targeted integration in rat and mouse embryos with zinc-finger nucleases. Nat Biotechnol. 2011;29(1):64–67. doi: 10.1038/nbt.1731. [DOI] [PubMed] [Google Scholar]
- 106.Boch J. TALEs of genome targeting. Nat Biotechnol. 2011;29(2):135–136. doi: 10.1038/nbt.1767. [DOI] [PubMed] [Google Scholar]
- 107.Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ. et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol. 2011;29(2):143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- 108.Tesson L, Usal C, Menoret S, Leung E, Niles BJ, Remy S, Santiago Y, Vincent AI, Meng X, Zhang L, Gregory PD, Anegon I, Cost GJ. Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol. 2011;29(8):695–696. doi: 10.1038/nbt.1940. [DOI] [PubMed] [Google Scholar]
- 109.Sung YH, Baek IJ, Seong JK, Kim JS, Lee HW. Mouse genetics: catalogue and scissors. BMB Rep. 2012;45(12):686–692. doi: 10.5483/BMBRep.2012.45.12.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Rooney DL, Zhang M, Ihrig MM, McManus MT, Gertler FB, Scott ML, Van Parijs L. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33(3):401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- 111.Tiscornia G, Singer O, Ikawa M, Verma IM. A general method for gene knockdown in mice by using lentiviral vectors expressing small interfering RNA. Proc Natl Acad Sci U S A. 2003;100(4):1844–1848. doi: 10.1073/pnas.0437912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rao DD, Vorhies JS, Senzer N, Nemunaitis J. siRNA vs. shRNA: similarities and differences. Adv Drug Deliv Rev. 2009;61(9):746–759. doi: 10.1016/j.addr.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 113.Joseph J, Bittner S, Kaiser FM, Wiendl H, Kissler S. IL-17 silencing does not protect nonobese diabetic mice from autoimmune diabetes. J Immunol. 2012;188(1):216–221. doi: 10.4049/jimmunol.1101215. [DOI] [PubMed] [Google Scholar]
- 114.Zheng P, Kissler S. PTPN22 Silencing in the NOD model indicates the type 1 diabetes-associated allele is not a loss-of-function variant. Diabetes. 2012 doi: 10.2337/db12-0929. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gerold KD, Zheng P, Rainbow DB, Zernecke A, Wicker LS, Kissler S. The soluble CTLA-4 splice variant protects from type 1 diabetes and potentiates regulatory T-cell function. Diabetes. 2011;60(7):1955–1963. doi: 10.2337/db11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kissler S, Stern P, Takahashi K, Hunter K, Peterson LB, Wicker LS. In vivo RNA interference demonstrates a role for Nramp1 in modifying susceptibility to type 1 diabetes. Nat Genet. 2006;38(4):479–483. doi: 10.1038/ng1766. [DOI] [PubMed] [Google Scholar]
- 117.Bettini ML, Bettini M, Vignali DA. T-cell receptor retrogenic mice: a rapid, flexible alternative to T-cell receptor transgenic mice. Immunology. 2012;136(3):265–272. doi: 10.1111/j.1365-2567.2012.03574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lennon GP, Bettini M, Burton AR, Vincent E, Arnold PY, Santamaria P, Vignali DA. T cell islet accumulation in type 1 diabetes is a tightly regulated, cell-autonomous event. Immunity. 2009;31(4):643–653. doi: 10.1016/j.immuni.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DA. Correction of multi-gene deficiency in vivo using a single 'self-cleaving' 2A peptide-based retroviral vector. Nat Biotechnol. 2004;22(5):589–594. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- 120.Majewski J, Schwartzentruber J, Lalonde E, Montpetit A, Jabado N. What can exome sequencing do for you? J Med Genet. 2011;48(9):580–589. doi: 10.1136/jmedgenet-2011-100223. [DOI] [PubMed] [Google Scholar]
- 121.Singleton AB. Exome sequencing: a transformative technology. Lancet Neurol. 2011;10(10):942–946. doi: 10.1016/S1474-4422(11)70196-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Funda DP, Kaas A, Bock T, Tlaskalova-Hogenova H, Buschard K. Gluten-free diet prevents diabetes in NOD mice. Diabetes Metab Res Rev. 1999;15(5):323–327. doi: 10.1002/(sici)1520-7560(199909/10)15:5<323::aid-dmrr53>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 124.Katahira M, Hanakita M, Ito T, Suzuki M, Segawa S. The age of onset of diabetes and glutamic acid decarboxylase titer measured long after diagnosis are associated with the clinical stage of slow-onset type 1 diabetes. Diabetes Res Clin Pract. 2012 doi: 10.1016/j.diabres.2012.11.017. In press. [DOI] [PubMed] [Google Scholar]
- 125.American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2012;35(Suppl 1):S64–S71. doi: 10.2337/dc12-s064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Valle A, Giamporcaro GM, Scavini M, Stabilini A, Grogan P, Bianconi E, Sebastiani G, Masini M, Maugeri N, Porretti L. et al. Reduced circulating neutrophils precedes and accompanies type 1 diabetes. Diabetes. 2013 doi: 10.2337/db12-1345. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bach JF. The natural history of islet-specific autoimmunity in NOD mice. In: Leiter EH, Atkinson MA, editors. NOD mice and related strains: research applications in diabetes, AIDS, cancer, and other diseases. Landes Bioscience Publishers; Austin: 1998. pp. 121–144. [Google Scholar]
- 128.Serreze DV, Chapman HD, Varnum DS, Hanson MS, Reifsnyder PC, Richard SD, Fleming SA, Leiter EH, Shultz LD. B lymphocytes are essential for the initiation of T cell mediated autoimmune diabetes: analysis of a new "speed congenic" stock of NOD.Ig µ null mice. J Exp Med. 1996;184:2049–2053. doi: 10.1084/jem.184.5.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Serreze DV, Fleming SA, Chapman HD, Richard SD, Leiter EH, Tisch RM. B lymphocytes are critical antigen-presenting cells for the initiation of T cell-mediated autoimmune diabetes in nonobese diabetic mice. J Immunol. 1998;161(8):3912–3918. [PubMed] [Google Scholar]
- 130.Harsunen MH, Puff R, D'Orlando O, Giannopoulou E, Lachmann L, Beyerlein A, von Meyer A, Ziegler AG. Reduced Blood Leukocyte and Neutrophil Numbers in the Pathogenesis of Type 1 Diabetes. Horm Metab Res. 2013 doi: 10.1055/s-0032-1331226. In press. [DOI] [PubMed] [Google Scholar]
- 131.Thayer TC, Delano M, Liu C, Chen J, Padgett LE, Tse HM, Annamali M, Piganelli JD, Moldawer LL, Mathews CE. Superoxide production by macrophages and T cells is critical for the induction of autoreactivity and type 1 diabetes. Diabetes. 2011;60(8):2144–2151. doi: 10.2337/db10-1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Beilke JN, Meagher CT, Hosiawa K, Champsaur M, Bluestone JA, Lanier LL. NK cells are not required for spontaneous autoimmune diabetes in NOD mice. PLoS One. 2012;7(4):e36011. doi: 10.1371/journal.pone.0036011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gomez-Diaz RA, Aguilar MV, Meguro EN, Marquez RH, Magana EG, Martinez-Garcia MC, Rodarte NW, Aguilar-Salinas CA, Canche-Pool E, Ortiz-Navarrete V. The role of natural killer T (NKT) cells in the pathogenesis of type 1 diabetes. Curr Diabetes Rev. 2011;7(4):278–283. doi: 10.2174/157339911796397839. [DOI] [PubMed] [Google Scholar]
- 134.Chen YG, Driver JP, Silveira PA, Serreze DV. Subcongenic analysis of genetic basis for impaired development of invariant NKT cells in NOD mice. Immunogenetics. 2007;59(9):705–712. doi: 10.1007/s00251-007-0236-4. [DOI] [PubMed] [Google Scholar]
- 135.Matsuki N, Stanic AK, Embers ME, Van Kaer L, Morel L, Joyce S. Genetic dissection of V alpha 14J alpha 18 natural T cell number and function in autoimmune-prone mice. J Immunol. 2003;170(11):5429–5437. doi: 10.4049/jimmunol.170.11.5429. [DOI] [PubMed] [Google Scholar]
- 136.Baxter AG, Cooke A. Complement lytic activity has no role in the pathogenesis of autoimmune diabetes in NOD mice. Diabetes. 1993;42(11):1574–1578. doi: 10.2337/diab.42.11.1574. [DOI] [PubMed] [Google Scholar]
- 137.Martin S, Wolf-Eichbaum D, Duinkerken G, Scherbaum WA, Kolb H, Noordzij JG, Roep BO. Development of type 1 diabetes despite severe hereditary B-lymphocyte deficiency. N Engl J Med. 2001;345(14):1036–1040. doi: 10.1056/NEJMoa010465. [DOI] [PubMed] [Google Scholar]
- 138.Todd JA, Aitman TJ, Cornall RJ, Ghosh S, Hall JR, Hearne CM, Knight AM, Love JM, McAleer MA, Prins JB. et al. Genetic analysis of autoimmune type 1 diabetes mellitus in mice. Nature. 1991;351:542–547. doi: 10.1038/351542a0. [DOI] [PubMed] [Google Scholar]
- 139.Pearce RB. Fine-mapping of the mouse T lymphocyte fraction (Tlf) locus on Chromosome 9: association with autoimmune diabetes. J Autoimm. 1998;10:1–15. doi: 10.3109/08916939808993843. [DOI] [PubMed] [Google Scholar]
- 140.McGuire HM, Vogelzang A, Hill N, Flodstrom-Tullberg M, Sprent J, King C. Loss of parity between IL-2 and IL-21 in the NOD Idd3 locus. Proc Natl Acad Sci U S A. 2009;106(46):19438–19443. doi: 10.1073/pnas.0903561106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ivakine EA, Mortin-Toth SM, Gulban OM, Valova A, Canty A, Scott C, Danska JS. The idd4 locus displays sex-specific epistatic effects on type 1 diabetes susceptibility in nonobese diabetic mice. Diabetes. 2006;55(12):3611–3619. doi: 10.2337/db06-0758. [DOI] [PubMed] [Google Scholar]
- 142.Litherland SA, Grebe KM, Belkin NS, Paek E, Elf J, Atkinson M, Morel L, Clare-Salzler MJ, McDuffie M. Nonobese diabetic mouse congenic analysis reveals chromosome 11 locus contributing to diabetes susceptibility, macrophage STAT5 dysfunction, and granulocyte-macrophage colony-stimulating factor overproduction. J Immunol. 2005;175(7):4561–4565. doi: 10.4049/jimmunol.175.7.4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wicker LS, Chamberlain G, Hunter K, Rainbow D, Howlett S, Tiffen P, Clark J, Gonzalez-Munoz A, Cumiskey AM, Rosa RL. et al. Fine mapping, gene content, comparative sequencing, and expression analyses support Ctla4 and Nramp1 as candidates for Idd5.1 and Idd5.2 in the nonobese diabetic mouse. J Immunol. 2004;173(1):164–173. doi: 10.4049/jimmunol.173.1.164. [DOI] [PubMed] [Google Scholar]
- 144.Hung MS, Avner P, Rogner UC. Identification of the transcription factor ARNTL2 as a candidate gene for the type 1 diabetes locus Idd6. Hum Mol Genet. 2006;15(18):2732–2742. doi: 10.1093/hmg/ddl209. [DOI] [PubMed] [Google Scholar]
- 145.Bergman ML, Duarte N, Campino S, Lundholm M, Motta V, Lejon K, Penha-Goncalves C, Holmberg D. Diabetes protection and restoration of thymocyte apoptosis in NOD Idd6 congenic strains. Diabetes. 2003;52(7):1677–1682. doi: 10.2337/diabetes.52.7.1677. [DOI] [PubMed] [Google Scholar]
- 146.Serreze DV, Choisy-Rossi CM, Grier AE, Holl TM, Chapman HD, Gahagan JR, Osborne MA, Zhang W, King BL, Brown A, Roopenian D, Marron MP. Through regulation of TCR expression levels, an Idd7 region gene(s) interactively contributes to the impaired thymic deletion of autoreactive diabetogenic CD8+ T cells in nonobese diabetic mice. J Immunol. 2008;180(5):3250–3259. doi: 10.4049/jimmunol.180.5.3250. [DOI] [PubMed] [Google Scholar]
- 147.Ghosh S, Palmer SM, Rodrigues NR, Cordell HJ, Hearne CM, Cornall RJ, Prins JB, McShane P, Lathrop GM, Peterson LB. et al. Polygenic control of autoimmune diabetes in nonobese diabetic mice. Nat Genet. 1993;4:404–409. doi: 10.1038/ng0893-404. [DOI] [PubMed] [Google Scholar]
- 148.Liston A, Lesage S, Gray DH, O'Reilly LA, Strasser A, Fahrer AM, Boyd RL, Wilson J, Baxter AG, Gallo EM, Crabtree GR, Peng K, Wilson SR, Goodnow CC. Generalized resistance to thymic deletion in the NOD mouse; a polygenic trait characterized by defective induction of Bim. Immunity. 2004;21(6):817–830. doi: 10.1016/j.immuni.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 149.Lyons PA, Hancock WW, Denny P, Lord CJ, Hill NJ, Armitage N, Siegmund T, Todd JA, Phillips MS, Hess JF, Chen SL, Fischer PA, Peterson LB, Wicker LS. The NOD Idd9 genetic interval influences the pathogenicity of insulitis and contains molecular variants of Cd30, Tnfr2, and Cd137. Immunity. 2000;13:107–115. doi: 10.1016/s1074-7613(00)00012-1. [DOI] [PubMed] [Google Scholar]
- 150.Siegmund T, Armitage N, Wicker LS, Peterson LB, Todd JA, Lyons PA. Analysis of the mouse CD30 gene: a candidate for the NOD mouse type 1 diabetes locus Idd9.2. Diabetes. 2000;49(9):1612–1616. doi: 10.2337/diabetes.49.9.1612. [DOI] [PubMed] [Google Scholar]
- 151.Cannons JL, Chamberlain G, Howson J, Smink LJ, Todd JA, Peterson LB, Wicker LS, Watts TH. Genetic and functional association of the immune signaling molecule 4-1BB (CD137/TNFRSF9) with type 1 diabetes. J Autoimmun. 2005;25(1):13–20. doi: 10.1016/j.jaut.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 152.Penha-Goncalves C, Moule C, Smink LJ, Howson J, Gregory S, Rogers J, Lyons PA, Suttie JJ, Lord CJ, Peterson LB, Todd JA, Wicker LS. Identification of a structurally distinct CD101 molecule encoded in the 950-kb Idd10 region of NOD mice. Diabetes. 2003;52(6):1551–1556. doi: 10.2337/diabetes.52.6.1551. [DOI] [PubMed] [Google Scholar]
- 153.Brodnicki TC, McClive P, Couper S, Morahan G. Localization of Idd11 using NOD congenic mouse strains: elimination of Slc9a1 as a candidate gene. Immunogenetics. 2000;51(1):37–41. doi: 10.1007/s002510050006. [DOI] [PubMed] [Google Scholar]
- 154.Morahan G, McClive P, Huang D, Little P, Baxter A. Genetic and physiological association of diabetes susceptibility with raised Na+/H+ exchange activity. Proc Natl Acad Sci U S A. 1994;91(13):5898–5902. doi: 10.1073/pnas.91.13.5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Serreze DV, Bridgett M, Chapman HD, Chen E, Richard SD, Leiter EH. Subcongenic analysis of the Idd13 locus in NOD/Lt mice: evidence for several susceptibility genes including a possible diabetogenic role for beta2-microglobulin. J Immunol. 1998;160:1472–1478. [PubMed] [Google Scholar]
- 156.Fletcher JM, Jordan MA, Snelgrove SL, Slattery RM, Dufour FD, Kyparissoudis K, Besra GS, Godfrey DI, Baxter AG. Congenic analysis of the NKT cell control gene Nkt2 implicates the peroxisomal protein Pxmp4. J Immunol. 2008;181(5):3400–3412. doi: 10.4049/jimmunol.181.5.3400. [DOI] [PubMed] [Google Scholar]
- 157.Hamilton-Williams EE, Serreze DV, Charlton B, Johnson EA, Marron MP, Mullbacher A, Slattery RM. Transgenic rescue implicates beta2-microglobulin as a diabetes susceptibility gene in NOD mice. Proc Natl Acad Sci USA. 2001;98:11533–11538. doi: 10.1073/pnas.191383798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Brodnicki TC, Quirk F, Morahan G. A susceptibility allele from a non-diabetes-prone mouse strain accelerates diabetes in NOD congenic mice. Diabetes. 2003;52(1):218–222. doi: 10.2337/diabetes.52.1.218. [DOI] [PubMed] [Google Scholar]
- 159.Deruytter N, Boulard O, Garchon HJ. Mapping non-class II H2-linked loci for type 1 diabetes in nonobese diabetic mice. Diabetes. 2004;53(12):3323–3327. doi: 10.2337/diabetes.53.12.3323. [DOI] [PubMed] [Google Scholar]
- 160.Podolin PL, Denny P, Lord CJ, Hill NJ, Todd JA, Peterson LB, Wicker LS, Lyons PA. Congenic mapping of the insulin-dependent diabetes (Idd) gene, Idd10, localizes two genes mediating the Idd10 effect and eliminates the candidate Fcgr1. J Immunol. 1997;159(4):1835–1843. [PubMed] [Google Scholar]
- 161.Lyons PA, Armitage N, Lord CJ, Phillips MS, Todd JA, Peterson LB, Wicker LS. Mapping by genetic interaction: high-resolution congenic mapping of the type 1 diabetes loci Idd10 and Idd18 in the NOD mouse. Diabetes. 2001;50(11):2633–2637. doi: 10.2337/diabetes.50.11.2633. [DOI] [PubMed] [Google Scholar]
- 162.Morin J, Boitard C, Vallois D, Avner P, Rogner UC. Mapping of the murine type 1 diabetes locus Idd20 by genetic interaction. Mamm Genome. 2006;17(11):1105–1112. doi: 10.1007/s00335-006-0076-9. [DOI] [PubMed] [Google Scholar]
- 163.Rogner UC, Boitard C, Morin J, Melanitou E, Avner P. Three loci on mouse chromosome 6 influence onset and final incidence of type I diabetes in NOD.C3H congenic strains. Genomics. 2001;74(2):163–171. doi: 10.1006/geno.2001.6508. [DOI] [PubMed] [Google Scholar]
- 164.Hollis-Moffatt JE, Hook SM, Merriman TR. Colocalization of mouse autoimmune diabetes loci Idd21.1 and Idd21.2 with IDDM6 (human) and Iddm3 (rat) Diabetes. 2005;54(9):2820–2825. doi: 10.2337/diabetes.54.9.2820. [DOI] [PubMed] [Google Scholar]
- 165.Reifsnyder PC, Li R, Silveira P, Churchill GA, Serreze DV, Leiter EH. Conditioning the genome identifies additional diabetes resistance loci in type 1 diabetes resistant NOR/Lt mice. Genes Immun. 2005;6(6):528–538. doi: 10.1038/sj.gene.6364241. [DOI] [PubMed] [Google Scholar]