

Abstract
Recent clinical trials, investigating type 1 diabetes (T1D), have focused mainly on newly diagnosed individuals who have developed diabetes. We need to continue our efforts to understand disease processes and to rationally design interventions that will be safe and specific for disease, but at the same time not induce undesirable immunosuppression. T cells are clearly involved in the pathogenesis of T1D, and have been a major focus for both antigen-specific and non-antigen-specific therapy, but thus far no single strategy has emerged as superior. As T1D is a multifactorial disease, in which multiple cell types are involved, some of these pathogenic and regulatory cell pathways may be important to consider. In this review, we examine evidence for whether monocytes, B cells, and innate lymphocytes, including natural killer cells, may be suitable targets for intervention.
Keywords: type 1 diabetes, B cell, NOD, Treg cell, dendritic cell, ATG, CD3, monoclonal antibody
Abbreviations: Ab – antibody; ADA – American Diabetes Association; APC – antigen-presenting cell; ATG – anti-thymocyte globulin; BAFF – B cell activating factor; BB – BioBreeding; BCMA – B cell maturation antigen; BCR – B cell receptor; BLyS – B lymphocyte stimulator; BMDC – bone marrow-derived dendritic cell; CCR5 – C-C chemokine receptor type 5 (CD195); CD11b – complement receptor 3; CTLA-4 – cytotoxic T lymphocyte antigen 4; DC – dendritic cell; ER – endoplasmic reticulum; FO – follicular; Foxp3 – forkhead box P3; GAD – glutamic acid decarboxylase; GM-CSF – granulocyte-macrophage colony-stimulating factor; HLA – human leukocyte antigen; HSP60 – heat shock protein 60; IAA – insulin auto-antibody; IDIN – IRF7-driven inflammatory gene network; IFNγ – interferon gamma; Ig – immunoglobulin; IL – interleukin; iNKT – invariant natural killer T; iNOS - inducible nitric oxide synthase; i.p. – intraperitoneal; IRF7 – interferon regulatory factor 7; KIR – killer cell immunoglobulin-like receptor; lip-Cl2MDP – liposome-encapsulated dichloromethylene diphosphonate; mAb – monoclonal antibody; MAP – mitogen-activated protein; MHC – major histocompatibility complex; MLR – mixed leukocyte reaction; MZ – marginal zone; MZB - marginal zone B (cell); NCR – natural cytotoxicity receptor; NF-κB – nuclear factor kappa B; NK – natual killer; NKT – natural killer T; NO – nitric oxide; NOD – non-obese diabetic; NOR – non-obese resistant; PAMPS – pathogen-associated molecular patterns; PBMC – peripheral blood mononuclear cell; PLN – pancreatic lymph node; RNA – ribonucleic acid; siRNA - small interfering RNA; SLC11A1 – human solute carrier family 11 member A1; T1D – type 1 diabetes; T2MZB – type 2 marginal zone B (cell); TCR – T cell receptor; TGF-β – transforming growth factor beta; TLR – toll-like receptor; TNF-α – tumor necrosis factor alpha; Treg – T regulatory; ZnT8 – zinc transporter 8
1. Introduction
At the time of diagnosis of type 1 diabetes (T1D), many insulin-producing β-cells have been damaged in a process that has taken weeks, months, or even years to develop. There are multiple genetic loci predisposing individuals to disease [1], interacting with unknown environmental factors, in a slow process. It is well known that in prediabetes, the presence of one or two autoantibodies does not inevitably lead to diabetes, but when there are multiple autoantibodies, progression to diabetes is very likely [2]. Studies in animal models of diabetes have pointed to diverse cellular pathways that may be involved, and some of these have shown distinct parallels with the human disease. The potentially long course of time over which damage and destruction of pancreatic islet β-cells occurs suggests that many approaches might be successful if intervention could take place at an early time point. However, by the time T1D manifests, therapy is likely to be much more challenging since the immune response has diversified, with many cell types and autoantigens recognized by memory T cells, even if there is substantial β-cell function remaining.
After a long time of research into T1D pathogenesis, which will help to focus on rational interventions, we should acknowledge treatments that have already made the long journey to the clinical trial. Of the various antigen-specific interventions that have been aimed at the tolerization of T cells, targeting proinsulin by the use of insulin peptide [3], or insulin B chain in tolerogenic adjuvant [4], have been tested in early-phase trials. GAD-alum had reached the phase III clinical trial, but unfortunately clinical efficacy could not be demonstrated [5-7]. A phase III trial of HSP60 in the form of DiaPep277 was reported, at the ADA 72nd Scientific Sessions 2012, to show a 2 year reduction in the loss of C-peptide on glucagon stimulation [8]. Another study using this approach is underway. Of the non-antigen-specific therapy, anti-CD3 mAb to target T cells (teplizumab, otelixizumab) was promising in the early trial stages [9-11], but stage III trials were terminated as the primary end-point was not met [12]. The reasons for this are discussed elsewhere in this special review series of The Review of Diabetic Studies [13, 14]. Anti-CTLA4 treatment (abatacept) showed initial delay in the loss of C-peptide over the first 6 months, but thereafter the decline in β-cell function was parallel to that seen in patients treated with placebo [15]. There are currently ongoing trials of anti-thymocyte globulin (ATG) and anti-CD2 which both target T cells [16].
How are we to decide what strategies are useful? For many years, animal models, both the non-obese diabetic (NOD) mouse and the BioBreeding (BB) rat have been used to test therapies. Some of these have moved into the clinical field and allow a semblance of rationality in the choice of therapy. It is clear that strategies for therapy that are successful in early stage disease, will not necessarily be translatable at this time, as most current clinical focus is on much later points in disease pathogenesis. We should hold this consideration in mind, should preventive measures for diabetes become more of a reality. However, therapies tested in disease models, which show promise at later phases in the disease models, could be useful. We need to build on these studies, with the investigation of the human immunology, and with the development and validation of biomarkers for efficacy. Clearly, having an outcome that will ultimately prevent T cells from causing damage to islet β-cells is the major goal. However, this does not mean that T cells are the only possible targets, as manipulations of other components of the immune response may aid in this process.
2. Monocytes, macrophages, and dendritic cells
Monocytes derive from hematopoietic stem cells in the bone marrow. They are precursors to both macrophages and myeloid dendritic cells in tissues where they are important in inflammation and defense against pathogens (reviewed by Auffray and colleagues [17]). There is considerable heterogeneity in the monocyte/macrophage subset of cells, with many surface markers defining the different types [18]. Dendritic cells (DCs) are key innate immune cells that direct the fate of T cells. The many subsets of DCs all have an input into activating immune responses [19]. As antigen-presenting cells (APCs), DCs have a central role in early innate immune responses and directing the adaptive immune response. Thus, they may have a number of roles in disease processes, and are potentially important targets for therapy.
2.1 Genetics
Interestingly, there is a genetic association in the diabetes susceptibility locus Idd5.2 in the NOD mouse, which encodes Slc11a1 (formerly called Nramp1). This gene codes for a lysosomal membrane protein that is involved in acidification within lysosomes, and therefore it is important in antigen presentation by APCs such as DCs. Silencing of the gene using lentivirus, encoding siRNA for the Slc11a1 gene, reduced diabetes incidence in an NOD mouse cohort, and recapitulated the effect of a natural mutation of Idd5.2 [20]. In humans, the orthologous region SLC11A1 encodes an evolutionary highly conserved protein. Although associations with a variety of immune-mediated diseases have been reported, a recent study did not observe changes in SLC11A1 expression at the RNA level in whole blood samples from patients with T1D. However, the study did not exclude the possibility that genetic effects of polymorphisms in the gene may be seen in purified monocyte or macrophage populations [21]. In rats, Heinig and colleagues have identified an interferon regulatory factor 7 (IRF7)-driven inflammatory gene network (IDIN) [22]. In humans, there is a conserved equivalent of the rat IDIN genes expressed in monocytes [22]. IRF7 regulates the type 1 interferon response that has been linked to T1D. The genes encode proteins that are highly expressed in cells of the immune system, and the investigators suggested that these genes may regulate the innate immune response in macrophages, contributing to the risk of T1D.
2.2 Studies in animal models
In animal models of autoimmune diabetes, macrophages are amongst the earliest cells that infiltrate islets in the BB rat [23-25]. Similarly, in the NOD mouse, characteristic patterns of dendritic-like cells and macrophages infiltrate into islets before lymphocytes [26]. The macrophages in NOD mice are reported to have a defect in phagocytosis of apoptotic cells [27, 28]. Moreover, they have an abnormal inflammatory response, producing increased amounts of inflammatory cytokines that include IL-1β and TNF-α when encountering apoptotic cells, compared with non-obese resistant (NOR) or C57BL/6 mice [29]. Targeting macrophages was one of the earliest therapeutic strategies shown to inhibit diabetes in the NOD mouse. Antibody against the complement receptor 3 (CD11b) expressed on macrophages prevented diabetes development in an adoptive transfer system in sublethally irradiated NOD mice [30]. Studies using silica, or liposome-encapsulated dichloromethylene diphosphonate (lip-Cl2MDP) to deplete macrophages also protected NOD mice from developing spontaneous diabetes [31]. The treatment reduced IL-12, IL-1β, and TNF-α in splenic macrophages, and there was a shift from a Th1 cytokine profile to Th2 in splenocytes and a reduction in the development of islet-reactive cytotoxic T cells [31].
Recent attempts at in situ macrophage-targeting using siRNA against Alox-15 have shown that this therapy is useful when given at an early phase in disease, similar to previous studies, but not effective when applied after 9 weeks of age [32].
2.3 Studies in humans
Macrophages are seen in human islet infiltrates in post-mortem sections of pancreas obtained from patients with diabetes, both at time of onset of disease [33] and later [34]. Furthermore, there have been many studies that focused on monocytes and their phenotypic changes in T1D. Differences in the maturation and function of monocyte-derived APCs have been suggested to be a reason for defective activation of regulatory cells in patients with diabetes [35]. In comparison with healthy first-degree relatives of T1D patients, there are raised serum cytokines from monocytes prior to the onset of diabetes [36]. Furthermore, monocytes from patients with T1D have an inflammatory phenotype, with increased IL-6 and IL-1β production. These cytokines can then stimulate the production of inflammatory IL-17-producing T cells [37]. The conversion to IL-17 cells in humans and mice appears to be different, with differentiation induced by IL-1β and IL-6, but not IL-12 or TGF-β, whereas TGF-β is critical for driving naïve CD4 T cells towards the Th17 lineage in mice [38].
Toll-like receptors (TLRs) are innate immune receptors that are expressed on both immune, especially APCs, and a variety of other cells in the body. They detect pathogen-associated molecular patterns (PAMPS). Interest has been generated in these receptors in connection with the activation of immune cells in diabetes. Altered surface expression of TLR2 (recognizes components of gram-positive bacteria) and TLR4 (recognizes lipopolysaccharide) has been reported in monocytes from patients with T1D [39]. The monocytes have been studied in conditions of raised blood glucose. Increased secretion of the chemokine IP-10 was found, which is important in the homing of T cells, and it is associated with diabetes [40]. It has also been suggested that signaling via the TLR pathways is aberrant in patients with T1D [41], with increased production of IL-1β from monocytes and decreased IL-6 from myeloid DCs, following stimulation of monocytes through TLR4. Similar alterations have been observed in autoantibody-positive individuals at risk of developing T1D compared with those who are autoantibody negative [42].
These phenotypic studies have become more complex and wide-ranging with our increased abilities to investigate a very large range of cellular constituents in terms of gene expression profiles. Using a novel assay to examine gene expression signatures, Wang and colleagues took sera from newly diagnosed patients with diabetes, and tested this by incubating it with unrelated peripheral blood mononuclear cells (PBMC) to look for a gene expression signature induced in the PBMC by serum from the new-onset T1D patients [43]. Comparison was made with serum taken from healthy control subjects and patients with long-standing diabetes. They found that a unique expression signature was induced by the serum of new-onset patients that was also found in a small number of autoantibody-positive siblings, and that it was no longer present in long-standing patients [43]. Altered genes included IL-1 cytokine family members and chemokines amongst other molecules [43]. This has been further elaborated to indicate that the patterns are distinct from other inflammatory conditions [44]. The investigators suggested that detection of these changes have the potential to be used as unique disease identifiers that could improve disease prediction [44].
Using purified CD14+ monocytes, Irvine and colleagues showed differences in monocyte gene expression patterns in children with newly-diagnosed diabetes compared with healthy control subjects, in addition to the finding of a reduction in CD14hiCD16+ monocytes and an increase in CD14lo/CD16- monocytes in these patients [45]. The differences of gene expression include upregulation of endoplasmic reticulum (ER)-nuclear signaling pathways, negative regulation of caspase activity, together with cell adhesion genes and downregulation of negative regulators of NF-κB, again pointing to a distinct molecular signature [45].
Thus, currently, there are a variety of observations that suggest that the monocyte/macrophage/dendritic cell pathways have altered activity in T1D. Some of these appear to be intrinsic, i.e. potentially genetically determined, while others may be a response to inflammatory and metabolic changes. However, there is clearly involvement of these cells in the pathogenic process, and this suggests that there may be merit in considering them as potential targets for therapy.
2.4 Potential targets
Cytokines produced by macrophages. How might these important accessory cells be targeted? Macrophages are major producers of IL-1β that triggers the NF-κB and MAP kinase signaling pathways in pancreatic islets. IL-1β is toxic to β-cells in vitro as shown by a number of studies, particularly in combination with IFN-γ and TNF-α. However, this cytokine cocktail had different effects on human islets compared to rodent islets [46, 47]. Furthermore, IL-1β plays a synergistic role with other molecules, inducing nitric oxide (NO) that also damages islets [48, 49]. IL-1β clearly has a role in a number of inflammatory diseases, including arthritis and autoinflammatory syndromes. Various means of antagonizing the effects of IL-1 have been developed, of which IL-1 receptor antagonist (anakinra) and IL-1 trap (rilonacept), a long acting IL-1 blocking agent, have been approved for human use. Some of the data from animal models have suggested that antagonizing IL-1 may be beneficial, and that this could be done with either soluble IL-1 receptor or IL1 receptor antagonist. However, it was not very effective in the NOD mouse, but improved efficacy was seen when it was used with low dose anti-CD3 treatment [50]. It has also been shown that knocking out IL-1 in NOD mice had little effect on diabetes development [51, 52]. However, there has been success in the use of IL-1 antagonism in a variety of clinical conditions. It has been shown to be safe [53]. Two clinical studies were recently completed that targeted IL-1, one using IL-1 receptor antagonist (anakinra) and the other using an antibody to IL-1 (canakinumab), and neither had shown any efficacy when used at the time of diabetes onset (reported at ADA 72nd Scientific Sessions 2012 and Immunology of Diabetes Society meeting 2012). It is not known whether there may have been a different outcome if these agents could have been used at an earlier stage of disease. This subject will be addressed in more detail in another review by Thomas Mandrup-Poulsen in this series [54].
In vivo treatment to generate tolerogenic antigen-presenting cells. If the cytokine products of macrophages cannot be effectively neutralized, another option would be to alter the phenotype of the monocytes and macrophages by mutually targeting the cells. The aim of this approach would be to generate APCs that are programmed to tolerize T cells and stimulate the production of regulatory cells. Given the importance of these APCs in immune responses, this goal of inducing immune tolerance to unwanted effects, while at the same time maintaining effectiveness in dealing with infections, is a considerable challenge.
Vitamin D has an impact on many cells as the vitamin D receptor is expressed in nucleated cells. The best known effects are on bone and calcium metabolism. However, there are also effects on immune cells, including DCs and macrophages, and these may in turn alter the activation of T cells [55]. Treatment of prediabetic NOD mice from an early age, with vitamin D and an analogue of Vitamin D that did not induce hypercalcemia, considerably reduced the incidence of autoimmune diabetes. Cells from treated mice could also suppress diabetes development induced in an immunosuppressed host by adoptive co-transfer [56, 57]. There has been considerable interest in the role of vitamin D and its effects in the generation of tolerogenic DCs. Mouse DCs, generated in the presence of 1,25 dihydroxy vitamin D3, the active form, expressed lower levels of MHC class II and costimulatory molecules, together with increased chemokine receptor CCR5 and antigen-uptake receptor DEC205, compared with those DCs that are matured in the absence of 1,25 dihydroxy vitamin D3 [58]. Vitamin D can also alter the macrophage phenotype as treatment of murine peritoneal macrophages with 1,25 dihydroxy vitamin D3 reduces proinflammatory cytokines and other mediators that include IL-12p40, inducible nitric oxide synthase (iNOS), and TNF-α upon antigen stimulation [59]. These macrophages have a reduced capacity to activate antigen-specific BDC2.5 T cells. This was suggested to be partly dependent on IL-10 [59].
The immunomodulatory effects of 1,25 dihydroxy vitamin D3 in humans have been known for many years. When monocytes are cultured in the presence of 1,25 dihydroxy vitamin D3, CD14 remained high but CD1a, CD83, and HLA-DR were expressed at a low level, with concomitant changes in costimulatory molecules. The phenotype of the treated monocytes appeared to resemble unstimulated monocytes, providing a population of relatively immature DCs, which were less able to stimulate T cell proliferation [60].
Wide-ranging effects of vitamin D include effects on pancreatic β-cells where a decrease in expression of chemokines and cytokines was seen when NOD mice were treated with 1,25 dihydroxy vitamin D3 [61]. Moreover, vitamin D3 protects pancreatic β-cells from apoptotic death through modulating inflammatory cytokines on the cells, induction of A20 which is an apoptotic protein, and the reduction of Fas on human islets [62, 63].
Given the potential for immunomodulation of both DCs and macrophages derived from monocytes by vitamin D metabolites, could these be targeted by vitamin D or an analogue? These DCs and macrophages may then be able to stimulate regulatory T cells. The studies using vitamin D in humans have been relatively small scale (reviewed in [64]). There have been some effects on reducing the risk of T1D when vitamin D supplements are given in the first year of life. In recently diagnosed T1D patients, however, there was no improvement or delay of the decline in C peptide. Given these small effects, it is likely that, if vitamin D is effective, this would be at the earlier stage in prevention. Only a large scale trial would be able to prove this, and it remains to be seen whether this will be a viable option.
Tolerogenic dendritic cells. Could the beneficial effects of tolerogenic monocyte-derived macrophages/DCs be harnessed as a cellular therapy? DCs targeted in NOD mice have also been shown to be of critical importance. The activation state of the DCs is of major importance in determining whether T cells become activated or tolerized [65]. Immature DCs with the ability to tolerize T cells have been used to stimulate and maintain regulatory T cells. This role has major importance in the consideration of the use of tolerogenic APCs in immunotherapy. A variety of DC-based therapies have been reported in NOD mice that have had varying degrees of success in preventing diabetes. The best success has been achieved when transferred early in the disease process. We have previously shown that IL-10 conditioning induces the differentiation of tolerogenic bone marrow-derived dendritic cells (BMDCs) that inhibit spontaneous diabetes, and that can protect against diabetes in the NOD mouse when administered very early in disease, before 7 weeks of age [66]. The IL-10-conditioned BMDCs reduced antigen-specific responses in vitro and in vivo. They stimulated the accumulation of B220+ plasmacytoid DCs in the islets, and reduced insulitis [66]. Others have also shown that the transfer of DCs stimulated with GM-CSF/IL4 was able to prevent diabetes, but again this required treatment to be given when the mice were 5 weeks old [67]. If the DCs were first transduced with IL-4, delivered in adenovirus vectors, treatment was more effective up to 10 weeks of age [67]. The IL-4-transduced DCs altered the intra-pancreatic cytokines.
A phase 1 study has been carried out where "immunosuppressive" DCs were generated using autologous monocytes from elutriation cultured in the presence of IL-4 and GM-CSF together with phosphorothioate-modified antisense oligonucleotides targeting CD40, CD80, and CD86, and then administered 4 times over 2 months intradermally. There were no significant adverse effects. Although this was primarily a safety study, it was noted that serum IL-4 and IL-10 were increased together with B220+ cells, involving a population that suppressed cytokine responses in mixed leukocyte reactions (MLRs). There was no generalized immunosuppression, as evidenced by the maintenance of responses in MLRs in vitro. PBMCs from the DC-treated patients responded in a similar manner to those seen at baseline, and there was no alteration of responses to viral peptide antigens [68]. This may be a promising treatment although it is still in an early phase, and its efficacy is not currently known. The treatment would require specialist preparation, and is much more costly. These cellular interventions, requiring the infusion of pre-prepared tolerogenic cells, could certainly hold promise for some patients, but compared with other strategies which exert their effects in vivo, they are likely to benefit a smaller number of patients overall.
Thus, at present, there are some potentially interesting options when considering monocyte, macrophage, and dendritic cell targets for therapy. These could include targeting innate immune receptors to reduce inflammatory responses, and means of skewing dendritic cells and macrophages towards a more "tolerogenic" phenotype. Currently, treatment of this nature may be very effective in vitro, but would require a more basic understanding of the effects in vivo before they are likely to be translatable to human therapy. Preclinical studies directed at monocyte, macrophage, and dendritic cell targets appear to be most effective early in the pathogenesis of disease, and it is possible that a therapy targeting these cells would be more appropriately used in prevention regimens.
3. B cells
3.1 Studies in animal models
The B cell is a target of therapy in T1D. Anti-B cell therapy has both delayed and prevented diabetes in NOD mice when given at early stages. It restored normoglycemia in a proportion of mice after the onset of diabetes. The strategies have included the use of anti-CD20 [69, 70], calicheamicin toxin-conjugated anti-CD22 [71], anti-BAFF [72], and BCMA-Fc [73], as summarized in Table 1.
Table 1. Summary of non-antigen specific B cell depletion studies in NOD mice.
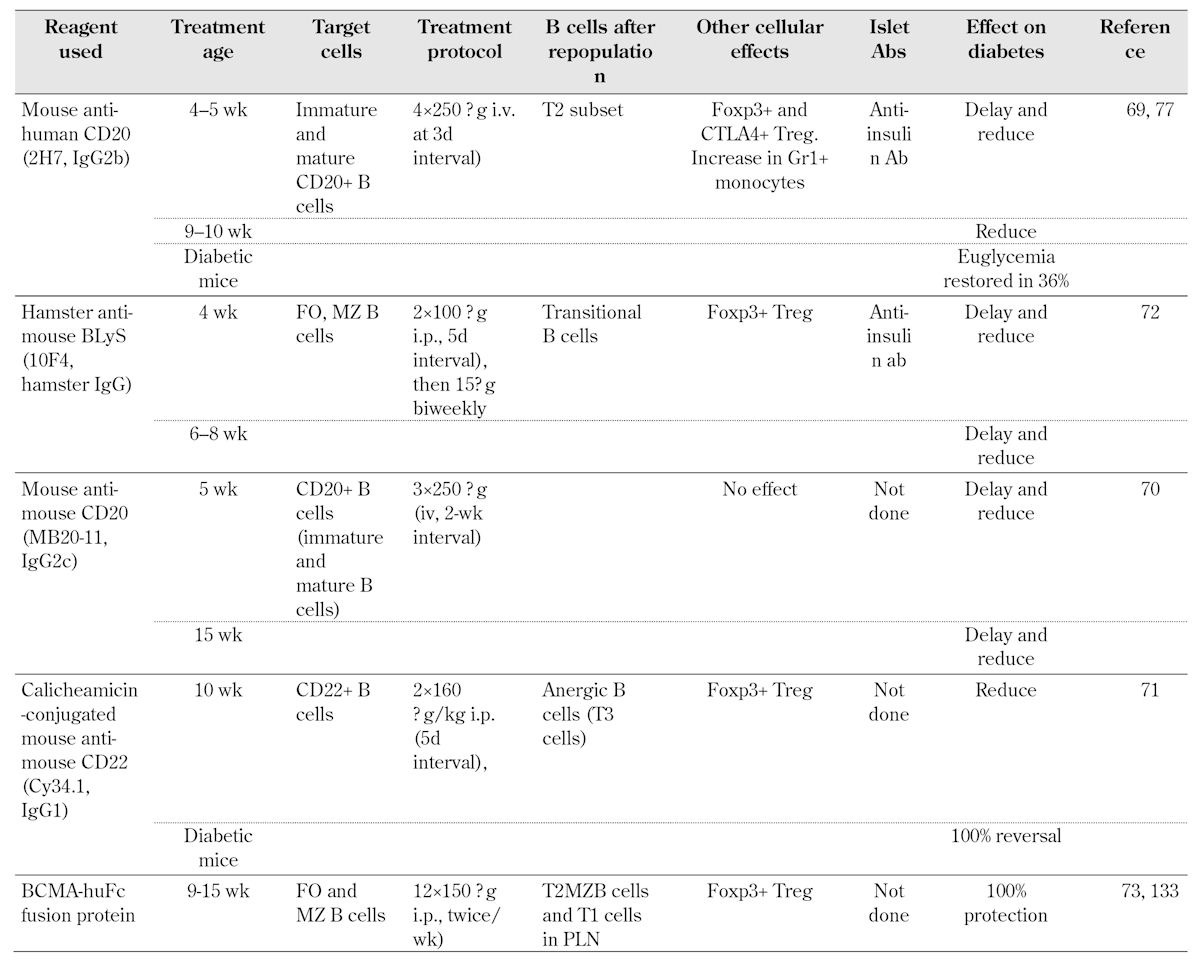
Legend: Ab – antibody, Foxp3 – forkhead box P3, CTLA-4 – cytotoxic T lymphocyte antigen 4, Treg – T regulatory, BCMA – B cell maturation antigen, BLyS – B lymphocyte stimulator, Ig – immunoglobulin, MZ – marginal zone, FO – follicular, T2MZB - type 2 marginal zone B (cell), i.p. – intraperitoneal, PLN – pancreatic lymph node.
3.2 Studies in humans
Depletion of B cells using anti-CD20 (rituximab) has already had partial success in early phase clinical trials [74]. It is now believed that many individuals presenting with diabetes may have a significant number of remaining β-cells. Whilst not producing sufficient insulin, these may be preserved at the time of onset of diabetes, and although not functioning normally, they are not destroyed. Within the first 3 months after rituximab treatment, there was a greater preservation of C-peptide responses in the Rituximab group, whereas the rate of decline after this time was parallel to control subjects [74]. Since the original studies, further investigation of the effects following treatment, as well as refinements and new treatments, have continued to maintain interest in the targeting of B cells as a potential therapy for T1D. However, there is need for caution.
What happens after B cell depletion using anti-CD20, during reconstitution, and when B cells are finally restored? In the phase II rituximab study in T1D [74], 87 patients with new onset T1D were treated with rituximab weekly over 4 weeks. There was a slower decline in mixed meal tolerance-stimulated C-peptide levels of the rituximab treated group compared with the placebo group. The B lymphocytes took many months to reconstitute. At 12 months after the treatment period, naïve B cells, defined as CD24+, IgD+ CD19+, CD38-, and CD10-, had returned to >80% of the baseline level. The switched memory B cells, defined as CD19+, CD27+, CD1c+/-, IgM+/-, and IgD-, remained reduced at an average of 40% of the initial baseline [75]. This has raised concerns about the level of immunosuppression using this treatment. In a follow-up study to assess the effects on antibody responses, patients from both active treatment and placebo arms of the study were immunized with diphtheria/tetanus and hepatitis A vaccines 12 months after the initial rituximab treatment, and pre-existing antibody responses to measles, mumps, and rubella were assessed [75]. In addition, bacteriophage phiX174 was given during the depletion phase at 2 weeks, after the end of the rituximab infusion period, and again at 6 weeks after this, to examine responses to a T lymphocyte-dependent antigen. Plasma cells do not express CD20, and as expected, there was no effect on the circulating antibody responses that had been induced prior to rituximab treatment. Responses to immunization with the diphtheria/tetanus and hepatitis A vaccines were seen in the rituximab-treated group, and were considered to give adequate protection, although they were lower than those achieved in the control group [75]. There was no response to phiX174 at the early time point after rituximab treatment, and a reduced response when the treatment was given after one year. However, the responses to immunization returned to normal upon B cell recovery [75]. These results certainly have important implications regarding concerns about the length of time that it takes to repopulate B cells. The resulting immunosuppression may preclude general introduction of this type of treatment to deplete B cells. Interestingly, rituximab treatment suppressed anti-insulin auto-antibodies (IAAs) more strongly than anti-GAD or anti-ZnT8 antibodies. This suppression continued for at least a year [76]. It was also observed that, independent of the treatment the subjects received, the level of IAAs was lower in those that maintained better C-peptide levels [76].
The effects on B cells immediately following depletion in the rituximab study have been well documented. Given the considerable interactions with T cells and other cells of the innate immune response, it is interesting to note other aspects of B cell treatment and effects after reconstitution. In mouse studies, following B cell depletion, when the B cells were restored, there was an increase in T regulatory cells [69, 71-73], a transitional population of B cells [69], and a regulatory population of monocyte-derived cells [77] (Table 1). In the rituximab trial, in those individuals who responded to the rituximab treatment, the number of CD4+ T cells had increased. Moreover, the number of regulatory T cells, defined as CD4+CD25+CD62L+ cells, was also greater 12 weeks after treatment, although this was not maintained over the first year. Interestingly, there was also a greater T cell proliferative response to 9 out of 12 antigens tested, which included neuronal, islet, and milk peptides in these individuals [78]. It is not clear in the human studies, what the underlying reason for this is. In the NOD mouse, when B cells were depleted in the TCR transgenic BDC2.5 mice (where the T cells recognize a peptide of chromogranin A), surprisingly, the BDC2.5 CD4+ T cells were more aggressive during the period when the B cells were depleted [79]. This may indicate that, in addition to the depletion of pathogenic B cells, B cells that have a regulatory function are also depleted. During this phase, there may be some expansion of autoreactive T cells, while B cells are depleted, and this may account for the fact that the improvement in C-peptide was not sustained in the human study.
3.3 Other strategies for targeting B cells
B cell signaling pathway. Following on from these B cell depletion experiments, there have also been some newer preclinical studies, with strategies that may hold some promise for therapeutic targeting of B cells. The spleen tyrosine kinase (Syk), which is important for B cell signaling and FcγR-mediated responses, can be targeted using a selective inhibitor R788 (fostamatinib). This is an orally administered small molecule that is converted to R406, which has been used in phase II clinical trials in rheumatoid arthritis [80] and immune thrombocytopenic purpura [81]. To test whether this agent may show promise in diabetes, in a study in NOD mice, R788 reduced B cells to nearly half the number one to three months after treatment, with reduced follicular B cells and correspondingly increased marginal zone B cells, while activated B cells and plasma cells were unchanged [82]. IL-10-producing B cells, which have been shown to have regulatory properties, were increased in spleen and peritoneal cavities, although the absolute numbers did not change. Coincident with these changes, DC numbers were decreased in the spleen and pancreatic lymph nodes, and in parallel, regulatory T cells appeared to be decreased. The treatment with R788 at 6 weeks of age in NOD mice delayed and prevented diabetes in a dose-dependent manner [82]. When initiated at a later stage, after the onset of glucose intolerance detected by intra-peritoneal glucose tolerance test, progression of diabetes was also delayed. However, once diabetes was established, the drug did not restore glucose tolerance [82]. Therefore, this treatment would hold more promise for an early stage therapeutic.
Antigen-specific B cell therapy. Development of antigen-specific T cell therapy is still a subject for investigation, as its specificity is attractive. Could the same apply for B cells and might antigen-specific B cell-targeted therapy be a viable option? Antigen-specific B cells, like antigen-specific T cells, are present at a low frequency, and B cells as a total population only make up 5-10% of peripheral blood mononuclear cells in humans. In NOD mice, Henry and colleagues showed that when antigen-specific B cells recognizing insulin were targeted with a specific monoclonal antibody, commencing treatment at 3 weeks of age, then diabetes incidence was considerably reduced [83]. It was not effective when the frequency of insulin-specific B cells was greater than the low frequency found in wild type NOD mice. This was illustrated by the use of insulin antibody depletion in mice transgenic for the heavy chain of the 125 insulin-specific B cell, or 125 transgenic mice expressing both heavy and light chains of the 125 B cell receptor. In the transgenic mice, the anti-insulin antibody treatment did not have any effect on diabetes, and was not sufficient to fully deplete the insulin-reactive B cells. It is not known whether this type of antigen-specific anti-B cell therapy would be efficient if commenced at a later time point [83].
Tolerogenic B cells. A B cell gene therapy approach has been used to induce antigen-specific tolerance, where B cells are transduced with a retroviral vector encoding a target peptide antigen fused with an IgG heavy chain carrier. These B cells have been shown to be tolerogenic in a variety of preclinical models of human autoimmune diseases, including autoimmune diabetes in NOD mice [84]. Treatment with transduced activated B cells reduced the incidence of diabetes when treatment was commenced at 7 and 10 weeks, with constructs expressing either GAD or insulin B chain amino acids 9-23 peptide [84]. However, if treatment was commenced later at 14 weeks, there was no protection from diabetes. Further experiments have shown that transduction of antigen-specific B cells does not have any effect on ameliorating the disease. The results indicate that this means of generating tolerogenic B cells does not overcome the pathogenicity of B cells which have pre-existing antigen specificity [85].
Are the results seen thus far with anti-B cell therapy sufficient to continue to explore this therapeutic avenue, or will the limited time period over which the treatment is effective and the safety issues preclude anti-B cell therapy? This depends on the individual anti-B cell therapy used, and the repopulation characteristics of the treatment. Moreover, it will be particularly important to continue to explore therapies against antigen-specific B cells. This would complement means of targeting pathogenic B cells, while preserving or boosting any regulatory activity that may be present, rather than total depletion of all B cells.
4. Gamma delta T cells
Gamma delta (γδ) T cells are a small subset of T cells that express a distinct T cell receptor (TCR) compared with the majority of T cells that express α and β TCR chains, hence, αβ T cells [86, 87]. Unlike αβ T cells, the antigen recognition of γδ T cells is mostly not restricted to antigen processing and presentation by classical MHC molecules on APCs [87]. It has been hypothesized that γδ T cells recognize antigen patterns, similar to innate immune cells [88]. γδ T cells are abundant in tissues at the interface with the external environment including skin, respiratory tract, and intestine [89-94], and they could be considered to be innate T cells. Studies have shown that γδ T cells contribute to the immunopathogenesis of autoimmune diseases including T1D in both mouse models and in patients [95-97].
It is interesting that insulin B9-23-reactive γδ T cell clones were isolated from spleen and pancreatic draining lymph node (PLN) of NOD mice [98]. Other investigations have shown elevated levels of γδ T cells in both NOD mice and patients with T1D [95-97], although it is not clear whether the γδ T cells, in particular insulin-reactive γδ T cells, are pathogenic or regulatory. However, since γδ T cells are abundant in mucosal tissue, Harrison and colleagues showed that delivery of proinsulin by the respiratory or digestive tracts could induce potent regulatory γδ T cells that prevented diabetes development in NOD mice [99-101]. Moreover, these regulatory γδ T cells could also prevent diabetes development induced by diabetogenic cells through adoptive transfer [99-101]. The regulatory mechanism of these γδ T cells is likely to be mediated by IL-10 as these cells strongly resemble the induced regulatory Tr1 αβ T cells [99]. It is conceivable that the regulatory γδ T cells provide a new therapeutic approach for prevention and treatment of T1D. However, it is clear that more studies are needed, particularly to define their biology, in order to discover whether they may be important in humans and if so, how to target such cells to increase their number.
5. Natural killer cells
Natural killer (NK) cells have been investigated at both cellular and molecular levels, and the number of studies in recent years has significantly increased. The discovery of activating receptors, natural cytotoxicity receptors (NCRs), and the inhibitory killer cell immunoglobulin-like receptors (KIRs), on NK cells has further promoted the research in NK cell biology [102-109]. NK cells are innate immune cells that do not express gene-rearranged receptors such as TCR and B cell receptors (BCR) for specific antigen recognition. The central function of the adaptive immune response mediated by T and B cells is to react against "non-self" recognized via TCR or BCR. However, the expression of NCRs and especially KIRs on NK cells enables them to react against "altered self" including virally-infected cells in the host [110]. It is also possible that pancreatic β-cells could express "altered self" caused by viral infection or environmental stress. Therefore, it is possible that NK cells play a critical role in immune tolerance to autoimmunity.
The role of NK cells in the immunopathogenesis of T1D has been studied in both mouse models and patients with T1D [111-114]. Most of these studies showed that NK cells indeed play an important role in immmunopathogenesis of T1D, although some studies suggested that NK cells were not required for the development of disease [115]. It is interesting that Mandelboim and colleagues recently demonstrated that both mouse and human islet β-cells express an, as yet unidentified, ligand for the NKp46 activating receptor on NK cells [116]. As blocking the binding of NKp46 to β-cells prevented diabetes development in NOD mice the investigators suggested that binding the ligand on β-cells would lead to NK cell activation and β-cell destruction [116, 117]. Their study suggested that targeting NK cells might be a new and an additional therapeutic approach for prevention and treatment of T1D development.
Human studies have revealed that patients with T1D feature a reduction in NK cell numbers and impaired NK cell functions [113, 114]. The cause of the altered phenotype of NK cells in T1D is unclear. In a longitudinal study, Gillespie and colleagues demonstrated that an increased frequency of human KIR and HLA-C group 1 was significantly correlated with early onset of T1D and a sharp rise in the incidence over the past half century [118].
An interesting study using a T cell transgenic mouse model of T1D revealed that regulatory T cells exert tight control of the expansion and function of diabetogenic NK cells. This was illustrated by removing Foxp3+ regulatory T cells which led to rapid diabetes development and considerable NK cell infiltration in pancreatic islets [119]. Regulatory T cell therapy is currently in clinical trial for treating patients with new-onset T1D [16]. Enhancing regulatory T cell number and function would also tame the diabetogenic NK cells. It will be interesting to observe whether NK cells are altered as a result of this treatment.
6. Natural killer T cells
There are a number of cells that fall into this category of innate lymphocytes. CD1d-restricted natural killer T (NKT) cells have functions different from cytotoxic NK cells. Type 1 or invariant NKT cells (iNKT) express a TCR using an invariant alpha chain (Vα24-Jα18 in humans and Vα14-Jα18 in mice) together with a restricted set of β chains, and they produce a large amount of IL-4 and other cytokines upon activation [120]. They recognize glycolipid antigens presented by CD1d. Type 2 NKT cells are also CD1d-restricted but have more diverse T cell receptors [121], and they target sulphatide [122] amongst other lipid antigens.
A number of studies have shown that NKT cells in NOD mice are impaired in both number and function (reviewed in [123, 124]). Improving the function of this subset protects against diabetes development in NOD mice [125-129]. Targeting invariant NKT cells is potentially an attractive approach for an alternative immunotherapy. Invariant NKT cells are stimulated by the glycolipid α-GalactosylCeramide (α-GalCer), and initially this was proposed to be a possible therapeutic agent to boost these cells [130]. Very recently, sulphatide that stimulates type II NKT cells has also been proposed as a possible therapy [131].
Whilst the promotion of cytokine production, particularly IL-4, may be attractive to counteract the Th1 dominance in T1D, inducing such a change in cytokine balance also raises the risk of enhancing Th2-mediated immunopathology including allergy. Possible applications of therapy that stimulates NKT cells in T1D and other autoimmune diseases, and potential problems of this immunotherapy, have been very recently reviewed by Simoni and colleagues [132]. Whilst preclinical studies suggest that therapy directed at NKT cells is of potential clinical interest, there are considerations of safety and efficacy which need to be further explored if it were to be used for human immunotherapy in T1D.
7. Concluding remarks
It is clear that there are several potential immune therapies beyond T cell therapy (see Figure 1). However, more basic and applied research is required to understand the biological processes and to test for the safety and efficacy of new therapies. Many treatments highlighted in preclinical studies are only effective in early stages of the disease process; they are likely to be more effective in the prevention of human T1D. There are still insufficient biomarkers, other than multiple autoantibodies, to predict the future development of disease. Clearly, research should continue to be focused on this area. The smaller number of therapies that are effective at later stages in the preclinical models will require rigorous testing and understanding of how their effects may differ in humans compared with rodents. The results of the recent clinical trials provide us with some important messages. Firstly, a combination of different interventions may be more effective as there is unlikely to be a "magic bullet" (i.e., a monotherapy) for a multifactoral disease such as T1D. Secondly, the approaches that target immune cells other than T cells currently are likely to be more effective in prevention of T1D. We will need to continue the search for safe and effective agents that can be used in both early and later phases of disease.
Figure 1. Summary of potential immune targets for immunotherapy in type 1 diabetes.
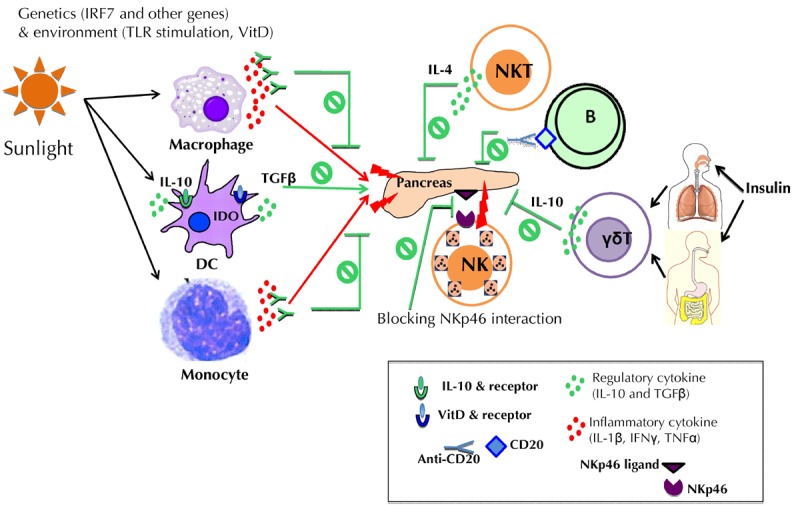
The figure summarizes the immune targets highlighted in the text that could be manipulated for immunotherapeutic purposes. These targets include antigen-presenting cells (monocytes), macrophages, and dendritic cells (DCs), as well as B cells, innate lymphocytes (γδT cells), natural killer (NK) cells, and NKT cells. Abbreviations: IDO – indolamin-2,3-dioxygenase, IFNγ – interferon gamma, IL – interleukin, IRF7 – interferon regulatory factor 7, NKp46 – natural killer cell p46-related protein, TGFβ – transforming growth factor beta, TLR – toll-like receptor, VitD – vitamin D.
Disclosure: The authors report no conflict of interests.
Acknowledgments
Studies in the laboratory of FSW are funded by the Medical Research Council (UK), Diabetes UK, the European Foundation for the Study of Diabetes, the Diabetes Research and Wellness Foundation, and the European Union. Studies in the laboratory of LW are funded by the NIH and Juvenile Diabetes Research Foundation.
References
- 1.Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C. et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41(6):703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gardner SG, Gale EA, Williams AJ, Gillespie KM, Lawrence KE, Bottazzo GF, Bingley PJ. Progression to diabetes in relatives with islet autoantibodies. Is it inevitable? Diabetes Care. 1999;22(12):2049–2054. doi: 10.2337/diacare.22.12.2049. [DOI] [PubMed] [Google Scholar]
- 3.Thrower SL, James L, Hall W, Green KM, Arif S, Allen JS, Van-Krinks C, Lozanoska-Ochser B, Marquesini L, Brown S, Wong FS, Dayan CM, Peakman M. Proinsulin peptide immunotherapy in type 1 diabetes: report of a first-in-man Phase I safety study. Clin Exp Immunol. 2009;155(2):156–165. doi: 10.1111/j.1365-2249.2008.03814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orban T, Farkas K, Jalahej H, Kis J, Treszl A, Falk B, Reijonen H, Wolfsdorf J, Ricker A, Matthews JB, Tchao N, Sayre P, Bianchine P. Autoantigen-specific regulatory T cells induced in patients with type 1 diabetes mellitus by insulin B-chain immunotherapy. J Autoimmun. 2010;34(4):408–415. doi: 10.1016/j.jaut.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ludvigsson J, Faresjo M, Hjorth M, Axelsson S, Cheramy M, Pihl M, Vaarala O, Forsander G, Ivarsson S, Johansson C, Lindh A, Nilsson NO, Aman J, Ortqvist E, Zerhouni P, Casas R. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N Engl J Med. 2008;359(18):1909–1920. doi: 10.1056/NEJMoa0804328. [DOI] [PubMed] [Google Scholar]
- 6.Ludvigsson J, Hjorth M, Cheramy M, Axelsson S, Pihl M, Forsander G, Nilsson NÖ, Samuelsson BO, Wood T, Aman J, Ortqvist E, Casas R. Extended evaluation of the safety and efficacy of GAD treatment of children and adolescents with recent-onset type 1 diabetes: a randomised controlled trial. Diabetologia. 2011;54(3):634–640. doi: 10.1007/s00125-010-1988-1. [DOI] [PubMed] [Google Scholar]
- 7.Ludvigsson J, Krisky D, Casas R, Battelino T, Castano L, Greening J, Kordonouri O, Otonkoski T, Pozzilli P, Robert JJ. et al. GAD65 antigen therapy in recently diagnosed type 1 diabetes mellitus. N Engl J Med. 2012;366(5):433–442. doi: 10.1056/NEJMoa1107096. [DOI] [PubMed] [Google Scholar]
- 8.Huurman VA, van der Meide PE, Duinkerken G, Willemen S, Cohen IR, Elias D, Roep BO. Immunological efficacy of heat shock protein 60 peptide DiaPep277 therapy in clinical type I diabetes. Clin Exp Immunol. 2008;152(3):488–497. doi: 10.1111/j.1365-2249.2008.03656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, Gitelman SE, Harlan DM, Xu D, Zivin RA, Bluestone JA. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346(22):1692–1698. doi: 10.1056/NEJMoa012864. [DOI] [PubMed] [Google Scholar]
- 10.Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, Gorus F, Goldman M, Walter M, Candon S. et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005;352(25):2598–2608. doi: 10.1056/NEJMoa043980. [DOI] [PubMed] [Google Scholar]
- 11.Keymeulen B, Walter M, Mathieu C, Kaufman L, Gorus F, Hilbrands R, Vandemeulebroucke E, Van de Velde U, Crenier L, De Block C, Candon S, Waldmann H, Ziegler AG, Chatenoud L, Pipeleers D. Four-year metabolic outcome of a randomised controlled CD3-antibody trial in recent-onset type 1 diabetic patients depends on their age and baseline residual beta cell mass. Diabetologia. 2010;53(4):614–623. doi: 10.1007/s00125-009-1644-9. [DOI] [PubMed] [Google Scholar]
- 12.Sherry N, Hagopian W, Ludvigsson J, Jain SM, Wahlen J, Ferry RJ Jr, Bode B, Aronoff S, Holland C, Carlin D. et al. Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial. Lancet. 2011;378(9790):487–497. doi: 10.1016/S0140-6736(11)60931-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chatenoud L, Waldman H. CD3 monoclonal antibodies: a first step towards operational immune tolerance in the clinic. Rev Diabet Stud. 2012;9(4):372–381. doi: 10.1900/RDS.2012.9.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nepom GT, Ehlers MR. Immune-directed therapy for type 1 diabetes at the clinical level: The Immune Tolerance Network (ITN) experience. Rev Diabet Stud. 2012;9(4):359–371. doi: 10.1900/RDS.2012.9.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, Gottlieb PA, Greenbaum CJ, Marks JB, Monzavi R. et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet. 2011;378(9789):412–419. doi: 10.1016/S0140-6736(11)60886-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Staeva TP, Chatenoud L, Insel R, Atkinson MA. Recent lessons learned from prevention and recent-onset type 1 diabetes immunotherapy trials. Diabetes. 2013;62(1):9–17. doi: 10.2337/db12-0562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol. 2009;27:669–692. doi: 10.1146/annurev.immunol.021908.132557. [DOI] [PubMed] [Google Scholar]
- 18.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5(12):953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 19.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392(6673):245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 20.Kissler S, Stern P, Takahashi K, Hunter K, Peterson LB, Wicker LS. In vivo RNA interference demonstrates a role for Nramp1 in modifying susceptibility to type 1 diabetes. Nat Genet. 2006;38(4):479–483. doi: 10.1038/ng1766. [DOI] [PubMed] [Google Scholar]
- 21.Yang JH, Downes K, Howson JM, Nutland S, Stevens HE, Walker NM, Todd JA. Evidence of association with type 1 diabetes in the SLC11A1 gene region. BMC Med Genet. 2011;12:59. doi: 10.1186/1471-2350-12-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heinig M, Petretto E, Wallace C, Bottolo L, Rotival M, Lu H, Li Y, Sarwar R, Langley SR, Bauerfeind A. et al. A trans-acting locus regulates an anti-viral expression network and type 1 diabetes risk. Nature. 2010;467(7314):460–464. doi: 10.1038/nature09386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee KU, Kim MK, Amano K, Pak CY, Jaworski MA, Mehta JG, Yoon JW. Preferential infiltration of macrophages during early stages of insulitis in diabetes-prone BB rats. Diabetes. 1988;37(8):1053–1058. doi: 10.2337/diab.37.8.1053. [DOI] [PubMed] [Google Scholar]
- 24.Walker R, Bone AJ, Cooke A, Baird JD. Distinct macrophage subpopulations in pancreas of prediabetic BB/E rats. Possible role for macrophages in pathogenesis of IDDM. Diabetes. 1988;37(9):1301–1304. doi: 10.2337/diab.37.9.1301. [DOI] [PubMed] [Google Scholar]
- 25.Voorbij HA, Jeucken PH, Kabel PJ, De Haan M, Drexhage HA. Dendritic cells and scavenger macrophages in pancreatic islets of prediabetic BB rats. Diabetes. 1989;38(12):1623–1629. doi: 10.2337/diab.38.12.1623. [DOI] [PubMed] [Google Scholar]
- 26.Jansen A, Homo-Delarche F, Hooijkaas H, Leenen PJ, Dardenne M, Drexhage HA. Immunohistochemical characterization of monocytes-macrophages and dendritic cells involved in the initiation of the insulitis and beta-cell destruction in NOD mice. Diabetes. 1994;43(5):667–675. doi: 10.2337/diab.43.5.667. [DOI] [PubMed] [Google Scholar]
- 27.O'Brien BA, Geng X, Orteu CH, Huang Y, Ghoreishi M, Zhang Y, Bush JA, Li G, Finegood DT, Dutz JP. A deficiency in the in vivo clearance of apoptotic cells is a feature of the NOD mouse. J Autoimmun. 2006;26(2):104–115. doi: 10.1016/j.jaut.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 28.O'Brien BA, Huang Y, Geng X, Dutz JP, Finegood DT. Phagocytosis of apoptotic cells by macrophages from NOD mice is reduced. Diabetes. 2002;51(8):2481–2488. doi: 10.2337/diabetes.51.8.2481. [DOI] [PubMed] [Google Scholar]
- 29.Stoffels K, Overbergh L, Giulietti A, Kasran A, Bouillon R, Gysemans C, Mathieu C. NOD macrophages produce high levels of inflammatory cytokines upon encounter of apoptotic or necrotic cells. J Autoimmun. 2004;23(1):9–15. doi: 10.1016/j.jaut.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 30.Hutchings P, Rosen H, O'Reilly L, Simpson E, Gordon S, Cooke A. Transfer of diabetes in mice prevented by blockade of adhesion-promoting receptor on macrophages. Nature. 1990;348(6302):639–642. doi: 10.1038/348639a0. [DOI] [PubMed] [Google Scholar]
- 31.Jun HS, Yoon CS, Zbytnuik L, van Rooijen N, Yoon JW. The role of macrophages in T cell-mediated autoimmune diabetes in nonobese diabetic mice. J Exp Med. 1999;189(2):347–358. doi: 10.1084/jem.189.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leconet W, Petit P, Peraldi-Roux S, Bresson D. Nonviral Delivery of Small Interfering RNA Into Pancreas-associated Immune Cells Prevents Autoimmune Diabetes. Mol Ther. 2012;20(12):2315–2325. doi: 10.1038/mt.2012.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uno S, Imagawa A, Okita K, Sayama K, Moriwaki M, Iwahashi H, Yamagata K, Tamura S, Matsuzawa Y, Hanafusa T, Miyagawa J, Shimomura I. Macrophages and dendritic cells infiltrating islets with or without beta cells produce tumour necrosis factor-alpha in patients with recent-onset type 1 diabetes. Diabetologia. 2007;50(3):596–601. doi: 10.1007/s00125-006-0569-9. [DOI] [PubMed] [Google Scholar]
- 34.Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol. 2009;155(2):173–181. doi: 10.1111/j.1365-2249.2008.03860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jansen A, van Hagen M, Drexhage HA. Defective maturation and function of antigen-presenting cells in type 1 diabetes. Lancet. 1995;345(8948):491–492. doi: 10.1016/s0140-6736(95)90586-3. [DOI] [PubMed] [Google Scholar]
- 36.Hussain MJ, Peakman M, Gallati H, Lo SS, Hawa M, Viberti GC, Watkins PJ, Leslie RD, Vergani D. Elevated serum levels of macrophage-derived cytokines precede and accompany the onset of IDDM. Diabetologia. 1996;39(1):60–69. doi: 10.1007/BF00400414. [DOI] [PubMed] [Google Scholar]
- 37.Bradshaw EM, Raddassi K, Elyaman W, Orban T, Gottlieb PA, Kent SC, Hafler DA. Monocytes from patients with type 1 diabetes spontaneously secrete proinflammatory cytokines inducing Th17 cells. J Immunol. 2009;183(7):4432–4439. doi: 10.4049/jimmunol.0900576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8(9):942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 39.Dasu MR, Devaraj S, Zhao L, Hwang DH, Jialal I. High glucose induces toll-like receptor expression in human monocytes: mechanism of activation. Diabetes. 2008;57(11):3090–3098. doi: 10.2337/db08-0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Devaraj S, Jialal I. Increased secretion of IP-10 from monocytes under hyperglycemia is via the TLR2 and TLR4 pathway. Cytokine. 2009;47(1):6–10. doi: 10.1016/j.cyto.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meyers AJ, Shah RR, Gottlieb PA, Zipris D. Altered Toll-like receptor signaling pathways in human type 1 diabetes. J Mol Med (Berl) 2010;88(12):1221–1231. doi: 10.1007/s00109-010-0666-6. [DOI] [PubMed] [Google Scholar]
- 42.Alkanani AK, Rewers M, Dong F, Waugh K, Gottlieb PA, Zipris D. Dysregulated Toll-like receptor-induced interleukin-1beta and interleukin-6 responses in subjects at risk for the development of type 1 diabetes. Diabetes. 2012;61(10):2525–2533. doi: 10.2337/db12-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang X, Jia S, Geoffrey R, Alemzadeh R, Ghosh S, Hessner MJ. Identification of a molecular signature in human type 1 diabetes mellitus using serum and functional genomics. J Immunol. 2008;180(3):1929–1937. doi: 10.4049/jimmunol.180.3.1929. [DOI] [PubMed] [Google Scholar]
- 44.Levy H, Wang X, Kaldunski M, Jia S, Kramer J, Pavletich SJ, Reske M, Gessel T, Yassai M, Quasney MW, Dahmer MK, Gorski J, Hessner MJ. Transcriptional signatures as a disease-specific and predictive inflammatory biomarker for type 1 diabetes. Genes Immun. 2012;13(8):593–604. doi: 10.1038/gene.2012.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Irvine KM, Gallego P, An X, Best SE, Thomas G, Wells C, Harris M, Cotterill A, Thomas R. Peripheral blood monocyte gene expression profile clinically stratifies patients with recent-onset type 1 diabetes. Diabetes. 2012;61(5):1281–1290. doi: 10.2337/db11-1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bendtzen K, Mandrup-Poulsen T, Nerup J, Nielsen JH, Dinarello CA, Svenson M. Cytotoxicity of human pI 7 interleukin-1 for pancreatic islets of Langerhans. Science. 1986;232(4757):1545–1547. doi: 10.1126/science.3086977. [DOI] [PubMed] [Google Scholar]
- 47.Delaney CA, Pavlovic D, Hoorens A, Pipeleers DG, Eizirik DL. Cytokines induce deoxyribonucleic acid strand breaks and apoptosis in human pancreatic islet cells. Endocrinology. 1997;138(6):2610–2614. doi: 10.1210/endo.138.6.5204. [DOI] [PubMed] [Google Scholar]
- 48.Corbett JA, Kwon G, Marino MH, Rodi CP, Sullivan PM, Turk J, McDaniel ML. Tyrosine kinase inhibitors prevent cytokine-induced expression of iNOS and COX-2 by human islets. Am J Physiol. 1996;270(6 Pt 1):C1581–C1587. doi: 10.1152/ajpcell.1996.270.6.C1581. [DOI] [PubMed] [Google Scholar]
- 49.Corbett JA, Kwon G, Misko TP, Rodi CP, McDaniel ML. Tyrosine kinase involvement in IL-1 beta-induced expression of iNOS by beta-cells purified from islets of Langerhans. Am J Physiol. 1994;267(1 Pt 1):C48–C54. doi: 10.1152/ajpcell.1994.267.1.C48. [DOI] [PubMed] [Google Scholar]
- 50.Ablamunits V, Henegariu O, Hansen JB, Opare-Addo L, Preston-Hurlburt P, Santamaria P, Mandrup-Poulsen T, Herold KC. Synergistic reversal of type 1 diabetes in NOD mice with anti-CD3 and interleukin-1 blockade: evidence of improved immune regulation. Diabetes. 2012;61(1):145–154. doi: 10.2337/db11-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thomas HE, Irawaty W, Darwiche R, Brodnicki TC, Santamaria P, Allison J, Kay TW. IL-1 receptor deficiency slows progression to diabetes in the NOD mouse. Diabetes. 2004;53(1):113–121. doi: 10.2337/diabetes.53.1.113. [DOI] [PubMed] [Google Scholar]
- 52.Wen L, Green EA, Stratmann T, Panosa A, Gomis R, Eynon EE, Flavell RA, Mezquita JA, Mora C. In vivo diabetogenic action of CD4+ T lymphocytes requires Fas expression and is independent of IL-1 and IL-18. Eur J Immunol. 2011;41(5):1344–1351. doi: 10.1002/eji.201041216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pickersgill LM, Mandrup-Poulsen TR. The anti-interleukin-1 in type 1 diabetes action trial--background and rationale. Diabetes Metab Res Rev. 2009;25(4):321–324. doi: 10.1002/dmrr.960. [DOI] [PubMed] [Google Scholar]
- 54.Mandrup-Poulsen T. Interleukin-1 antagonists and other cytokine blockade strategies for Type 1 diabetes. Rev Diabet Stud. 2012;9(4):338–347. doi: 10.1900/RDS.2012.9.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bouillon R, Verstuyf A, Verlinden L, Allewaert K, Branisteanu D, Mathieu C, van Baelen H. Non-hypercalcemic pharmacological aspects of vitamin D analogs. Biochem Pharmacol. 1995;50(5):577–583. doi: 10.1016/0006-2952(95)00121-f. [DOI] [PubMed] [Google Scholar]
- 56.Mathieu C, Waer M, Laureys J, Rutgeerts O, Bouillon R. Prevention of autoimmune diabetes in NOD mice by 1,25 dihydroxyvitamin D3. Diabetologia. 1994;37(6):552–558. doi: 10.1007/BF00403372. [DOI] [PubMed] [Google Scholar]
- 57.Mathieu C, Waer M, Casteels K, Laureys J, Bouillon R. Prevention of type I diabetes in NOD mice by nonhypercalcemic doses of a new structural analog of 1,25-dihydroxyvitamin D3, KH1060. Endocrinology. 1995;136(3):866–872. doi: 10.1210/endo.136.3.7867594. [DOI] [PubMed] [Google Scholar]
- 58.Ferreira GB, van Etten E, Verstuyf A, Waer M, Overbergh L, Gysemans C, Mathieu C. 1,25-Dihydroxyvitamin D3 alters murine dendritic cell behaviour in vitro and in vivo. Diabetes Metab Res Rev. 2011;27(8):933–941. doi: 10.1002/dmrr.1275. [DOI] [PubMed] [Google Scholar]
- 59.Korf H, Wenes M, Stijlemans B, Takiishi T, Robert S, Miani M, Eizirik DL, Gysemans C, Mathieu C. 1,25-Dihydroxyvitamin D3 curtails the inflammatory and T cell stimulatory capacity of macrophages through an IL-10-dependent mechanism. Immunobiology. 2012;217(12):1292–1300. doi: 10.1016/j.imbio.2012.07.018. [DOI] [PubMed] [Google Scholar]
- 60.Canning MO, Grotenhuis K, de Wit H, Ruwhof C, Drexhage HA. 1-alpha,25-Dihydroxyvitamin D3 (1,25(OH)(2)D(3)) hampers the maturation of fully active immature dendritic cells from monocytes. Eur J Endocrinol. 2001;145(3):351–357. doi: 10.1530/eje.0.1450351. [DOI] [PubMed] [Google Scholar]
- 61.Gysemans CA, Cardozo AK, Callewaert H, Giulietti A, Hulshagen L, Bouillon R, Eizirik DL, Mathieu C. 1,25-Dihydroxyvitamin D3 modulates expression of chemokines and cytokines in pancreatic islets: implications for prevention of diabetes in nonobese diabetic mice. Endocrinology. 2005;146(4):1956–1964. doi: 10.1210/en.2004-1322. [DOI] [PubMed] [Google Scholar]
- 62.Riachy R, Vandewalle B, Kerr Conte J, Moerman E, Sacchetti P, Lukowiak B, Gmyr V, Bouckenooghe T, Dubois M, Pattou F. 1,25-dihydroxyvitamin D3 protects RINm5F and human islet cells against cytokine-induced apoptosis: implication of the antiapoptotic protein A20. Endocrinology. 2002;143(12):4809–4819. doi: 10.1210/en.2002-220449. [DOI] [PubMed] [Google Scholar]
- 63.Riachy R, Vandewalle B, Moerman E, Belaich S, Lukowiak B, Gmyr V, Muharram G, Kerr Conte J, Pattou F. 1,25-Dihydroxyvitamin D3 protects human pancreatic islets against cytokine-induced apoptosis via down-regulation of the Fas receptor. Apoptosis. 2006;11(2):151–159. doi: 10.1007/s10495-006-3558-z. [DOI] [PubMed] [Google Scholar]
- 64.Takiishi T, Gysemans C, Bouillon R, Mathieu C. Vitamin D and diabetes. Rheum Dis Clin North Am. 2012;38(1):179–206. doi: 10.1016/j.rdc.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 65.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 66.Tai N, Yasuda H, Xiang Y, Zhang L, Rodriguez-Pinto D, Yokono K, Sherwin R, Wong FS, Nagata M, Wen L. IL-10-conditioned dendritic cells prevent autoimmune diabetes in NOD and humanized HLA-DQ8/RIP-B7.1 mice. Clin Immunol. 2011;139(3):336–349. doi: 10.1016/j.clim.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 67.Feili-Hariri M, Falkner DH, Gambotto A, Papworth GD, Watkins SC, Robbins PD, Morel PA. Dendritic cells transduced to express interleukin-4 prevent diabetes in nonobese diabetic mice with advanced insulitis. Hum Gene Ther. 2003;14(1):13–23. doi: 10.1089/10430340360464679. [DOI] [PubMed] [Google Scholar]
- 68.Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care. 2011;34(9):2026–2032. doi: 10.2337/dc11-0472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hu CY, Rodriguez-Pinto D, Du W, Ahuja A, Henegariu O, Wong FS, Shlomchik MJ, Wen L. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J Clin Invest. 2007;117(12):3857–3867. doi: 10.1172/JCI32405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xiu Y, Wong CP, Bouaziz JD, Hamaguchi Y, Wang Y, Pop SM, Tisch RM, Tedder TF. B lymphocyte depletion by CD20 monoclonal antibody prevents diabetes in nonobese diabetic mice despite isotype-specific differences in Fc gamma R effector functions. J Immunol. 2008;180(5):2863–2875. doi: 10.4049/jimmunol.180.5.2863. [DOI] [PubMed] [Google Scholar]
- 71.Fiorina P, Vergani A, Dada S, Jurewicz M, Wong M, Law K, Wu E, Tian Z, Abdi R, Guleria I, Rodig S, Dunussi-Joannopoulos K, Bluestone J, Sayegh MH. Targeting CD22 reprograms B-cells and reverses autoimmune diabetes. Diabetes. 2008;57(11):3013–3024. doi: 10.2337/db08-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zekavat G, Rostami SY, Badkerhanian A, Parsons RF, Koeberlein B, Yu M, Ward CD, Migone TS, Yu L, Eisenbarth GS, Cancro MP, Naji A, Noorchashm H. In vivo BLyS/BAFF neutralization ameliorates islet-directed autoimmunity in nonobese diabetic mice. J Immunol. 2008;181(11):8133–8144. doi: 10.4049/jimmunol.181.11.8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marino E, Villanueva J, Walters S, Liuwantara D, Mackay F, Grey ST. CD4(+)CD25(+) T-cells control autoimmunity in the absence of B-cells. Diabetes. 2009;58(7):1568–1577. doi: 10.2337/db08-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, Gottlieb PA, Marks JB, McGee PF, Moran AM. et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361(22):2143–2152. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pescovitz MD, Torgerson TR, Ochs HD, Ocheltree E, McGee P, Krause-Steinrauf H, Lachin JM, Canniff J, Greenbaum C, Herold KC, Skyler JS, Weinberg A. Effect of rituximab on human in vivo antibody immune responses. J Allergy Clin Immunol. 2011;128(6):1295–1302 e1295. doi: 10.1016/j.jaci.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu L, Herold K, Krause-Steinrauf H, McGee PL, Bundy B, Pugliese A, Krischer J, Eisenbarth GS Type 1 Diabetes TrialNet Anti-CD20 Study Group. Rituximab selectively suppresses specific islet antibodies. Diabetes. 2011;60(10):2560–2565. doi: 10.2337/db11-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hu C, Du W, Zhang X, Wong FS, Wen L. The role of Gr1+ cells after anti-CD20 treatment in type 1 diabetes in nonobese diabetic mice. J Immunol. 2012;188(1):294–301. doi: 10.4049/jimmunol.1101590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Herold KC, Pescovitz MD, McGee P, Krause-Steinrauf H, Spain LM, Bourcier K, Asare A, Liu Z, Lachin JM, Dosch HM. Increased T cell proliferative responses to islet antigens identify clinical responders to anti-CD20 monoclonal antibody (rituximab) therapy in type 1 diabetes. J Immunol. 2011;187(4):1998–2005. doi: 10.4049/jimmunol.1100539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xiang Y, Peng J, Tai N, Hu C, Zhou Z, Wong FS, Wen L. The dual effects of B cell depletion on antigen-specific T cells in BDC2.5NOD mice. J Immunol. 2012;188(10):4747–4758. doi: 10.4049/jimmunol.1103055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Genovese MC, Kavanaugh A, Weinblatt ME, Peterfy C, DiCarlo J, White ML, O'Brien M, Grossbard EB, Magilavy DB. An oral Syk kinase inhibitor in the treatment of rheumatoid arthritis: a three-month randomized, placebo-controlled, phase II study in patients with active rheumatoid arthritis that did not respond to biologic agents. Arthritis Rheum. 2011;63(2):337–345. doi: 10.1002/art.30114. [DOI] [PubMed] [Google Scholar]
- 81.Podolanczuk A, Lazarus AH, Crow AR, Grossbard E, Bussel JB. Of mice and men: an open-label pilot study for treatment of immune thrombocytopenic purpura by an inhibitor of Syk. Blood. 2009;113(14):3154–3160. doi: 10.1182/blood-2008-07-166439. [DOI] [PubMed] [Google Scholar]
- 82.Colonna L, Catalano G, Chew C, D'Agati V, Thomas JW, Wong FS, Schmitz J, Masuda ES, Reizis B, Tarakhovsky A, Clynes R. Therapeutic targeting of syk in autoimmune diabetes. J Immunol. 2010;185(3):1532–1543. doi: 10.4049/jimmunol.1000983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Henry RA, Kendall PL, Thomas JW. Autoantigen-specific B-cell depletion overcomes failed immune tolerance in type 1 diabetes. Diabetes. 2012;61(8):2037–2044. doi: 10.2337/db11-1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Melo ME, Qian J, El-Amine M, Agarwal RK, Soukhareva N, Kang Y, Scott DW. Gene transfer of Ig-fusion proteins into B cells prevents and treats autoimmune diseases. J Immunol. 2002;168(9):4788–4795. doi: 10.4049/jimmunol.168.9.4788. [DOI] [PubMed] [Google Scholar]
- 85.Zhang AH, Li X, Onabajo OO, Su Y, Skupsky J, Thomas JW, Scott DW. B-cell delivered gene therapy for tolerance induction: Role of autoantigen-specific B cells. J Autoimmun. 2010;35(2):107–113. doi: 10.1016/j.jaut.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hayday AC, Saito H, Gillies SD, Kranz DM, Tanigawa G, Eisen HN, Tonegawa S. Structure, organization, and somatic rearrangement of T cell gamma genes. Cell. 1985;40(2):259–269. doi: 10.1016/0092-8674(85)90140-0. [DOI] [PubMed] [Google Scholar]
- 87.Hayday AC. Gamma delta cells: a right time and a right place for a conserved third way of protection. Annu Rev Immunol. 2000;18:975–1026. doi: 10.1146/annurev.immunol.18.1.975. [DOI] [PubMed] [Google Scholar]
- 88.Witherden DA, Havran WL. Molecular aspects of epithelial gammadelta T cell regulation. Trends Immunol. 2011;32(6):265–271. doi: 10.1016/j.it.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.de Saint-Basile G, Le Deist F, de Villartay JP, Cerf-Bensussan N, Journet O, Brousse N, Griscelli C, Fischer A. Restricted heterogeneity of T lymphocytes in combined immunodeficiency with hypereosinophilia (Omenn's syndrome) J Clin Invest. 1991;87(4):1352–1359. doi: 10.1172/JCI115139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mallick-Wood CA, Pao W, Cheng AM, Lewis JM, Kulkarni S, Bolen JB, Rowley B, Tigelaar RE, Pawson T, Hayday AC. Disruption of epithelial gamma delta T cell repertoires by mutation of the Syk tyrosine kinase. Proc Natl Acad Sci U S A. 1996;93(18):9704–9709. doi: 10.1073/pnas.93.18.9704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Minagawa M, Ito A, Shimura H, Tomiyama K, Ito M, Kawai K. Homogeneous epithelial gamma delta T cell repertoire of the skin is shaped through peripheral selection. J Dermatol Sci. 2001;25(2):150–155. doi: 10.1016/s0923-1811(00)00119-5. [DOI] [PubMed] [Google Scholar]
- 92.Modlin RL, Pirmez C, Hofman FM, Torigian V, Uyemura K, Rea TH, Bloom BR, Brenner MB. Lymphocytes bearing antigen-specific gamma delta T-cell receptors accumulate in human infectious disease lesions. Nature. 1989;339(6225):544–548. doi: 10.1038/339544a0. [DOI] [PubMed] [Google Scholar]
- 93.Tigelaar RE, Lewis JM, Bergstresser PR. TCR gamma/delta+ dendritic epidermal T cells as constituents of skin-associated lymphoid tissue. J Invest Dermatol. 1990;94(6 Suppl):58S–63S. doi: 10.1111/1523-1747.ep12875138. [DOI] [PubMed] [Google Scholar]
- 94.Maki K, Sunaga S, Komagata Y, Kodaira Y, Mabuchi A, Karasuyama H, Yokomuro K, Miyazaki JI, Ikuta K. Interleukin 7 receptor-deficient mice lack gammadelta T cells. Proc Natl Acad Sci U S A. 1996;93(14):7172–7177. doi: 10.1073/pnas.93.14.7172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Funda D, Stenvang JP, Buschard K. Age-related changes in T gamma delta cells of NOD mice. Immunol Lett. 1995;45(3):179–184. doi: 10.1016/0165-2478(95)00003-n. [DOI] [PubMed] [Google Scholar]
- 96.Lang FP, Pollock BH, Riley WJ, Maclaren NK, Barrett DJ. The temporal association between gamma delta T cells and the natural history of insulin-dependent diabetes. J Autoimmun. 1993;6(1):107–119. doi: 10.1006/jaut.1993.1009. [DOI] [PubMed] [Google Scholar]
- 97.Lang FP, Schatz DA, Pollock BH, Riley WJ, Maclaren NK, Dumont-Driscoll M, Barrett DJ. Increased T lymphocytes bearing the gamma-delta T cell receptor in subjects at high risk for insulin dependent diabetes. J Autoimmun. 1991;4(6):925–933. doi: 10.1016/0896-8411(91)90055-h. [DOI] [PubMed] [Google Scholar]
- 98.Zhang L, Jin N, Nakayama M, O'Brien RL, Eisenbarth GS, Born WK. Gamma delta T cell receptors confer autonomous responsiveness to the insulin-peptide B:9-23. J Autoimmun. 2010;34(4):478–484. doi: 10.1016/j.jaut.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Harrison LC, Dempsey-Collier M, Kramer DR, Takahashi K. Aerosol insulin induces regulatory CD8 gamma delta T cells that prevent murine insulin-dependent diabetes. J Exp Med. 1996;184(6):2167–2174. doi: 10.1084/jem.184.6.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Harrison LC, Solly NR, Martinez NR. (Pro)insulin-specific regulatory T cells. Novartis Found Symp. 2003;252:132–141. [PubMed] [Google Scholar]
- 101.Locke NR, Stankovic S, Funda DP, Harrison LC. TCR gamma delta intraepithelial lymphocytes are required for self-tolerance. J Immunol. 2006;176(11):6553–6559. doi: 10.4049/jimmunol.176.11.6553. [DOI] [PubMed] [Google Scholar]
- 102.Castriconi R, Della Chiesa M, Moretta A. Shaping of adaptive immunity by innate interactions. C R Biol. 2004;327(6):533–537. doi: 10.1016/j.crvi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 103.Djeu JY, Jiang K, Wei S. A view to a kill: signals triggering cytotoxicity. Clin Cancer Res. 2002;8(3):636–640. [PubMed] [Google Scholar]
- 104.Fauriat C, Just-Landi S, Mallet F, Arnoulet C, Sainty D, Olive D, Costello RT. Deficient expression of NCR in NK cells from acute myeloid leukemia: Evolution during leukemia treatment and impact of leukemia cells in NCRdull phenotype induction. Blood. 2007;109(1):323–330. doi: 10.1182/blood-2005-08-027979. [DOI] [PubMed] [Google Scholar]
- 105.Ferlazzo G, Thomas D, Lin SL, Goodman K, Morandi B, Muller WA, Moretta A, Münz C. The abundant NK cells in human secondary lymphoid tissues require activation to express killer cell Ig-like receptors and become cytolytic. J Immunol. 2004;172(3):1455–1462. doi: 10.4049/jimmunol.172.3.1455. [DOI] [PubMed] [Google Scholar]
- 106.Foster CE, Colonna M, Sun PD. Crystal structure of the human natural killer (NK) cell activating receptor NKp46 reveals structural relationship to other leukocyte receptor complex immunoreceptors. J Biol Chem. 2003;278(46):46081–46086. doi: 10.1074/jbc.M308491200. [DOI] [PubMed] [Google Scholar]
- 107.Middleton D, Curran M, Maxwell L. Natural killer cells and their receptors. Transpl Immunol. 2002;10(2-3):147–164. doi: 10.1016/s0966-3274(02)00062-x. [DOI] [PubMed] [Google Scholar]
- 108.Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331(6013):44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Thielens A, Vivier E, Romagne F. NK cell MHC class I specific receptors (KIR): from biology to clinical intervention. Curr Opin Immunol. 2012;24(2):239–245. doi: 10.1016/j.coi.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 110.Jaeger BN, Vivier E. Natural killer cell tolerance: control by self or self-control? Cold Spring Harb Perspect Biol. 2012;4(3) doi: 10.1101/cshperspect.a007229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ogasawara K, Hamerman JA, Hsin H, Chikuma S, Bour-Jordan H, Chen T, Pertel T, Carnaud C, Bluestone JA, Lanier LL. Impairment of NK cell function by NKG2D modulation in NOD mice. Immunity. 2003;18(1):41–51. doi: 10.1016/s1074-7613(02)00505-8. [DOI] [PubMed] [Google Scholar]
- 112.Ogasawara K, Hamerman JA, Ehrlich LR, Bour-Jordan H, Santamaria P, Bluestone JA, Lanier LL. NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity. 2004;20(6):757–767. doi: 10.1016/j.immuni.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 113.Rodacki M, Svoren B, Butty V, Besse W, Laffel L, Benoist C, Mathis D. Altered natural killer cells in type 1 diabetic patients. Diabetes. 2007;56(1):177–185. doi: 10.2337/db06-0493. [DOI] [PubMed] [Google Scholar]
- 114.Qin H, Lee IF, Panagiotopoulos C, Wang X, Chu AD, Utz PJ, Priatel JJ, Tan R. Natural killer cells from children with type 1 diabetes have defects in NKG2D-dependent function and signaling. Diabetes. 2011;60(3):857–866. doi: 10.2337/db09-1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Beilke JN, Meagher CT, Hosiawa K, Champsaur M, Bluestone JA, Lanier LL. NK cells are not required for spontaneous autoimmune diabetes in NOD mice. PLoS One. 2012;7(4):e36011. doi: 10.1371/journal.pone.0036011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gur C, Porgador A, Elboim M, Gazit R, Mizrahi S, Stern-Ginossar N, Achdout H, Ghadially H, Dor Y, Nir T, Doviner V, Hershkovitz O, Mendelson M, Naparstek Y, Mandelboim O. The activating receptor NKp46 is essential for the development of type 1 diabetes. Nat Immunol. 2010;11(2):121–128. doi: 10.1038/ni.1834. [DOI] [PubMed] [Google Scholar]
- 117.Gur C, Enk J, Kassem SA, Suissa Y, Magenheim J, Stolovich-Rain M, Nir T, Achdout H, Glaser B, Shapiro J, Naparstek Y, Porgador A, Dor Y, Mandelboim O. Recognition and killing of human and murine pancreatic beta cells by the NK receptor NKp46. J Immunol. 2011;187(6):3096–3103. doi: 10.4049/jimmunol.1101269. [DOI] [PubMed] [Google Scholar]
- 118.Mehers KL, Long AE, van der Slik AR, Aitken RJ, Nathwani V, Wong FS, Bain S, Gill G, Roep BO, Bingley PJ, Gillespie KM. An increased frequency of NK cell receptor and HLA-C group 1 combinations in early-onset type 1 diabetes. Diabetologia. 2011;54(12):3062–3070. doi: 10.1007/s00125-011-2299-x. [DOI] [PubMed] [Google Scholar]
- 119.Feuerer M, Shen Y, Littman DR, Benoist C, Mathis D. How punctual ablation of regulatory T cells unleashes an autoimmune lesion within the pancreatic islets. Immunity. 2009;31(4):654–664. doi: 10.1016/j.immuni.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bendelac A, Lantz O, Quimby ME, Yewdell JW, Bennink JR, Brutkiewicz RR. CD1 recognition by mouse NK1+ T lymphocytes. Science. 1995;268(5212):863–865. doi: 10.1126/science.7538697. [DOI] [PubMed] [Google Scholar]
- 121.Rolf J, Berntman E, Stenstrom M, Smith EM, Mansson R, Stenstad H, Yamagata T, Agace W, Sigvardsson M, Cardell SL. Molecular profiling reveals distinct functional attributes of CD1d-restricted natural killer (NK) T cell subsets. Mol Immunol. 2008;45(9):2607–2620. doi: 10.1016/j.molimm.2007.12.022. [DOI] [PubMed] [Google Scholar]
- 122.Jahng A, Maricic I, Aguilera C, Cardell S, Halder RC, Kumar V. Prevention of autoimmunity by targeting a distinct, noninvariant CD1d-reactive T cell population reactive to sulfatide. J Exp Med. 2004;199(7):947–957. doi: 10.1084/jem.20031389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Baxter AG, Kinder SJ, Hammond KJ, Scollay R, Godfrey DI. Association between alphabetaTCR+CD4-CD8- T-cell deficiency and IDDM in NOD/Lt mice. Diabetes. 1997;46(4):572–582. doi: 10.2337/diab.46.4.572. [DOI] [PubMed] [Google Scholar]
- 124.Godfrey DI, Kinder SJ, Silvera P, Baxter AG. Flow cytometric study of T cell development in NOD mice reveals a deficiency in alphabetaTCR+CDR-CD8- thymocytes. J Autoimmun. 1997;10(3):279–285. doi: 10.1006/jaut.1997.0129. [DOI] [PubMed] [Google Scholar]
- 125.Laloux V, Beaudoin L, Jeske D, Carnaud C, Lehuen A. NK T cell-induced protection against diabetes in V alpha 14-J alpha 281 transgenic nonobese diabetic mice is associated with a Th2 shift circumscribed regionally to the islets and functionally to islet autoantigen. J Immunol. 2001;166(6):3749–3756. doi: 10.4049/jimmunol.166.6.3749. [DOI] [PubMed] [Google Scholar]
- 126.Lehuen A, Lantz O, Beaudoin L, Laloux V, Carnaud C, Bendelac A, Bach JF, Monteiro RC. Overexpression of natural killer T cells protects Valpha14- Jalpha281 transgenic nonobese diabetic mice against diabetes. J Exp Med. 1998;188(10):1831–1839. doi: 10.1084/jem.188.10.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ly D, Mi QS, Hussain S, Delovitch TL. Protection from type 1 diabetes by invariant NK T cells requires the activity of CD4+CD25+ regulatory T cells. J Immunol. 2006;177(6):3695–3704. doi: 10.4049/jimmunol.177.6.3695. [DOI] [PubMed] [Google Scholar]
- 128.Poulton LD, Smyth MJ, Hawke CG, Silveira P, Shepherd D, Naidenko OV, Godfrey DI, Baxter AG. Cytometric and functional analyses of NK and NKT cell deficiencies in NOD mice. Int Immunol. 2001;13(7):887–896. doi: 10.1093/intimm/13.7.887. [DOI] [PubMed] [Google Scholar]
- 129.Wang B, Geng YB, Wang CR. CD1-restricted NK T cells protect nonobese diabetic mice from developing diabetes. J Exp Med. 2001;194(3):313–320. doi: 10.1084/jem.194.3.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sharif S, Arreaza GA, Zucker P, Mi QS, Sondhi J, Naidenko OV, Kronenberg M, Koezuka Y, Delovitch TL, Gombert JM. et al. Activation of natural killer T cells by alpha-galactosylceramide treatment prevents the onset and recurrence of autoimmune type 1 diabetes. Nat Med. 2001;7(9):1057–1062. doi: 10.1038/nm0901-1057. [DOI] [PubMed] [Google Scholar]
- 131.Subramanian L, Blumenfeld H, Tohn R, Ly D, Aguilera C, Maricic I, Mansson JE, Buschard K, Kumar V, Delovitch TL. NKT cells stimulated by long fatty acyl chain sulfatides significantly reduce the incidence of type 1 diabetes in nonobese diabetic mice (corrected) Plos One. 2012;7(5):e37771. doi: 10.1371/journal.pone.0037771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Simoni Y, Diana J, Ghazarian L, Beaudoin L, Lehuen A. Therapeutic manipulation of natural killer (NK) T cells in autoimmunity: are we close to reality? Clin Exp Immunol. 2013;171(1):8–19. doi: 10.1111/j.1365-2249.2012.04625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Marino E, Tan B, Binge L, Mackay CR, Grey ST. B-cell cross-presentation of autologous antigen precipitates diabetes. Diabetes. 2012;61(11):2893–2905. doi: 10.2337/db12-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]