

Abstract
Protein tyrosine phosphatases (PTPs) play a central role in modulating the transduction of cellular signals, including the cells of the immune system. Several PTPs, PTPN22, PTPN2, and UBASH3A, have been associated with risk of type 1 diabetes (T1D) by genome wide association studies. Based on the current understanding of PTPs, it is clear that these variants impact antigen receptor signaling and cytokine signaling. This impact likely contributes to the development and progression of autoimmunity through multiple mechanisms, including failures of central and peripheral tolerance and the promotion of proinflammatory T cell responses. In this review, we discuss the genetic and functional implications of two of these PTPs, PTPN22 and PTPN2, in the development of T1D. We describe the known roles of these proteins in immune function, and how the expression and function of these proteins is altered by the genetic variants associated with T1D. Yet, there are still controversies in the field that require further study and the development of new approaches to extend our understanding of these PTP variants, with the goal of using the information gained to improve our ability to predict and cure T1D.
Keywords: type 1 diabetes, genome-wide association study, PTPN22, PTPN2, LYP, TCPTP
Abbreviations: BCL2 – B cell lymphoma 2; BCR – B cell receptor; BIM – BCL2-interacting mediator; C57BL/6 – inbred strain C57 black; c-CBL – Casitas B-lineage lymphoma proto-oncogene; cDNA – complementary DNA; CEU – Centre d'Etude du Polymorphisme Humain from Utah (Utah residents of northern European ancestry); CLL - chronic lymphoid leukemia; CSF-1 - colony stimulating factor 1; CSK – C-terminal Src kinase; DAISY – Diabetes Autoimmunity Study in the Young; EGF – epidermal growth factor; ERK – extracellular signal-regulated kinase; GRB2 – growth factor receptor-bound protein 2; GWAS – genome-wide association study; HapMap – Haplotype Map (project); HLA – human leukocyte antigen; IFNγ – interferon gamma; IFNGR2 – interferon gamma receptor 2; Ig – immunoglobulin; IL – interleukin; IRβ – insulin receptor beta; JAK – Janus kinase; LYP – lymphocyte tyrosine phosphatase; MAPK – mitogen-activated protein kinase; OR – odds ratio; PBMC – peripheral blood mononuclear cell; PEP – PEST domain phosphatase; PEST – Pro-Glu-Ser-Thr domain phosphatase; PI3K - phosphatidylinositol 3'-kinase; PLCγ2 - phospholipase Cγ2; PTK – protein tyrosine kinase; PTP – protein tyrosine phosphatase; PTPRC – protein tyrosine phosphatase, receptor type C; PTPN22 – protein tyrosine phosphatase, non-receptor type 22; RA – rheumatoid arthritis; SHIP - Src homology 2-containing inositol 5'-phosphatase; siRNA – small interfering RNA; SLE – systemic lupus erythematosus; SNP – single nucleotide polymorphism; STAT - signal transducer and activator of transcription; SYK – spleen tyrosine kinase; T1D – type 1 diabetes; Teff – effector T cell; TNFα – tumor necrosis factor alpha; Treg – regulatory T cell; TCPTP – T cell protein tyrosine phosphatase; TCR – T cell receptor; TRAF2 – TNF receptor-associated factor 2; VCP – valosin-containing protein; ZAP70 – zeta-chain-associated protein kinase 70
1. Introduction
Genetic studies have led to the identification of over 50 genomic regions containing single nucleotide polymorphisms (SNPs) that are associated with type 1 diabetes (T1D) (www.t1dbase.org). Many of these genes participate in intracellular signaling, including a group of protein tyrosine phosphatases. Tyrosine phosphorylation plays a central role in the transduction and regulation of intracellular signals, including the regulation of antigen receptor signaling, cytokine-induced differentiation in lymphocytes, and insulin signaling. Protein tyrosine kinases (PTKs) potentiate the phosphorylation of tyrosine residues, while protein tyrosine phosphatases (PTPs) dephosphorylate tyrosine residues on proteins, thereby mediating positive or negative regulation of target pathways. Over 45 PTPs are expressed in lymphocytes, each having activities within signaling pathways. Therefore, alterations in the level of expression or function of PTPs have the potential to alter the function and fate of cells, including those involved in the immune response. Currently, three PTP genes have been associated with T1D, PTPN22, PTPN2, and UBASH3A [1]. Understanding the functional impact of these genetic variations in PTPs associated with T1D is likely to shed light on the mechanisms that drive disease development and assist in creating therapies to stop or reverse this process. In this review, we discuss variants of two PTPs which are associated with T1D, PTPN22 and PTPN2, and their potential impact on disease.
2. PTPN22
2.1 Genetic studies and broad association of PTPN22 1858T with autoimmunity
PTPN22 encodes the protein tyrosine phosphatase N22 (PTPN22), also referred to as lymphocyte tyrosine phosphatase (LYP) [2]. A single-nucleotide polymorphism (SNP) c.1858C>T (rs2476601) of PTPN22 was found to be associated with T1D in 2004 [3]. Multiple subsequent studies have confirmed the association of this SNP with T1D, with a recent genome wide meta-analysis reporting a p-value of 5.93x10-80 and an odds ratio (OR) of 1.96 [4]. This variant is associated with multiple autoimmune diseases, including rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), Graves' disease, and myasthenia gravis [5-8]. The 1858T variant is a missense mutation of the coding region, in which cytosine is replaced by thymidine at position 1858, resulting in a change from an arginine (R) at position 620 of the protein to tryptophan (W). The R620W variant is located within the P1 proline-rich repeat in the PTPN22 SH3 domain of the protein that mediates interaction with the C-terminal Src kinase (CSK), as described below, making it likely that this SNP is functionally significant. In addition to rs2476601, other coding variants within PTPN22 have been identified in T1D subjects, indicating the importance of this gene in disease development [9].
2.2 Known function of lymphocyte tyrosine phosphatase (LYP)
LYP is expressed in all hematopoietic cells and is part of the Pro-Glu-Ser-Thr domain phosphatase (PEST) group of nonreceptor classical class I PTPs in human cell lines. LYP has been shown to be a negative regulator of T cell receptor (TCR) signal transduction via its interactions with the activating tyrosines of LCK (Y394), FYN (Y427), and zeta-chain-associated protein kinase 70 (ZAP70) as well as phospho-sites on TCRζ, CD3ε, VAV, and valosin-containing protein (VCP) [10-12]. Yu et al. have demonstrated that the phosphatase function of LYP is regulated by phosphorylation at Ser-35. The phosphorylation at this residue alters the conformational state of LYP and impairs its phosphatase activity resulting in an augmentation of TCR signal transduction [13]. The role of LYP as a regulator of TCR signaling is also bolstered by the development of a salicylic acid-based LYP inhibitor which binds to the catalytic site and enhances TCR signaling [13]. LYP has additional binding partners, including the adaptor molecule GRB2 (growth factor receptor-bound protein 2) and the E3 ligase c-CBL (Casitas B-lineage lymphoma proto-oncogene) [2], indicating that LYP may act alone or via interactions with other partners or substrates downstream of the TCR, B cell receptor (BCR), and other immune receptors.
2.3 Dissecting the functional impact of LYP R620W on lymphocytes
Cell lines have been used to understand better the function of LYP in T cells. Studies in Jurkat cells have shown that LYP's inhibition of TCR signal transduction is enhanced when it interacts with the PTK CSK [10]. This interaction occurs in the region of LYP which includes amino acid residue 620. Owing to the position of the LYP risk variant within the P1 proline-rich region, which interacts with the SH3 domain of CSK, several groups have investigated the impact of the LYP R620W variant on LYP/CSK interaction. Jurkat cell lines transduced to express LYP 620W and CSK demonstrate a loss of interaction between LYP and CSK and an increase in TCR responses [14, 15]. However, when Jurkat cells are transduced to express LYP 620W alone, a blunting of TCR signal transduction is seen [16]. More recent studies have shown that LYP inhibits TCR-induced signaling after dissociation from CSK, and is recruited to lipid rafts; in this way a more rapid dissociation of the LYP-CSK complex could result in an increase in LYP 620W availability to lipid rafts and an increase in its inhibitory activity [17].
2.4 Function of the LYP R620W variant deduced from murine models of PEP and PEP R619W
Studies of the role of PEP (PEST domain phosphatase), the murine homologue of LYP, have shed light on its multiplicity of functions in the immune response. PEP and LYP share 89% identity at the PTP domain, and 61% identity in the non-catalytic domains. The majority of these studies have been performed in mice where the Ptpn22 gene is knocked out [18-20], but more recently, a model in which Ptpn22 is inducibly knocked down has also been described [21]. Ptpn22 deficiency has been shown to enhance signaling through the TCR, as measured by calcium flux and phosphorylation of LCK, ZAP70, and extracellular signal-regulated kinase (ERK) (Table 1) [15, 18]. These alterations are most pronounced in memory T cells. They also result in increased proliferation of effector T cells and a general expansion of the T cell compartment over time. Regulatory T cells (Tregs) are also altered in these animals, with increased Treg number, IL-10 production, and suppressive function [19, 20]. Ptpn22 deficiency has also been shown to cause an expansion of germinal centers and an increase of immunoglobulin production [18]. With respect to a disease phenotype, Ptpn22-/- effector T cells (Teffs) are more potent mediators of colitis upon cell transfer. However, these Teffs can be controlled by co-transfer of Ptpn22-/- Tregs (but not wild type Tregs) [20]. Ptpn22-/- mice are resistant to EAE [19] and NOD mice, in which Ptpn22 is knocked down, have a decreased incidence of diabetes [21].
Table 1. Phenotypes associated with variants in the PTPN22 gene in mouse models and human cells.

These studies indicate that PEP plays multiple roles in the murine immune response, likely regulating both the effector and regulatory T cell compartments. These findings are consistent with the role of the LYP R620W variant in human T1D, in that it alone is not sufficient to drive autoimmunity. Zikherman et al. explored the question of whether Ptpn22-/- mice would develop autoimmunity if additional factors that contribute to loss of tolerance are present. To this end, they crossed the Ptpn22-/- mouse onto a B6 mouse strain with the Ptprc (CD45) E613R mutation, a mutation known to cause a loss of B cell tolerance [15]. They observed that these animals had both enhanced T cell responses in the periphery and the thymus as well as an increase in B cell response to activation. Finally, the animals developed lupus-like disease with anti-nuclear antibodies, and lymphoproliferative disease.
These studies are informative as to the multiplicity of functions of this PTP in murine T cells, but limited in that they do not directly address the impact of the risk variant, which may lead to an alteration but not a loss of function. To address this question, Zhang et al. engineered a mouse to express the PEP R619W variant which is similar to the R620W variant found in human autoimmunity [22]. These animals did not develop autoimmunity, confirming a need for additional triggers in the process of autoimmunity. However, the animals had an increase in the number of dendritic cells, hyper-responsive T cell signaling and an increase in antibody production in response to vaccination. The authors attributed these changes to a relative decrease in expression of PEP 619W through enhanced calpain-mediated degradation of the PEP 619W protein. They concluded that in humans the likely functional implications of the variant was a loss-of-function of LYP owing to decreased protein levels in T and B cells. These findings contradict multiple studies in human B and T cells which find no difference in LYP protein based on the 1858T genotype. A second group has independently generated a PEP R619W knock-in-mouse and found normal stability of the PEP 619W protein. Like Zhang et al., they found enhanced antigen receptor signaling in lymphocytes, but in contrast to Zhang et al., loss of tolerance and autoimmunity have been observed [23].
2.5 Assessment of the role of LYP and the LYP R620W variant in human cells
The studies of cell lines and murine models have helped to define, at least in part, how PTPN22 participates in immune function in humans. However, these approaches have limitations due to the possibility that a cell line or an animal may not reflect the intracellular signaling environment or functional outcomes that are found in human cells. To fully understand how LYP and its variants impact immune function and human disease, studies with human lymphocytes have been performed.
Vang et al. were the first to examine the functional impact of LYP R620W on human T cells. They found that T cells from T1D subjects heterozygous for the 1858T allele, displayed decreased IL-2 production upon stimulation with anti-CD3/CD28 (Table 1) [16]. A second study with T1D 1858T carriers also found a decrease in T cell proliferation and IL-2 production upon CD3/CD28 stimulation [24]. In further studies, healthy individuals who carry the 1858T variant allele were shown to have an expanded CD4 memory compartment and a blunted response to stimulation via CD3, as measured by Ca+ flux, IL-2, and IL-10 production, but no alteration in proliferation or production of the proinflammatory cytokines tumor necrosis factor alpha (TNFα) and interferon gamma (IFNγ) [25]. A decrease in IL-10 production was also described in 1858T carriers with ANCA vasculitis [26]. These studies seem to suggest that LYP R620W is a gain of function variant, leading to an increased inhibition of TCR signaling. Yet, the more recent study of the murine knock-in of PEP 619W by Zhang et al. suggested that the R>W substitution causes a loss of function due to decreased protein levels of PEP [22]. As part of that study, T cells from healthy and RA subjects were examined for LYP expression using a flow cytometric based assay. Using this approach, decreased expression of LYP was identified in individuals with the 1858T variant, and these subjects also had an increase in phosphorylation of ERK upon TCR stimulation [22]. Both the murine and human findings in this study seem to contradict the previous work using human peripheral blood mononuclear cells (PBMCs). The finding of decreased LYP expression in human lymphocytes has not been observed by other groups who have utilized Western blot, a more rigorous assay for protein levels [16, 27]. Also, phosphorylation of ERK has not been reported by other studies that have typically assessed more proximal aspects of TCR signaling in human samples. The differences between mouse and human primary T cells raise concerns about whether murine systems can sufficiently model LYP 620W function in human T cells.
LYP is also expressed in B cells. In chronic lymphoid leukemia (CLL), LYP is overexpressed resulting in a blunted BCR signal via spleen tyrosine kinase (SYK) which leads to a decrease in phosphorylation of phospholipase Cγ2 (PLCγ2), p38 MAPK, and ERK. However, the increase in LYP also leads to the inhibition of LYN and Src homology 2-containing inositol 5'-phosphatase (SHIP) which inhibit signaling via phosphatidylinositol 3'-kinase (PI3K) and AKT, leading to an increase in AKT activity [28]. This results in an enhancement of the prosurvival signals GSK3 (glycogen synthase kinase 3) and FOXO (forkhead box protein O), with the result that CLL cells escape BCR-mediated cell death [28]. LYP 620W has also been shown to impact primary B cells; expression of LYP is unaffected by the variant, but BCR signaling via PLCγ2 is decreased in naïve and memory B cells, suggesting a gain of function similar to that seen with overexpression of LYP in CLL [27, 29]. An extension of these studies to LYN, SHIP, and AKT has not yet been performed. However, studies have linked LYP 620W with alterations in B cell development and tolerance. It has been shown that such alterations can be associated with a resistance to BCR-mediated cell death, an expansion of transitional and anergic B cells, a decrease in circulating memory B cells, and an increase in the escape of autoreactive B cells into the transitional and mature naïve B cell compartment [29, 30]. These findings indicate that the LYP 620W variant likely contributes to the development of autoimmunity and autoantibodies via a B cell-intrinsic mechanism in addition to its impact on Teffs and Tregs.
2.6 Caveats related to studies of PTPN22 in human lymphocytes
The in vitro studies conducted in primary human lymphocytes have produced findings that partly differ from those conducted in immortalized cell lines or murine cells. Additional efforts are necessary to clarify the reasons for this discrepancy, to validate the present findings, and to reveal the true functions of PTPN22 in human lymphocytes. Human immune responses reflect a combination of genetic background, environment, and previous immune activation over years. Thus, the function of human primary lymphocytes not only reflects the direct impact of the variant itself, but may also be the result of compensatory mechanisms that have developed over time as a result of the presence of the genetic variant. In order to model this in animals, studies may need to follow up the animals as they age, and to determine impacts upon exposure to foreign antigens and pathogens. The influence of PTPN22 c.1858C>T on the development of autoimmunity may in fact be due to both types of immune alterations, through genetic variation and compensatory mechanisms, as disease develops over time. Thus, both should be addressed as we move forward with studies of PTPN22.
2.7 LYP's role in the development of T1D
Studies that have assessed the impact of the PTPN22 1858T variant on disease development have indicated that carriers of the variant are younger at diagnosis of T1D [31]. In contrast, another study found the age at onset to be older among 1858T/T subjects [32]. In addition, PTPN22 1858T has been shown to be a predictor of more rapid progression to T1D among islet autoantibody-positive at-risk relatives [33]. Modeling of known genetic factors has shown that combinations of genes which include PTPN22 can be used to predict both islet autoantibody and diabetes development [34, 35]. Furthermore, the PTPN22 risk variant has been linked to the development of autoantibodies when combined with environmental triggers [36]. These studies support the concept that the PTPN22 1858T variant contributes to a failure in tolerance which leads to the development of autoantibodies and disease.
3. PTPN2
3.1 Genetic association of PTPN2 with autoimmunity
The PTPN2 gene was first associated with autoimmunity in the Wellcome Trust Case Control Study in 2007 [37]; an SNP in the intergenic region 5.5kb downstream of the PTPN2 gene, rs2542151, was found to be associated with susceptibility to T1D, Crohn's disease, and weakly with RA. This finding was replicated and refined by Todd et al. [38], who identified two non-coding SNPs in PTPN2 that are independently associated with T1D: rs1893217 in intron 7 (p = 1.16x10-11, OR = 1.3, 95% CI: 1.21-1.41) and rs478582 in intron 3 (p = 9.15x10-9, OR = 0.82, 95% CI: 0.77-0.87). In the largest meta-analysis of T1D cases (11,781), control subjects (13,715), and family trios (4,342) to date, the association of PTPN2 rs1893217 with T1D was highly significant (p = 3.6x10-15) [1]. Note that rs1893217 in intron 7 is in complete linkage disequilibrium (LD, D'=1, r2=1) with rs2542151 located 3' of the PTPN2 gene, and thus their genotypes are interchangeable.
Like PTPN22, the PTPN2 gene is associated with multiple autoimmune diseases, suggesting that it contributes to common mechanisms in the development and progression of autoimmune disease. Association of PTPN2 rs1893217 has been confirmed for Crohn's disease (p = 1.29x10-14, OR = 1.25, 95% CI: 1.18-1.32) [39], celiac disease (p = 2.5x10-10, OR = 1.17, 95% CI: 1.12-1.23) [40], and, with weaker association, ulcerative colitis (p = 4.78x10-5, OR = 1.12) [41] and RA (p = 2.4x10-5) [42]. An initial association with Grave's disease [38] has not been replicated.
3.2 Known function of PTPN2
The PTPN2 gene encodes a non-receptor protein tyrosine phosphatase and was originally cloned from a human T cell cDNA library, hence the alternative name T cell protein tyrosine phosphatase (TCPTP) [43]. While expression of PTPN2 is highest in cells of the hematopoietic lineage, it is expressed ubiquitously throughout the body and as early as day e8.5 of development in mouse embryos [43, 44]. Two isoforms of PTPN2 are expressed in human cells via alternative splicing; a 45kD variant (387 amino acids) which results from correct splicing of exon 9 to the terminal exon 10, and a 48kD variant which does not splice exon 9 to exon 10, but rather continues translation into intron 9, where a stop codon terminates the protein at amino acid 415 [43, 44]. The subcellular localizations of the 45kD and 48kD variants differ as a result of these alternative C-terminal sequences, and thus impact their substrate selection [45]. The 45kD variant is found primarily in the nucleus in resting cells due to a bipartite nuclear localization sequence, but can diffuse into the cytoplasm upon stimulation. In the cytoplasm, it can interact with cytoplasmic substrates and intracellular domains of transmembrane-bound receptors and their associated signaling proteins [46]. The 48kD variant contains a hydrophobic transmembrane domain and is targeted to the endoplasmic reticulum. In contrast to human cells, mouse cells express primarily the 45kD isoform of PTPN2 [47].
Structurally, the PTPN2 protein contains an N-terminal phosphatase domain and a C-terminal non-catalytic domain involved in autoregulation of catalytic activity and determination of subcellular localization, as described above [45, 48]. PTPN2 is closely related to the intracellular phosphatase PTP1B (PTPN1), with 74% identity in the catalytic domains [49]. Both PTPN2 and PTP1B prefer tandem phospho-tyrosine residues and share some substrates. Indeed, synthesis of inhibitors specific for each phosphatase has been challenging [50]. However, despite their relatedness, Ptpn2 deficiency in mice results in defects in lymphopoiesis and systemic inflammation (discussed in detail below), whereas Ptpn1 deficiency results in increased insulin sensitivity and protection from diet-induced obesity, reflecting their relative contributions to cytokine/antigen receptor signaling versus insulin receptor signaling [51].
Numerous substrates have been identified for PTPN2 that reveal roles in regulating signaling in response to growth factors, cytokines, hormones, and antigens. Specific targets have been identified through in vitro phosphatase assays and expression of substrate trapping mutants of PTPN2. Functional confirmation of substrates has been achieved using Ptpn2+/+ and Ptpn2-/- fibroblasts and lymphocytes and PTPN2 siRNA in specific cell types. Using such techniques, PTPN2 has been shown to dephosphorylate the epidermal growth factor (EGF) receptor and p52Shc in response to EGF, impacting association of p52Shc with GRB2 [52] and the colony stimulating factor 1 (CSF-1) receptor which mediates downstream ERK activation [53]. The Janus kinases JAK1 and JAK3 are dephosphorylated by PTPN2, resulting in modulation of IL-2, IFNγ, and IFNα signaling [54], while the transcription factors STAT1, STAT3, and STAT6 are targets of PTPN2 in response to IFNγ, IL-6, and IL-4, respectively [55-57]. TNF has been shown to stimulate PTPN2 interaction with the adaptor protein TNF receptor-associated factor 2 (TRAF2), with subsequent dephosphorylation of c-SRC and regulation of ERK activation [58]. Consistent with Src family kinases being substrates, PTPN2 was recently shown to dephosphorylate LCK and FYN following T cell receptor engagement, impacting activation of the downstream signaling molecules ZAP70, PLCγ1, and ERK [59].
PTPN2 has also been shown to play a role in regulating the response to the hormones prolactin, leptin, and insulin. The transcription factors STAT5A and STAT5B appear to be substrates for PTPN2 upon prolactin binding the prolactin receptor [60], and STAT3 is dephosphorylated by PTPN2 in response to leptin receptor engagement in the hypothalamus, attenuating leptin signaling [61]. In response to insulin, PTPN2 binds and dephosphorylates the insulin receptor beta (IRβ) in cooperation with PTP1B [47]. PTPN2 has been shown to control specifically the duration of downstream AKT phosphorylation, but not ERK phosphorylation, in PTP1B deficient and sufficient cells [47, 62]. Given the ubiquitous expression of PTPN2, it is likely that additional PTPN2 substrates will be identified that impact other pathways.
3.3 Functional impact of the PTPN2 risk variant
PTPN2 is the only gene within the T1D-associated LD block on chromosome 18, making it a likely candidate gene for the genetic association. No common coding variants in LD with either rs2542151 or rs1893217 have been identified, although rare coding changes in PTPN2 have been identified through the 1000 Genomes Project (www.1000genomes.org). Thus, PTPN2 expression differences may contribute to T1D susceptibility. A modest but significant reduction in PTPN2 RNA levels with the rs1893217 risk allele have been observed in memory CD4 T cells from the peripheral blood of genotyped control subjects. This was confirmed in an independent data set generated from HapMap CEU EBV-transformed B cells [63]. However, neither rs1893217 nor rs2542151 overlap DNase I hypersensitivity sites, transcription factor binding sites, or microRNA target sites (genome.ucsc.edu), although binding sites for the vitamin D receptor have been reported in intron 7 near rs1893217 [64]. Thus, it seems likely that rs1893217 and rs2542151 are not directly responsible for alterations in PTPN2 expression, but may be found in LD with a variant that does alter expression.
3.4 Role of PTPN2 in autoimmunity deduced from murine models of PTPN2 deficiency
To understand better the role of PTPN2 in vivo, the Ptpn2 gene has been disrupted globally in all mouse tissues of BALB/c and C57BL/6 mouse strains by homologous recombination [65] and Cre/LoxP recombination [66]. In all cases, Ptpn2 deficiency results in Mendelian ratios of offspring that appear normal at birth. By 2 weeks of age, Ptpn2-/- mice begin to display growth retardation, and at 3-5 weeks of age, mice develop severe defects in B cell lymphopoiesis and erythropoiesis, resulting in death in 100% of animals. In contrast, mice heterozygous for the null allele display no obvious phenotype. On a BALB/c genetic background, Ptpn2-/- deficiency also results in a systemic inflammatory disease characterized by increased levels of IL-12p40, IFNγ, TNFα, iNOS, and mononuclear infiltrates in the spleen and non-lymphoid tissues [65, 67]. This inflammatory phenotype is not observed when Ptpn2 deficiency is present on the more autoimmune resistant C57BL/6 genetic background [66].
Histologically, Ptpn2-/- mice are characterized by decreased bone marrow cellularity over time, with reduced numbers of progenitor B cells, pre B cells, immature IgM+ B cells, and mature IgD+ recirculating B cells, which is reflected in decreased peripheral B cell subsets [66, 68]. The thymus also has decreased cellularity over time due to a reduction in CD4+CD8+ double positive thymocytes, double negative thymocytes, and CD4+ and CD8+ single positive thymocytes, resulting in decreased numbers of CD4+ and CD8+ peripheral T cells [65, 66]. These B and T cell phenotypes result from both cell intrinsic and extrinsic factors, as evidenced by decreased B cell colony forming ability in Ptpn2-/- bone marrow compared to Ptpn2+/+ bone marrow when grown on wild type stromal cells, both of which are reduced when cultured on Ptpn2-/- stromal cells [68]. Extrinsically, cells in Ptpn2-/- mice are exposed to increased levels of inflammatory cytokines, while intrinsically, Ptpn2 deficiency exacerbates signaling in response to growth factors, cytokines, and antigen receptor stimulation, since PTPN2 negatively regulates these pathways, as described above.
Recent experiments that specifically knocked out Ptpn2 in CD4+ T cells using a floxed Ptpn2 locus and Lck-Cre mice have further defined the role of PTPN2 in T cell development, thymic selection, and response to antigen [59]. Consistent with LCK and FYN being direct targets of PTPN2, Ptpn2fl/fl/Lck-Cre thymocytes and peripheral T cells display increased LCK and FYN phosphorylation, as well as increased phosphorylation of downstream TCR signaling pathway molecules following stimulation with anti-CD3. This translates into increased proliferation, and lowers the threshold for TCR-induced proliferation, antigen affinity, and need for costimulation via CD28 both in vitro and in vivo. The thymocytes of Ptpn2fl/fl/Lck-Cre mice show evidence of increased positive selection, resulting in increased numbers of CD4+ and CD8+ thymocytes and peripheral T cells, particularly peripheral CD8+ effector memory T cells. There is also increased responsiveness to IL-2 in Ptpn2fl/fl/Lck-Cre T cells. Interestingly, at 48 weeks of age, Ptpn2fl/fl/Lck-Cre mice develop inflammation and autoimmunity that is characterized by increased levels of circulating IL-6, TNF, and IFNγ, high levels of anti-nuclear antibodies, and infiltration of CD8+ effector memory T cells in lymphoid and non-lymphoid tissues that can transfer disease.
Taken together, the phenotypes observed in Ptpn2-/- and Ptpn2fl/fl/Lck-Cre mice provide clues to how decreased PTPN2 expression in humans may contribute to the loss of T cell tolerance and the development of inflammation, leading to autoimmunity. While conditional deletion of Ptpn2 in T cells reveals T cell-intrinsic effects in the absence of non-T cell extrinsic factors, mice with global Ptpn2 deficiency may model human PTPN2 deficiency more accurately since expression of PTPN2 is ubiquitous. Thus, cell-extrinsic effects of PTPN2 deficiency are likely to play a role in human disease settings, as they do in mouse models.
3.5 The role of PTPN2 in the development and progression of T1D
The role of PTPN2 in T1D is likely to be complex because of the ubiquitous expression of PTPN2 in both hematopoietic cells and non-hematopoietic cells, including β-cells in pancreatic islets. Moreover, based on in vivo mouse models, PTPN2 deficiency is likely to result in both cell-extrinsic and cell-intrinsic effects. Although the specific role of PTPN2 in T1D has only been studied in the last 5 years, pleiotropic functions are apparent, including effects on the maintenance of Tregs, β-cell apoptosis, and the response to insulin, as illustrated in Figure 1.
Figure 1. Potential roles for PTPN2 in the development of T1D.
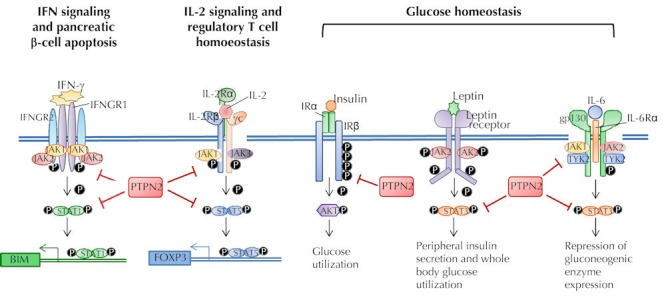
Experimental evidence indicates that PTPN2 regulates apoptosis of pancreatic β-cells upon exposure to inflammatory cytokines by modulating phosphorylation of STAT1, which induces the mitochondrial apoptotic pathway. The T1D-associated variant rs1893217 in the PTPN2 gene has been associated with reduced IL-2 receptor signaling in response to IL-2, impacting the maintenance of FOXP3+ Tregs in T1D subjects. PTPN2 has also been shown to influence glucose homeostasis by dephosphorylating the insulin receptor b-chain in conjunction with the PTP1B phosphatase. In addition, PTPN2 impacts glucose levels by modulating STAT3 signaling downstream of the leptin receptor in the hypothalamus and in response to IL-6, repressing hepatic gluconeogenesis.
The presence of the risk genotype at PTPN2 has been examined for its impact on the development of islet autoimmunity and T1D in several studies. A case-control study of European T1D cases reported that the frequency of carriers of the PTPN2 rs2542151 G risk allele was significantly higher in T1D cases with an earlier onset (≤16y) compared with later onset T1D or controls [69]. This finding is not necessarily surprising since GWAS typically include T1D subjects with an age of onset <17y, which would enrich for genetic loci contributing to early onset disease. PTPN2 rs1893217 risk genotype has also been studied in the BABYDIAB study in Europe, which has followed 1650 children of T1D parents, and the DAISY study in the US, following ~1700 children with a first-degree relative with T1D or with high-risk HLA DR3/4-DQ8 genotype [34, 35, 70, 71]. These children have been followed prospectively over time for the development of islet autoantibodies and progression to T1D. The advantage of these valuable cohorts is the ability to assess the predictive value of non-HLA genes to the initiation and progression of T1D. The disadvantage is that a small number of subjects in the cohort develop islet autoantibodies and progress to T1D, limiting the statistical power of the analysis. Nonetheless, Steck et al. reported a weak association of the PTPN2 rs1893217 risk genotype with the development of islet autoimmunity, which remained significant after adjustment for family history and high-risk HLA genotype (p = 0.04, HR = 1.42, 95% CI: 1.02-1.99) [34]. Other studies have not detected a significant association of the PTPN2 genotype with the development of islet autoantibodies [35, 70, 71]. No association of the PTPN2 rs1893217 risk genotype with progression to T1D has been detected in these studies or others [34, 70-72], although an interaction between the PTPN2 rs1893217 protective genotype and the vitamin D receptor SNP rs1544410 was associated with decreased risk of T1D in the DAISY cohort (p = 0.0004, HR = 0.24, 95% CI:0.11-0.53) [71]. Taken together, these studies indicate that the PTPN2 rs1893217 genotype alone does not appear to be a significant risk factor for the development of T1D, but PTPN2 may influence the development of islet autoimmunity and age at onset, suggesting a role for PTPN2 in disease initiation rather than progression.
Altered PTPN2 expression has been correlated with defects in IL-2 signaling and altered FOXP3 expression in Tregs in T1D. Consistent with JAK1/3 and STAT5 being substrates for PTPN2, the risk allele at the T1D-associated SNP PTPN2 rs1893217 was correlated with decreased IL-2 signaling in peripheral CD4+ T cells from genotyped control subjects (Figure 1) [63]. This phenotype was not due to altered expression of the IL-2 receptor, JAK1, JAK3, or STAT5; however, the rs1893217 genotype correlated with reduced expression of PTPN2 in CD4+ memory cells, suggesting that PTPN2 may indirectly modulate IL-2 responsiveness in primary human T cells. In subjects with T1D, IL-2 signaling is further decreased compared with controls, indicating that the autoimmune environment acts coordinately with the PTPN2 genotype to reduce IL-2 signaling ([73] and unpublished data). Interestingly, PTPN2 expression was increased in T1D subjects compared with control subjects, most likely due to inflammatory cytokines which have been shown to induce PTPN2 expression [74, 75]. Functionally, diminished IL-2 signaling is associated with reduced maintenance of FOXP3 expression in natural Tregs from subjects with T1D and decreased FOXP3 expression in induced Tregs [73]. Thus, the effect of PTPN2 on IL-2 responsiveness may contribute to the development of T1D through an impact on Treg homeostasis, consistent with alterations in IL-2 and Tregs in NOD mice [76, 77]. Recent experiments have shown that the PTPN2 rs1893217 risk variant acts independently of T1D risk variants in the IL2RA gene, which encodes the high-affinity IL-2 receptor, CD25, to reduce IL-2 responsiveness in T cells, potentially further impacting Treg persistence (unpublished data).
PTPN2 may also modulate pancreatic β-cell apoptosis in response to inflammatory cytokines. Knockdown of PTPN2 expression in rat or human β-cells enhances apoptosis upon treatment with IFN-α, IFN-γ, IL-1β+IFN-γ, or TNF+IFN-γ (Figure 1) [74, 78]. Decreased PTPN2 expression is correlated with an increase in phosphorylation of STAT1 in rat insulinoma cells, and amelioration of STAT1 expression with a STAT1-specific siRNA protected cells from apoptosis. Thus, if the PTPN2 rs1893217 risk genotype is associated with reduced PTPN2 expression in pancreatic β-cells, as found in lymphocytes, then it may predispose β-cells to apoptosis upon exposure to inflammatory cytokines via increased phosphorylation of STAT1.
Lastly, the role of PTPN2 in regulating insulin and leptin signaling may impact the development of hyperglycemia in T1D. The function of PTP1B and PTPN2 in these pathways has been a focus for type 2 diabetes (T2D) which is associated with the PTPN1 gene [79-81]. Although the PTPN2 gene is not associated with susceptibility to T2D, altered PTPN2 expression can impact hepatic gluconeogenesis. Heterozygous Ptpn2+/- mice fed a high fat diet display reduced fasting glucose levels compared with Ptpn2+/+ littermates, which correlates with decreased fasting hepatic gluconeogenesis [82]. Decreased hepatic glucose output is driven by the mechanism of increased STAT3 phosphorylation downstream of IL-6 signaling in the liver, which suppresses expression of gluconeogenic enzymes (Figure 1). Peripheral insulin sensitivity and whole body glucose utilization is also impacted by PTPN2 expression via leptin signaling. Deletion of the Ptpn2 gene specifically in neurons results in sustained STAT3 phosphorylation downstream of the leptin receptor in the hypothalamus of mice on a high fat diet, correlating with decreased diet-induced obesity, and reduced fasting plasma insulin and glucose [61]. These results suggest that decreased PTPN2 expression may protect from hyperglycemia by enhancing phosphorylation of STAT3 in response to IL-6 or leptin, whereas increased PTPN2 expression in the context of T1D could potentially contribute to increased glucose levels by attenuation of STAT3 phosphorylation.
4. Conclusions
The importance of the protein tyrosine phosphatases, PTPN2 and PTPN22, in T1D was first identified through genetic association studies. This has led to a series of further genetic and immunologic investigations which have enlightened us on the potential role of these proteins in the development and progression of T1D. In the case of each PTP, studies have uncovered alterations in multiple signaling pathways within multiple cell types caused by the genetic variants, leading to failures of central and peripheral tolerance, the promotion of proinflammatory T cell responses, and even the loss of regulation of glucose homeostasis. These findings have challenged our motivation to identify a single mechanism by which these variants lead to disease, or by extension a single pathway to target for therapeutic intervention. Further study is needed to determine if there is a single dominant immunologic alteration that contributes to disease. Alternatively, the study of these PTPs may demonstrate that the ubiquitous nature of these variants and their association with many autoimmune diseases is due, in part, to the multiplicity of ways in which these variants subtly alter the immune response.
Disclosure: The authors report no conflict of interests.
Acknowledgments
This work was supported by grants from NIAID and JDRF.
References
- 1.Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C. et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41(6):703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen S, Dadi H, Shaoul E, Sharfe N, Roifman CM. Cloning and characterization of a lymphoid-specific, inducible human protein tyrosine phosphatase, Lyp. Blood. 1999;93(6):2013–2024. [PubMed] [Google Scholar]
- 3.Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, MacMurray J, Meloni GF, Lucarelli P, Pellecchia M. et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet. 2004;36(4):337–338. doi: 10.1038/ng1323. [DOI] [PubMed] [Google Scholar]
- 4.Bradfield JP, Qu HQ, Wang K, Zhang H, Sleiman PM, Kim CE, Mentch FD, Qiu H, Glessner JT, Thomas KA. et al. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. Plos Genet. 2011;7(9):e1002293. doi: 10.1371/journal.pgen.1002293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kyogoku C, Langefeld CD, Ortmann WA, Lee A, Selby S, Carlton VE, Chang M, Ramos P, Baechler EC, Batliwalla FM. et al. Genetic association of the R620W polymorphism of protein tyrosine phosphatase PTPN22 with human SLE. Am J Hum Genet. 2004;75(3):504–507. doi: 10.1086/423790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, Tsao BP, Vyse TJ, Langefeld CD, Nath SK. et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40(2):204–210. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Velaga MR, Wilson V, Jennings CE, Owen CJ, Herington S, Donaldson PT, Ball SG, James RA, Quinton R, Perros P. et al. The codon 620 tryptophan allele of the lymphoid tyrosine phosphatase (LYP) gene is a major determinant of Graves' disease. J Clin Endocrinol Metab. 2004;89(11):5862–5865. doi: 10.1210/jc.2004-1108. [DOI] [PubMed] [Google Scholar]
- 8.Vandiedonck C, Capdevielle C, Giraud M, Krumeich S, Jais JP, Eymard B, Tranchant C, Gajdos P, Garchon HJ. Association of the PTPN22*R620W polymorphism with autoimmune myasthenia gravis. Ann Neurol. 2006;59(2):404–407. doi: 10.1002/ana.20751. [DOI] [PubMed] [Google Scholar]
- 9.Onengut-Gumuscu S, Buckner JH, Concannon P. A haplotype-based analysis of the PTPN22 locus in type 1 diabetes. Diabetes. 2006;55(10):2883–2889. doi: 10.2337/db06-0225. [DOI] [PubMed] [Google Scholar]
- 10.Cloutier JF, Veillette A. Cooperative inhibition of T-cell antigen receptor signaling by a complex between a kinase and a phosphatase. J Exp Med. 1999;189(1):111–121. doi: 10.1084/jem.189.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gjorloff-Wingren A, Saxena M, Williams S, Hammi D, Mustelin T. Characterization of TCR-induced receptor-proximal signaling events negatively regulated by the protein tyrosine phosphatase PEP. Eur J Immunol. 1999;29(12):3845–3854. doi: 10.1002/(SICI)1521-4141(199912)29:12<3845::AID-IMMU3845>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 12.Wu J, Katrekar A, Honigberg LA, Smith AM, Conn MT, Tang J, Jeffery D, Mortara K, Sampang J, Williams SR. et al. Identification of substrates of human protein-tyrosine phosphatase PTPN22. J Biol Chem. 2006;281(16):11002–11010. doi: 10.1074/jbc.M600498200. [DOI] [PubMed] [Google Scholar]
- 13.Yu X, Sun JP, He Y, Guo X, Liu S, Zhou B, Hudmon A, Zhang ZY. Structure, inhibitor, and regulatory mechanism of Lyp, a lymphoid-specific tyrosine phosphatase implicated in autoimmune diseases. Proc Natl Acad Sci U S A. 2007;104(50):19767–19772. doi: 10.1073/pnas.0706233104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, Ardlie KG, Huang Q, Smith AM, Spoerke JM. et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet. 2004;75(2):330–337. doi: 10.1086/422827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zikherman J, Hermiston M, Steiner D, Hasegawa K, Chan A, Weiss A. PTPN22 deficiency cooperates with the CD45 E613R allele to break tolerance on a non-autoimmune background. J Immunol. 2009;182(7):4093–4106. doi: 10.4049/jimmunol.0803317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vang T, Congia M, Macis MD, Musumeci L, Orru V, Zavattari P, Nika K, Tautz L, Tasken K, Cucca F. et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet. 2005;37(12):1317–1319. doi: 10.1038/ng1673. [DOI] [PubMed] [Google Scholar]
- 17.Vang T, Liu WH, Delacroix L, Wu S, Vasile S, Dahl R, Yang L, Musumeci L, Francis D, Landskron J. et al. LYP inhibits T-cell activation when dissociated from CSK. Nat Chem Biol. 2012;8(5):437–446. doi: 10.1038/nchembio.916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hasegawa K, Martin F, Huang G, Tumas D, Diehl L, Chan AC. PEST domain-enriched tyrosine phosphatase (PEP) regulation of effector/memory T cells. Science. 2004;303(5658):685–689. doi: 10.1126/science.1092138. [DOI] [PubMed] [Google Scholar]
- 19.Maine CJ, Hamilton-Williams EE, Cheung J, Stanford SM, Bottini N, Wicker LS, Sherman LA. PTPN22 alters the development of regulatory T cells in the thymus. J Immunol. 2012;188(11):5267–5275. doi: 10.4049/jimmunol.1200150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brownlie RJ, Miosge LA, Vassilakos D, Svensson LM, Cope A, Zamoyska R. Lack of the Phosphatase PTPN22 Increases Adhesion of Murine Regulatory T Cells to Improve Their Immunosuppressive Function. Sci Signal. 2012;5(252):ra87. doi: 10.1126/scisignal.2003365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng P, Kissler S. PTPN22 Silencing in the NOD Model Indicates the Type 1 Diabetes-Associated Allele Is Not a Loss-of-Function Variant. Diabetes. 2012 doi: 10.2337/db12-0929. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, Zahir N, Jiang Q, Miliotis H, Heyraud S, Meng X, Dong B, Xie G, Qiu F, Hao Z. et al. The autoimmune disease-associated PTPN22 variant promotes calpain-mediated Lyp/Pep degradation associated with lymphocyte and dendritic cell hyperresponsiveness. Nat Genet. 2011;43(9):902–907. doi: 10.1038/ng.904. [DOI] [PubMed] [Google Scholar]
- 23.Dai X, James R, Habib T, Singh S, Jackson S, Khim S, Moon RT, Liggitt D, Wolf-Yadlin A, Buckner JH. et al. The PTPN22 risk variant promotes systemic autoimmunity in murine models. J Clin Invest. 2013 doi: 10.1172/JCI66963. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aarnisalo J, Treszl A, Svec P, Marttila J, Oling V, Simell O, Knip M, Korner A, Madacsy L, Vasarhelyi B. et al. Reduced CD4+T cell activation in children with type 1 diabetes carrying the PTPN22/Lyp 620Trp variant. J Autoimmun. 2008;31(1):13–21. doi: 10.1016/j.jaut.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 25.Rieck M, Arechiga A, Onengut-Gumuscu S, Greenbaum C, Concannon P, Buckner JH. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J Immunol. 2007;179(7):4704–4710. doi: 10.4049/jimmunol.179.7.4704. [DOI] [PubMed] [Google Scholar]
- 26.Cao Y, Yang J, Colby K, Hogan SL, Hu Y, Jennette CE, Berg EA, Zhang Y, Jennette JC, Falk RJ. et al. High basal activity of the PTPN22 gain-of-function variant blunts leukocyte responsiveness negatively affecting IL-10 production in ANCA vasculitis. Plos One. 2012;7(8):e42783. doi: 10.1371/journal.pone.0042783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arechiga AF, Habib T, He Y, Zhang X, Zhang ZY, Funk A, Buckner JH. Cutting edge: The PTPN22 allelic variant associated with autoimmunity impairs B cell signaling. J Immunol. 2009;182(6):3343–3347. doi: 10.4049/jimmunol.0713370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Negro R, Gobessi S, Longo PG, He Y, Zhang ZY, Laurenti L, Efremov DG. Overexpression of the autoimmunity-associated phosphatase PTPN22 promotes survival of antigen-stimulated CLL cells by selectively activating AKT. Blood. 2012;119(26):6278–6287. doi: 10.1182/blood-2012-01-403162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Habib T, Funk A, Rieck M, Brahmandam A, Dai X, Panigrahi AK, Luning Prak ET, Meyer-Bahlburg A, Sanda S, Greenbaum C. et al. Altered B cell homeostasis is associated with type I diabetes and carriers of the PTPN22 allelic variant. J Immunol. 2012;188(1):487–496. doi: 10.4049/jimmunol.1102176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Menard L, Saadoun D, Isnardi I, Ng YS, Meyers G, Massad C, Price C, Abraham C, Motaghedi R, Buckner JH. et al. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J Clin Invest. 2011;121(9):3635–3644. doi: 10.1172/JCI45790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kordonouri O, Hartmann R, Badenhoop K, Kahles H, Ilonen J. PTPN22 1858T allele is associated with younger age at onset of type 1 diabetes and is not related to subsequent thyroid autoimmunity. Hum Immunol. 2010;71(7):731–732. doi: 10.1016/j.humimm.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 32.Okruszko A, Szepietowska B, Wawrusiewicz-Kurylonek N, Gorska M, Kretowski A, Szelachowska M. HLA-DR, HLA-DQB1 and PTPN22 gene polymorphism: association with age at onset for autoimmune diabetes. Arch Med Sci. 2012;8(5):874–878. doi: 10.5114/aoms.2012.31619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lempainen J, Hermann R, Veijola R, Simell O, Knip M, Ilonen J. Effect of the PTPN22 and INS risk genotypes on the progression to clinical type 1 diabetes after the initiation of beta-cell autoimmunity. Diabetes. 2012;61(4):963–966. doi: 10.2337/db11-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steck AK, Wong R, Wagner B, Johnson K, Liu E, Romanos J, Wijmenga C, Norris JM, Eisenbarth GS, Rewers MJ. Effects of non-HLA gene polymorphisms on development of islet autoimmunity and type 1 diabetes in a population with high-risk HLA-DR,DQ genotypes. Diabetes. 2012;61(3):753–758. doi: 10.2337/db11-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winkler C, Krumsiek J, Lempainen J, Achenbach P, Grallert H, Giannopoulou E, Bunk M, Theis FJ, Bonifacio E, Ziegler AG. A strategy for combining minor genetic susceptibility genes to improve prediction of disease in type 1 diabetes. Genes Immun. 2012;13(7):549–555. doi: 10.1038/gene.2012.36. [DOI] [PubMed] [Google Scholar]
- 36.Lempainen J, Vaarala O, Makela M, Veijola R, Simell O, Knip M, Hermann R, Ilonen J. Interplay between PTPN22 C1858T polymorphism and cow's milk formula exposure in type 1 diabetes. J Autoimmun. 2009;33(2):155–164. doi: 10.1016/j.jaut.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 37.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F. et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39(7):857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R. et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010;42(12):1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dubois PC, Trynka G, Franke L, Hunt KA, Romanos J, Curtotti A, Zhernakova A, Heap GA, Adany R, Aromaa A. et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010;42(4):295–302. doi: 10.1038/ng.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anderson CA, Boucher G, Lees CW, Franke A, D'Amato M, Taylor KD, Lee JC, Goyette P, Imielinski M, Latiano A. et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43(3):246–252. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cotsapas C, Voight BF, Rossin E, Lage K, Neale BM, Wallace C, Abecasis GR, Barrett JC, Behrens T, Cho J. et al. Pervasive sharing of genetic effects in autoimmune disease. Plos Genet. 2011;7(8):e1002254. doi: 10.1371/journal.pgen.1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cool DE, Tonks NK, Charbonneau H, Walsh KA, Fischer EH, Krebs EG. cDNA isolated from a human T-cell library encodes a member of the protein-tyrosine-phosphatase family. Proc Natl Acad Sci U S A. 1989;86(14):5257–5261. doi: 10.1073/pnas.86.14.5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mosinger B Jr, Tillmann U, Westphal H, Tremblay ML. Cloning and characterization of a mouse cDNA encoding a cytoplasmic protein-tyrosine-phosphatase. Proc Natl Acad Sci U S A. 1992;89(2):499–503. doi: 10.1073/pnas.89.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lorenzen JA, Dadabay CY, Fischer EH. COOH-terminal sequence motifs target the T cell protein tyrosine phosphatase to the ER and nucleus. J Cell Biol. 1995;131(3):631–643. doi: 10.1083/jcb.131.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lam MH, Michell BJ, Fodero-Tavoletti MT, Kemp BE, Tonks NK, Tiganis T. Cellular stress regulates the nucleocytoplasmic distribution of the protein-tyrosine phosphatase TCPTP. J Biol Chem. 2001;276(40):37700–37707. doi: 10.1074/jbc.M105128200. [DOI] [PubMed] [Google Scholar]
- 47.Galic S, Klingler-Hoffmann M, Fodero-Tavoletti MT, Puryer MA, Meng TC, Tonks NK, Tiganis T. Regulation of insulin receptor signaling by the protein tyrosine phosphatase TCPTP. Mol Cell Biol. 2003;23(6):2096–2108. doi: 10.1128/MCB.23.6.2096-2108.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hao L, Tiganis T, Tonks NK, Charbonneau H. The noncatalytic C-terminal segment of the T cell protein tyrosine phosphatase regulates activity via an intramolecular mechanism. J Biol Chem. 1997;272(46):29322–29329. doi: 10.1074/jbc.272.46.29322. [DOI] [PubMed] [Google Scholar]
- 49.Iversen LF, Moller KB, Pedersen AK, Peters GH, Petersen AS, Andersen HS, Branner S, Mortensen SB, Moller NP. Structure determination of T cell protein-tyrosine phosphatase. J Biol Chem. 2002;277(22):19982–19990. doi: 10.1074/jbc.M200567200. [DOI] [PubMed] [Google Scholar]
- 50.Zhang S, Chen L, Luo Y, Gunawan A, Lawrence DS, Zhang ZY. Acquisition of a potent and selective TC-PTP inhibitor via a stepwise fluorophore-tagged combinatorial synthesis and screening strategy. J Am Chem Soc. 2009;131(36):13072–13079. doi: 10.1021/ja903733z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tiganis T, Bennett AM. Protein tyrosine phosphatase function: the substrate perspective. Biochem J. 2007;402(1):1–15. doi: 10.1042/BJ20061548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tiganis T, Bennett AM, Ravichandran KS, Tonks NK. Epidermal growth factor receptor and the adaptor protein p52Shc are specific substrates of T-cell protein tyrosine phosphatase. Mol Cell Biol. 1998;18(3):1622–1634. doi: 10.1128/mcb.18.3.1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Simoncic PD, Bourdeau A, Lee-Loy A, Rohrschneider LR, Tremblay ML, Stanley ER, McGlade CJ. T-cell protein tyrosine phosphatase (Tcptp) is a negative regulator of colony-stimulating factor 1 signaling and macrophage differentiation. Mol Cell Biol. 2006;26(11):4149–4160. doi: 10.1128/MCB.01932-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Simoncic PD, Lee-Loy A, Barber DL, Tremblay ML, McGlade CJ. The T cell protein tyrosine phosphatase is a negative regulator of janus family kinases 1 and 3. Curr Biol. 2002;12(6):446–453. doi: 10.1016/s0960-9822(02)00697-8. [DOI] [PubMed] [Google Scholar]
- 55.ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, Zhu W, Tremblay M, David M, Shuai K. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol. 2002;22(16):5662–5668. doi: 10.1128/MCB.22.16.5662-5668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamamoto T, Sekine Y, Kashima K, Kubota A, Sato N, Aoki N, Matsuda T. The nuclear isoform of protein-tyrosine phosphatase TC-PTP regulates interleukin-6-mediated signaling pathway through STAT3 dephosphorylation. Biochem Biophys Res Commun. 2002;297(4):811–817. doi: 10.1016/s0006-291x(02)02291-x. [DOI] [PubMed] [Google Scholar]
- 57.Lu X, Chen J, Sasmono RT, Hsi ED, Sarosiek KA, Tiganis T, Lossos IS. T-cell protein tyrosine phosphatase, distinctively expressed in activated-B-cell-like diffuse large B-cell lymphomas, is the nuclear phosphatase of STAT6. Mol Cell Biol. 2007;27(6):2166–2179. doi: 10.1128/MCB.01234-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Vliet C, Bukczynska PE, Puryer MA, Sadek CM, Shields BJ, Tremblay ML, Tiganis T. Selective regulation of tumor necrosis factor-induced Erk signaling by Src family kinases and the T cell protein tyrosine phosphatase. Nat Immunol. 2005;6(3):253–260. doi: 10.1038/ni1169. [DOI] [PubMed] [Google Scholar]
- 59.Wiede F, Shields BJ, Chew SH, Kyparissoudis K, van VC, Galic S, Tremblay ML, Russell SM, Godfrey DI, Tiganis T. T cell protein tyrosine phosphatase attenuates T cell signaling to maintain tolerance in mice. J Clin Invest. 2011;121(12):4758–4774. doi: 10.1172/JCI59492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aoki N, Matsuda T. A nuclear protein tyrosine phosphatase TC-PTP is a potential negative regulator of the PRL-mediated signaling pathway: dephosphorylation and deactivation of signal transducer and activator of transcription 5a and 5b by TC-PTP in nucleus. Mol Endocrinol. 2002;16(1):58–69. doi: 10.1210/mend.16.1.0761. [DOI] [PubMed] [Google Scholar]
- 61.Loh K, Fukushima A, Zhang X, Galic S, Briggs D, Enriori PJ, Simonds S, Wiede F, Reichenbach A, Hauser C. et al. Elevated hypothalamic TCPTP in obesity contributes to cellular leptin resistance. Cell Metab. 2011;14(5):684–699. doi: 10.1016/j.cmet.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Galic S, Hauser C, Kahn BB, Haj FG, Neel BG, Tonks NK, Tiganis T. Coordinated regulation of insulin signaling by the protein tyrosine phosphatases PTP1B and TCPTP. Mol Cell Biol. 2005;25(2):819–829. doi: 10.1128/MCB.25.2.819-829.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Long SA, Cerosaletti K, Wan JY, Ho JC, Tatum M, Wei S, Shilling HG, Buckner JH. An autoimmune-associated variant in PTPN2 reveals an impairment of IL-2R signaling in CD4(+) T cells. Genes Immun. 2011;12(2):116–125. doi: 10.1038/gene.2010.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ramagopalan SV, Heger A, Berlanga AJ, Maugeri NJ, Lincoln MR, Burrell A, Handunnetthi L, Handel AE, Disanto G, Orton SM. et al. A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res. 2010;20(10):1352–1360. doi: 10.1101/gr.107920.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.You T, Muise ES, Itie A, Michaliszyn E, Wagner J, Jothy S, Lapp WS, Tremblay ML. Impaired bone marrow microenvironment and immune function in T cell protein tyrosine phosphatase-deficient mice. J Exp Med. 1997;186(5):683–693. doi: 10.1084/jem.186.5.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wiede F, Chew SH, van VC, Poulton IJ, Kyparissoudis K, Sasmono T, Loh K, Tremblay ML, Godfrey DI, Sims NA. et al. Strain-dependent differences in bone development, myeloid hyperplasia, morbidity and mortality in ptpn2-deficient mice. PLoS One. 2012;7(5):e36703. doi: 10.1371/journal.pone.0036703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heinonen KM, Nestel FP, Newell EW, Charette G, Seemayer TA, Tremblay ML, Lapp WS. T-cell protein tyrosine phosphatase deletion results in progressive systemic inflammatory disease. Blood. 2004;103(9):3457–3464. doi: 10.1182/blood-2003-09-3153. [DOI] [PubMed] [Google Scholar]
- 68.Bourdeau A, Dube N, Heinonen KM, Theberge JF, Doody KM, Tremblay ML. TC-PTP-deficient bone marrow stromal cells fail to support normal B lymphopoiesis due to abnormal secretion of interferon-gamma. Blood. 2007;109(10):4220–4228. doi: 10.1182/blood-2006-08-044370. [DOI] [PubMed] [Google Scholar]
- 69.Espino-Paisan L, De La Calle H, Fernandez-Arquero M, Figueredo MA, de la Concha EG, Urcelay E, Santiago JL. A polymorphism in PTPN2 gene is associated with an earlier onset of type 1 diabetes. Immunogenetics. 2011;63(4):255–258. doi: 10.1007/s00251-010-0500-x. [DOI] [PubMed] [Google Scholar]
- 70.Winkler C, Lauber C, Adler K, Grallert H, Illig T, Ziegler AG, Bonifacio E. An interferon-induced helicase (IFIH1) gene polymorphism associates with different rates of progression from autoimmunity to type 1 diabetes. Diabetes. 2011;60(2):685–690. doi: 10.2337/db10-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Frederiksen B, Liu E, Romanos J, Steck AK, Yin X, Kroehl M, Fingerlin TE, Erlich H, Eisenbarth GS, Rewers M. et al. Investigation of the vitamin D receptor gene (VDR) and its interaction with protein tyrosine phosphatase, non-receptor type 2 gene (PTPN2) on risk of islet autoimmunity and type 1 diabetes: The Diabetes Autoimmunity Study in the Young (DAISY) J Steroid Biochem Mol Biol. 2013;133:51–57. doi: 10.1016/j.jsbmb.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Butty V, Campbell C, Mathis D, Benoist C. Impact of diabetes susceptibility loci on progression from pre-diabetes to diabetes in at-risk individuals of the diabetes prevention trial-type 1 (DPT-1) Diabetes. 2008;57(9):2348–2359. doi: 10.2337/db07-1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Long SA, Cerosaletti K, Bollyky PL, Tatum M, Shilling H, Zhang S, Zhang ZY, Pihoker C, Sanda S, Greenbaum C. et al. Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4+CD25+ regulatory T cells of type 1 diabetic subjects. Diabetes. 2010;59(2):407–415. doi: 10.2337/db09-0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moore F, Colli ML, Cnop M, Igoillo EM, Cardozo AK, Cunha DA, Bugliani M, Marchetti P, Eizirik DL. PTPN2, a candidate gene for type 1 diabetes, modulates interferon-gamma-induced pancreatic beta-cell apoptosis. Diabetes. 2009;58(6):1283–1291. doi: 10.2337/db08-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Scharl M, Paul G, Weber A, Jung BC, Docherty MJ, Hausmann M, Rogler G, Barrett KE, McCole DF. Protection of epithelial barrier function by the Crohn's disease associated gene protein tyrosine phosphatase n2. Gastroenterology. 2009;137(6):2030–2040. doi: 10.1053/j.gastro.2009.07.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yamanouchi J, Rainbow D, Serra P, Howlett S, Hunter K, Garner VE, Gonzalez-Munoz A, Clark J, Veijola R, Cubbon R. et al. Interleukin-2 gene variation impairs regulatory T cell function and causes autoimmunity. Nat Genet. 2007;39(3):329–337. doi: 10.1038/ng1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, Piccirillo CA, Salomon BL, Bluestone JA. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity. 2008;28(5):687–697. doi: 10.1016/j.immuni.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Santin I, Moore F, Colli ML, Gurzov EN, Marselli L, Marchetti P, Eizirik DL. PTPN2, a candidate gene for type 1 diabetes, modulates pancreatic beta-cell apoptosis via regulation of the BH3-only protein Bim. Diabetes. 2011;60(12):3279–3288. doi: 10.2337/db11-0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Palmer ND, Bento JL, Mychaleckyj JC, Langefeld CD, Campbell JK, Norris JM, Haffner SM, Bergman RN, Bowden DW. Association of protein tyrosine phosphatase 1B gene polymorphisms with measures of glucose homeostasis in Hispanic Americans: the insulin resistance atherosclerosis study (IRAS) family study. Diabetes. 2004;53(11):3013–3039. doi: 10.2337/diabetes.53.11.3013. [DOI] [PubMed] [Google Scholar]
- 80.Bento JL, Palmer ND, Mychaleckyj JC, Lange LA, Langefeld CD, Rich SS, Freedman BI, Bowden DW. Association of protein tyrosine phosphatase 1B gene polymorphisms with type 2 diabetes. Diabetes. 2004;53(11):3007–3012. doi: 10.2337/diabetes.53.11.3007. [DOI] [PubMed] [Google Scholar]
- 81.Di PR, Frittitta L, Miscio G, Bozzali M, Baratta R, Centra M, Spampinato D, Santagati MG, Ercolino T, Cisternino C. et al. A variation in 3' UTR of hPTP1B increases specific gene expression and associates with insulin resistance. Am J Hum Genet. 2002;70(3):806–812. doi: 10.1086/339270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fukushima A, Loh K, Galic S, Fam B, Shields B, Wiede F, Tremblay ML, Watt MJ, Andrikopoulos S, Tiganis T. T-cell protein tyrosine phosphatase attenuates STAT3 and insulin signaling in the liver to regulate gluconeogenesis. Diabetes. 2010;59(8):1906–1914. doi: 10.2337/db09-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]