

Abstract
Reestablishing immune tolerance in type 1 diabetes (T1D), a chronic autoimmune disease, is a major goal. The Immune Tolerance Network (ITN) has initiated eight clinical trials of immunomodulatory therapies in recent-onset T1D over the past decade. Results have been mixed in terms of clinical efficacy, but the studies have provided valuable mechanistic insight that are enhancing our understanding of the disease and guiding the design of future trials. Trials of non-Fc-binding anti-CD3 mAbs have revealed that modulation of this target leads to partial responses, and ITN's AbATE trial led to identification of a robust responder group that could be distinguished from non-responders by baseline metabolic and immunologic features. A pilot study of the combination of IL-2 and rapamycin gave the first demonstration that frequency and function of regulatory T cells (Tregs) can be enhanced in T1D subjects, although the therapy triggered the activation of effectors with transient β-cell dysfunction. Similarly, therapy with anti-thymocyte globulin led to substantial lymphocyte depletion, but also to the activation of the acute-phase response with no clinical benefit during preliminary analyses. These and other results provide mechanistic tools that can be used as biomarkers for safety and efficacy in future trials. Furthermore, our results, together with those of other organizations, notably TrialNet, delineate the roles of the major components of the immune response in T1D. This information is setting the stage for future combination therapy trials. The development of disease-relevant biomarkers will also enable the implementation of innovative trial designs, notably adaptive trials, which will increase efficiencies in terms of study duration and sample size, and which will expedite the conduct of trials in which there are uncertainties about dose response and effect size.
Keywords: AbATE, adaptive trials, alefacept, alpha-1 antitrypsin, anti-CD3, antithymocyte globulin, autoimmunity, beta-cell, combination therapy, IL-2/rapa, immune tolerance, new onset type 1 diabetes, RETAIN, START, T1DAL
Abbreviations: AAT - alpha-1 antitrypsin; AbATE – Autoimmunity-Blocking Antibody for Tolerance in Recently Diagnosed Type 1 Diabetes; AE - adverse event; APC – antigen-presenting cell; ATG – antithymocyte globulin; AUC – area under the curve; CRS - cytokine release syndrome; CTLA4 – cytotoxic T lymphocyte antigen 4; DCCT – Diabetes Control and Complications Trial; EBV – Epstein-Barr virus; FoxP3 – forkhead box P3; GAD – glutamate decarboxylase; G-CSF – granulocyte colony-stimulating factor; HbA1c – glycosylated hemoglobin; HCV – hepatitis C virus; Ig – immunoglobulin; IL – interleukin; JDRF – Juvenile Diabetes Research Foundation; sIL-2Rα – soluble IL-2 receptor alpha; ITN – Immune Tolerance Network; LFA-3 – lymphocyte function-associated antigen; mAb – monoclonal antibody; MMTT – mixed-meal tolerance test; mTOR – mammalian target of rapamycin; NF-κB – nuclear factor kappa B; NK – natural killer; NOD – non-obese diabetic; pAkt – phosphorylated Akt; PCR – polymerase chain reaction; PK – pharmacokinetics; pS6 – phosphorylated ribosomal protein S6; pSTAT5 – phosphorylated signal transducer and activator of transcription 5; Rapa – rapamycin; PI-3 – phosphatidylinositide 3; RETAIN - Research Trial of Aralast NP in New-Onset Type 1 Diabetes; STAT5 – signal transducer and activator of transcription 5; T1D – type 1 diabetes; T1DAL – Inducing Remission in Type 1 Diabetes with Alefacept; Teff – effector T cell; TGF-β – transforming growth factor beta; Th – T helper; TNFα – tumor necrosis factor alpha; Treg – regulatory T cell
1. Introduction
Over the past decade, the Immune Tolerance Network (ITN) has initiated eight clinical trials in recent-onset type 1 diabetes (T1D), several of which are currently underway. Others are in various stages of planning and development, as the ITN continues to seek effective immunomodulatory strategies with durable clinical benefit. In this review, we describe the main findings from the recent trials, discuss mechanistic insights, challenges and lessons learned, and outline new approaches going forward.
T1D results from a progressive autoimmune destruction of the islet β-cells, the end result of a period of preclinical autoimmunity of variable length, during which pancreatic β-cells are silently destroyed [1]. This characteristic timeline provides several potential opportunities for disease-modifying interventions, including the following:
1. Prevention prior to the initiation of autoimmunity
2. Intervention after the onset of autoimmunity, but prior to clinical onset
3. Intervention during the new-onset period following diagnosis, in which preservation of remaining β-cells is clinically advantageous [2].
The goal of T1D studies by the ITN is to demonstrate successful intervention therapy that will not only inhibit ongoing immune destruction of β-cells, but also provide durable immune tolerance, such that therapy can be discontinued without recurrence of disease. While such a therapy is desirable in any of the stages of T1D progression, the ITN studies focus on clinical trials aimed at reestablishing tolerance in recent-onset disease, primarily because the decline of β-cell function—measured as loss of C-peptide secretion—is predictable at this stage. This allows a feasible, well-powered study design to be established. While this issue constrains the present trials to a fairly small subset of T1D subjects, therapies aimed at preserving β-cell function in recent-onset disease are excellent candidates for broader trials that include a more diverse population, including high-risk non-diabetic individuals with early signs of islet autoimmunity [3].
A comprehensive model for T1D pathogenesis implicates a dysregulated innate immune platform supporting a chronic inflammatory response that drives autoreactive effector T and B cells. Paired with defects in regulatory cells, such a scenario results in the breakdown of immune homeostasis. The resulting pathogenic chronic autoimmune response elicits progressive β-cell destruction. Based on this framework, there are multiple potential immune pathways that are therapeutic targets, broadly grouped into:
1. Inflammation and innate immunity
2. Effector T cells, including Th1, Th17, and CD8+ cytotoxic cells
3. Regulatory T cells (Tregs)
4. Cytokines and cytokine receptors
5. Antigen-specific approaches, including modulation of antigen-presenting cells (APCs) [1, 2]
One of the major challenges facing the design of clinical trials is the realization that several of these pathways simultaneously contribute to progressive disease, but very few (if any) single-agent therapies can effectively target more than one pathway (Table 1). As a consequence, we consider the current era of monotherapy trials to be the "first act" in a long-term program, designed to identify candidates with partial responses and well-justified mechanisms of action that will ultimately be combined to achieve a more comprehensive and successful clinical outcome, addressing all facets of the disease (Figure 1).
Table 1. Proposed effects of major types of immunomodulatory drugs in recent and planned new-onset T1D trials.
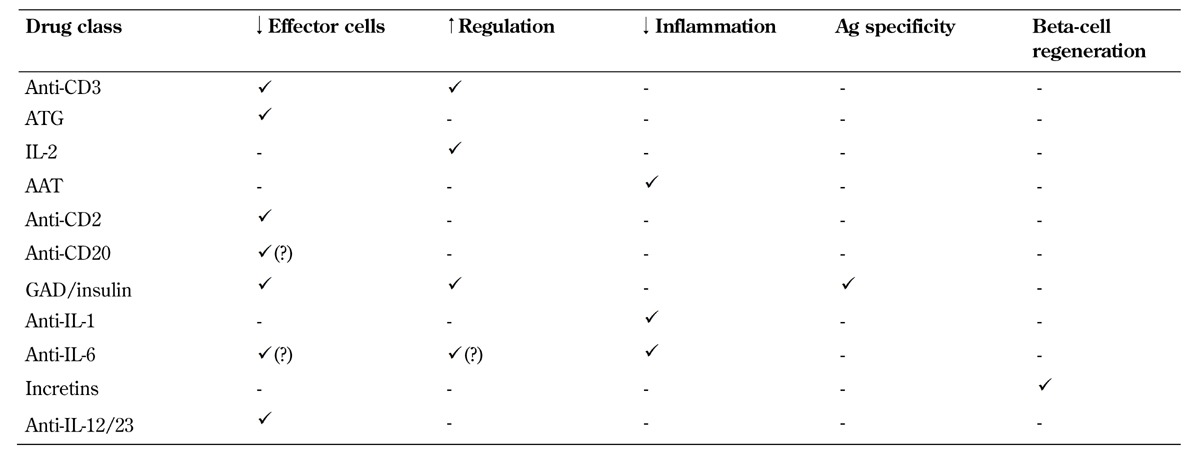
Legend: AAT – alpha-1 antitrypsin, ATG – antithymocyte globulin, IL – interleukin.
Figure 1. Challenges to successful tolerance immunotherapy in T1D.
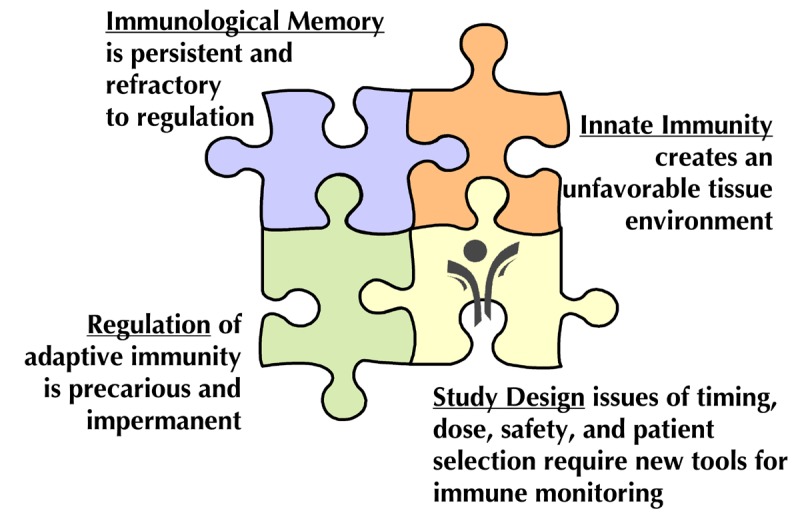
Biological challenges include multiple facets of the autoimmune response, and operational challenges include a need for improved study designs that build upon the lessons from recent trials.
2. ITN trials in new-onset T1D
For the purpose of clinical trials, the "new-onset" period is generally defined as the first 3 months after diagnosis, while intervention by blockade of cytokines such as TNFα and IL-1 may be limited to the first month due to their disappearance after that period [4]. This is an attractive window for intervention because there is active ongoing destruction of β-cells, but there is also a sizeable residual islet mass (20-40% of normal) that is "worth saving". Evidence from the DCCT indicates that preservation of residual islet cell mass, even if only temporary, can improve long-term clinical outcomes [5]. Therefore, in the new-onset period, T1D patients have measurable islet function, which undergoes a predictable decline, and which is potentially modifiable by an effective intervention [6]. Moreover, preservation of residual β-cell function is expected to confer clinical benefit, justifying the evaluation of novel interventions that may carry a degree of risk. These considerations have led to a standardized trial design that has been broadly implemented by ITN and TrialNet and, with some variation, by other organizations [7]. The standard design is a phase-2, two-arm, double-blind, placebo-controlled, 2:1-randomized (drug-placebo) trial that enrolls 60-90 subjects (Figure 2). The primary endpoint, generally at 1 year, is the change from baseline in the C-peptide area under the curve (AUC) during a mixed-meal tolerance test (MMTT). MMTT-stimulated C-peptide responses are an accurate measure of endogenous insulin secretion, and have been validated as a useful surrogate for islet cell function [8].
Figure 2. The standard study design for proof-of-concept trials of novel interventions in new-onset T1D.
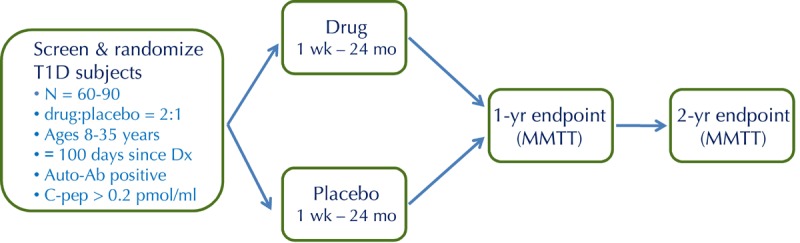
This is a randomized, placebo-controlled, double-blind phase 2 trial, with 2:1 randomization (drug to placebo). Key inclusion criteria are as shown, including ≤100 days since diagnosis (Dx). The primary endpoint, generally at 1 year, is the change from baseline in C-peptide area under the curve (AUC) following a mixed-meal tolerance test (MMTT).
ITN trials in new-onset T1D have focused on interventions that offer the prospect of inducing immune tolerance to pancreatic islets. To date, this objective has been explored with non-antigen-specific immune-modulating agents, for which there was a strong scientific rationale, preclinical data (usually in the NOD mouse), and sometimes clinical data from other autoimmune diseases.
2.1 The AbATE trial (anti-CD3 mAb)
The utility of non-Fc receptor-binding anti-CD3 mAb in halting the destruction of β-cells and inducing tolerance in T1D has been an active area of clinical investigation for the past 10-15 years [9]. The publication by Herold and colleagues in 2002 was a landmark in the field of T1D interventions because it was the first demonstration that a short course of targeted immunomodulation―as opposed to chronic immunosuppression―could preserve islet function for a period of 1-2 years in a proportion of subjects with new-onset T1D [10]. A subsequent report from a European consortium using a different anti-CD3 mAb confirmed these encouraging findings [11]. However, two issues were identified that required further investigation: preservation of C-peptide secretion was temporary and started to decline again 1-2 years after treatment; and there were dose-related adverse events (AEs), primarily cytokine release and Epstein-Barr virus (EBV) reactivation, which were considered unacceptable.
To address the question of the variability and durability of response, the ITN initiated the AbATE trial, in which the anti-CD3 mAbteplizumab was administered as two 14-day courses one year apart, the first at study entry and the second at 12 months. Data from the original US study with teplizumab as well as the European experience with otilixizumab suggested that there may be an optimal therapeutic window, with higher doses leading to greater EBV reactivation and cytokine release (reviewed in [2] and [9]). Prior to initiation of AbATE, a pilot clinical trial was completed better to understand safety, pharmacokinetics (PK), and dose response, and this led to adoption of the dosing regimen used in AbATE (described in [12]). The design of the AbATE trial followed the standard design described above (Figure 2) except that subjects randomized to the control group did not receive placebo and the study was not blinded. However, all subjects received intensive diabetes management with the goal of achieving HbA1c < 7.5%, and the study investigators were blinded to C-peptide results until the primary endpoint was reached after 2 years.
The trial met its primary endpoint, with the following principal results:
1. Teplizumab treatment led to a significantly better preservation of C-peptide secretion at 2 years in the intent-to-treat subjects than in controls [13].
2. No new safety concerns emerged; among subjects who were EBV-seropositive at randomization, ∼45% had modest, transient increases in EBV viral loads detected by polymerase chain reaction (PCR) 1 month after drug treatment, but by 2 months, EBV viral loads were undetectable in all subjects [13].
The pattern of C-peptide responses appeared similar to earlier trials with a single course of treatment; the treatment effect was most pronounced in the first 6-12 months, after which the slope of decline in the treatment group was similar to that of the control group, which cast doubt on whether the second course of teplizumab conferred any additional effect. However, an analysis of responder rates identified a subgroup of treated subjects (22 of the 45 who received the drug) whose C-peptide responses were significantly different from both controls and "non-responders", with almost complete preservation at 2 years. By contrast, mean C-peptide secretion of the non-responders was virtually indistinguishable from that of the controls [13]. Further analysis revealed that, compared to non-responders, the responders as a group had significantly lower insulin use and HbA1c levels at baseline, whereas C-peptide secretion at baseline did not differ. Moreover, flow cytometry performed at baseline demonstrated differences in specific lymphocyte subsets between responders and non-responders. It is significant that, in another recent trial in T1D using teplizumab―the DELAY trial, in which subjects were beyond the new-onset period, i.e. 4-12 months since diagnosis―the treatment response was also significantly associated with lower HbA1c levels at baseline [12]. It is also important that, in the European otelixizumab study, drug response appeared to be dependent on baseline C-peptide secretion [11].
The AbATE result is exciting because it shows that a responder group exhibits a drug response, which can be evaluated for qualitative and quantitative differences relative to non-responders and untreated controls. Interestingly, the responder status appears to be determined by baseline metabolic and immunologic features that confer the responsiveness to anti-CD3 therapy. While the mechanism of response remains to be determined, it is important to find out whether the metabolic health of islets at the time of treatment is decisive in enabling functional recovery. Furthermore, we need to clarify the following two questions:
1. Is the state of the immune reactivity at baseline, including ratios of Tregs to effector T cells (Teffs) and the general inflammatory milieu, important for governing response?
2. Are there distinct properties of autoreactive T-cells in the non-responders that may account for a resistance to therapy?
These and other questions are the subject of ongoing mechanistic analyses of AbATE samples. We will use the results to guide future trials with anti-CD3 mAb and other immunomodulatory agents.
2.2 The IL-2/rapa trial
In recent years, there has been growing interest in the use of interleukin-2 (IL-2) to boost Treg populations in diverse clinical settings, including autoimmunity and graft versus host disease. Treg cells are strongly dependent on IL-2 signaling, and express high levels of CD25, the IL-2 receptor α chain. IL-2 (proleukin) is used for the treatment of solid tumors such as melanoma and renal cell carcinoma on the basis that it expands activated T cells. However, it has been suggested that IL-2 at lower doses could have a preferential effect on Treg cells. Indeed, two recent pilot studies of low-dose IL-2 found clinical improvements and significant increases in peripheral Treg cells in hepatitis C virus (HCV)-induced vasculitis [14] and graft versus host disease [15].
The ITN conducted a pilot study with nine T1D subjects, who were beyond the new-onset period (4-48 months since diagnosis), but still had significant C-peptide secretion (≥0.4 pmol/ml). The subjects received a combination of IL-2 (1 month) and rapamycin (3 months), with intensive monitoring during the first year and continued follow-up until 3 years after treatment. IL-2 (proleukin) was given subcutaneously at a low-to-moderate dose (4.5 x 106 IU 3 x/wk). Additionally, rapamycin, an inhibitor of mTOR, was given to block effector cell expansion and potentially boost Treg generation. Unexpectedly, all subjects had a marked decrease in MMTT-stimulated C-peptide secretion at 3 months. However, in almost all subjects, the subsequent C-peptide levels increased by 6 months, and then showed a stable, expected rate of decline at 1 year, suggesting that the acute β-cell impairment was transient [16]. All subjects experienced transient eosinophilia, IL-2 injection site reactions, and flu-like symptoms during the first month of treatment. Detailed mechanistic studies revealed robust 2-3-fold elevations in FoxP3+ Tregs coincident with IL-2 treatment and enhanced IL-2 responsiveness in CD25+ cells as measured by pSTAT5, which persisted at 1 year, suggesting that IL-2/rapa treatment boosted Treg numbers and responsiveness in peripheral blood [15].
Given the favorable effects on Tregs, what is the explanation for the impairment in β-cell function? Further analysis revealed that, in addition to the transient eosinophilia, IL-2/rapa treatment also led to significant increases in CD56+ NK cells and sIL-2Rα levels as well as a significant decrease in TGF-β levels, suggesting that this treatment led to transient immune activation, which may have contributed to the β-cell impairment [16]. Many of the mechanistic changes observed in the trial were coincident with the period of IL-2 administration. The contribution of rapamycin to these effects is uncertain. We observed a modulation of pAkt but not pS6 in T cells, suggesting that the combination did affect the Akt/PI-3 kinase pathway, a known rapamycin target. The effects of rapamycin in T1D are controversial; some reports indicate a benefit, while others suggest that the drug is toxic to β-cells and anti-tolerogenic [17-19]. The role of rapamycin in the treatment of T1D requires further careful evaluation.
A key message delivered by this trial is the need to separate therapeutic expansion of Tregs from deleterious expansion of effector populations. This was the first demonstration that robust expansion of Treg number and responsiveness is possible in new-onset T1D, thereby correcting what many consider to be a key immunologic defect in this disease. However the expansion of effector cells and concomitant immune activation likely negated the favorable effects on Tregs, leading to a net impairment of islet function. The challenge now is to find a regimen that leads to a sustained enhancement of the Treg compartment without expansion of effector and inflammatory cells. This may require different dosing, formulations, or timing of IL-2, perhaps in combination with adoptive transfer of ex vivo-expanded autologous Tregs, although the potential for effector cell expansion will require close monitoring and innovative study designs to minimize adverse outcomes.
2.3 The START trial using ATG
Lymphocyte depletion and modulation has a long history in the context of allogeneic transplantation, often using antithymocyte globulin (ATG). ATG is a polyclonal antiserum raised in rabbits against human thymocytes and is known to target a wide range T and B cell surface antigens. ATG has also shown efficacy in certain autoimmune diseases, notably aplastic anemia, and has been used in two pilot human trials in T1D [20, 21]. An aggressive combination regimen including cyclophosphamide, G-CSF, and ATG was given to a small number of T1D subjects, and resulted in insulin independence for an average of 16 months, although severe toxicities were observed [22, 23]. The therapeutic mechanism is thought to be depletion of T effector cells with relative sparing of Tregs and/or selective expansion of Tregs during homeostatic proliferation.
The ITN is conducting a trial of ATG monotherapy in new-onset T1D subjects who recently reached its 1-year primary endpoint. The trial conformed to the standard design shown in Figure 2, and 58 subjects were randomized. Enrolment was slow, most likely because of the nature of the regimen, which required 5 days of hospitalization and intravenous infusion on 4 consecutive days. All subjects experienced cytokine release syndrome (CRS), which was controlled with intravenous corticosteroids, and serum sickness 7-10 days after the course of ATG infusions, requiring oral steroids [24]. The high rate of serum sickness was unexpected as this is not the experience in transplantation, but might have resulted from the absence of additional immunosuppression (other than short courses of steroids); it may also have been a feature of the study population with active underlying autoimmunity.
Analysis of the primary endpoint (MMTT-stimulated C-peptide AUC at 12 months) showed no difference between the ATG group and the placebo group. Data analyses are ongoing, but inspection of the C-peptide response curves revealed an accelerated loss of C-peptide during the first 6 months in the ATG group (compared to average rates of decline in this study population [25]), which then recovered during the second 6 months. This suggests that there may have been an early worsening of islet function after treatment, followed by recovery and stabilization, which is reminiscent of the experience in the IL-2/rapa trial. Mechanistic analyses confirmed the expected depletion of CD4 and CD8 T cells, which was prolonged, and which only partially recovered by month 12 [24]. Preliminary results suggest that there may have been an unfavorable balance between Tregs and effector cells during the first 12 months of the trial, as well as an activation of the acute-phase response during treatment with ATG, but further work is required to define these changes more completely. Together, the results to date suggest that ATG therapy may have resulted in acute activation of pro-inflammatory events together with an unfavorable balance between activated effector cells and Tregs. This likely contributed to the absence of a treatment effect at the 12-month endpoint. However, the trial is ongoing and will reach secondary endpoints at 24 months, at which time a more complete evaluation of the effects of ATG therapy in T1D will be possible.
Although the primary endpoint result is disappointing, the START trial promises to be a rich source of mechanistic information about immunologic surrogates that are potentially linked to unwanted outcomes in intervention trials in new-onset T1D. Once complete, ratios of lymphocyte subsets, expression of activation markers, and changes in serum cytokines and acute-phase reactants may serve as a potential set of biomarkers that correlate with treatment effects, and help to focus attention on gaps in our current immunotherapeutic options.
2.4 The RETAIN trial (α1-antitrypsin)
In addition to modulating Teff and Treg subsets, altering innate and inflammatory responses is a major goal in T1D interventions since inflammation plays a critical role in shaping the autoreactive adaptive immune response. Alpha-1 antitrypsin (AAT), a major circulating serine protease inhibitor, has a physiological role, inhibiting neutrophil elastase in the lung and other organs, and is regularly used for the treatment of emphysema related to congenital AAT deficiency [26, 27]. More recently, AAT was shown to have broader anti-inflammatory and anti-apoptotic effects that may or may not be mediated by the protease inhibitory activity of the protein [28]. This prompted investigation in models relevant to T1D, and AAT was shown to prevent cytokine- or toxin-induced β-cell apoptosis [29, 30], inhibit production of pro-inflammatory cytokines by monocytes [31], protect islet allografts in rodents [32], and reverse diabetes in the NOD mouse [33].
The ITN initiated an exploration of the potential use of AAT in T1D through the design of a multi-stage trial known as RETAIN. Major gaps in knowledge, including mechanistic biomarkers, safety, and dosing in pediatric populations, are being evaluated prior to initiating a randomized placebo-controlled trial conforming to the standard T1D design. To date, AAT infusions have been well tolerated and no safety concerns have emerged [34]. To develop suitable biomarkers for therapeutic monitoring and to determine optimal dosing, the ITN is conducting a separate, ongoing pilot study, designed to measure an immunological effect of AAT through the analysis of whole blood drawn from treated subjects. Preliminary studies using blood from untreated T1D subjects spiked with a range of AAT concentrations and incubated ex vivo in the presence or absence of lipopolysaccharide are now underway; these samples are assayed for production of pro-inflammatory cytokines and expression of genes in the NF-κB and apoptosis pathways. These studies illustrate the stepwise ITN approach to selecting a rational dosing and monitoring plan prior to enrolment of additional subjects for the proof-of-concept trial.
2.5 The T1DAL trial with alefacept
Anti-CD3 mAb and ATG are T cell-directed therapies that are thought to affect both Teff and Treg subsets. To date, a drug that selectively targets effector-memory T cells has not been evaluated in new-onset T1D. CD2, a surface antigen that binds LFA-3, is broadly expressed on CD4 and CD8 cells, but the highest levels of expression are on CD45RO+ effector-memory cells. Alefacept is an LFA-3-IgG1 fusion protein that selectively targets CD2. It was approved for the treatment of psoriasis. Preclinical and clinical data indicate that alefacept moderately depletes CD4 and CD8 cells, with an emphasis on effector-memory populations. Clinically, the drug has a slow onset of action, but induces long-lasting remission in a proportion of psoriasis patients, suggesting that induction of tolerance may be possible in a subset of responders (reviewed in [35]).
The T1DAL trial was initiated to evaluate the safety and efficacy of alefacept in preserving C-peptide secretion in new-onset T1D. The design of the trial conforms to the standard model (Figure 2), and the primary endpoint will be reached in the 2nd quarter of 2013. To date, no undue safety concerns have emerged.
3. What have we learned: insights and challenges
During the past decade, new-onset T1D trials conducted by ITN, TrialNet, and the industry have produced mixed results. On the positive side, phase 2 trials of anti-CD3 mAb, anti-CD20 mAb (rituximab) [36], and CTLA4-Ig fusion protein (abatacept) [37] have shown modest efficacy in delaying the loss of islet function for a period of time (6-24 months). On the other hand, none of these therapies achieved durable responses in T1D subjects. Phase 2 trials of anti-IL-1 therapies (canakinumab and anakinra) [38, 39], GAD-alum vaccination [40], and ATG showed no efficacy, while the pilot study of IL-2/rapa even led to transient islet impairment [16]. This body of work has provided important insight, has identified key challenges, and should be studied thoroughly to provide guidance on the way forward.
Notwithstanding the failure of two phase 3 trials (in part due to trial design and dosing issues [41]), trials of anti-CD3 mAb have consistently shown preservation of C-peptide secretion. The AbATE trial has shown that C-peptide preservation is particularly striking in a responder subgroup with baseline differences in metabolic and immunologic features, including lower insulin use and HbA1c levels and differences in lymphocyte subsets at baseline. While the immunologic mechanism in responders remains to be elucidated, identification of the role of glycemic control prior to treatment may be helpful for future trial design as a correlate of response; HbA1c and insulin use at baseline may be considered as inclusion/exclusion criteria and/or stratification during randomization.
Similarly, the partial responses to anti-CD20 mAb and CTLA4-Ig treatment indicate that modulation of the B cell compartment and T cell costimulatory blockade, respectively, play a role in altering immune responses in T1D. However, additional work is required to define the exact nature of the role. The failure of anti-IL-1 to modify the course of C-peptide decline was unexpected, given a strong scientific rationale implicating this cytokine in driving the inflammatory response, and evidence that IL-1 is directly toxic to β-cells. It is possible that the main potential benefit of anti-IL-1, suppression of intra-islet inflammation, is largely negated in patients with tight glucose control because hyperglycemia is an important driver of inflammation and β-cell apoptosis. It remains to be seen whether an alternative approach to controlling islet inflammation and apoptosis, AAT, is effective.
The IL-2/rapa and START trials sounded a note of caution. Mechanistic analyses have begun to identify a common theme, namely that stimulation of an acute inflammatory response and emergence of activated effectors, even in the presence of boosted Tregs, are unfavorable and can lead to transient disease exacerbation. While a variety of potential strategies are being considered to boost regulatory responses, it is essential to consider concomitant measures to ablate Teffs or prevent their inadvertent activation. The biomarkers of immunologic changes in these two trials that correlated with undesirable outcomes should now be incorporated in future trials.
4. Future directions
4.1 Combination therapies
In T1D, as in other chronic autoimmune disorders, the defects in immunity involve multiple arms of the immune system, and it may be unrealistic to expect a single therapy to restore normal immune function (Table 1). A joint JDRF-ITN therapy assessment group recently proposed a combination-based strategy, and identified a large number of possible combinations that could be evaluated [42]. The current first act in the era of T1D clinical trials was designed to identify components of these future therapies that have safe and plausible mechanisms of action and that are candidates for synergistic benefit when used in combination with other treatments (Table 1).
Recent reports of combination therapies in the NOD mouse model have indicated that synergies can be achieved when drugs from different mechanistic classes are combined. These combinations include anti-CD3 plus anti-IL-1 [43], anti-CD3 plus nasal insulin [44], and ATG plus G-CSF [45]. However, not all combinations will be useful, as we have already learnt in clinical practice from trials of anti-CD25 plus mycophenolatemofetil [46] and IL-2/rapa [16], and in the NOD with anti-CD20 plus insulin [47]. Thus, combination therapies that are to be translated into clinical practice must be based on a sound scientific rationale, excellent safety, and strong supporting evidence from relevant animal models. Other limitations in translating therapeutic combinations into clinical practice include regulatory and commercial hurdles.
Another proof-of-concept approach is to utilize cell-based interventions, notably adoptive transfer of ex vivo-expanded autologous Tregs, as a first stage towards gaining a better understanding of the essential balance between effector and regulatory compartments of the autoreactive repertoire. Preclinical data in several models of autoimmunity, including the NOD mouse and models of alloreactivity (organ transplantation), have highlighted the promise of this approach, and have prompted pilot clinical trials [48-50]. The initial focus is on polyclonal Tregs, which are postulated to control organ-specific autoimmunity via a process of bystander suppression and "infectious tolerance" [48]. It may be possible to refine these procedures to produce antigen-specific Tregs with exquisite specificity for autoreactive Teffs in T1D. The latter will require resolution of several challenges, including the identification of the appropriate islet autoantigens and maintenance of Treg stability following reinfusion to prevent conversion to antigen-specific effectors that can exacerbate disease. Treg stability is also an issue with polyclonal cells, and has led to the suggestion that adoptively transferred Tregs can be maintained in vivo by cotreatment with low-dose IL-2 which, as discussed above, is a critical growth factor for Tregs [51].
4.2 Novel trial designs
The standard clinical trial design shown in Figure 2 has served the new-onset T1D research community well over the past decade, but it does have its limitations, principally regarding duration and sample size. Even when enrolment is rapid and can be completed within 6 months or less, as has been achieved by TrialNet in recent trials (e.g., the GAD-alum and abatacept studies [37, 40]), trials typically require 3-5 years to complete, because enrolment may be staggered for safety reasons, and can extend over 12-24 months. However, the primary endpoint is usually at 1 year after study entry, and continued follow-up extends to 2 years or longer. Sample size is in the range of 60-90 subjects, which seems modest, but can be a challenge because of the requirement to enroll patients within 100 days of diagnosis. In the US, there are only 20,000-30,000 new-onset T1D patients per year, 10% of whom are eligible, interested, and referred to a clinical trial site within the 100-day window. When multiple new-onset trials are in progress simultaneously, the number of eligible subjects becomes a constraint. Given the fact that ITN has identified more than 40 potential combination therapy approaches of interest [42], trial designs need to become more efficient.
For these reasons, we have explored alternative trial designs at ITN, most notably adaptive designs. An adaptive trial is "a study that includes a prospectively planned opportunity for modification of one or more specified aspects of the study design" [52]. Adaptive trials offer the prospect of significant efficiencies in terms of study duration and sample size, and hence have attracted a lot of attention from both industry and academia. Adaptive designs are particularly well suited for optimizing exploratory, proof-of-concept trials in which there are uncertainties about dose response, endpoints, and effect sizes [53].
A critical component of an adaptive trial is the availability of a surrogate endpoint or biomarker that is informative about drug effect at early time points and that enables prospectively planned interim analyses that, in turn, drive study adaptations during the conduct of the trial (Figure 3). This remains a significant challenge in T1D trials because the field lacks robust biomarkers informative about β-cell-specific autoimmunity. The current gold-standard endpoint, MMTT-stimulated C-peptide secretion, is a good surrogate of β-cell function, but does not provide information about β-cell viability or immune responses, and generally gives a readout only after 1 year from the start of treatment. Therefore, MMTT-stimulated C-peptide secretion is not a useful surrogate to drive decisions in an adaptive trial. However, as already discussed, recent ITN trials have provided insight into drug mechanisms that may enable adaptive trial designs.
Figure 3. Adaptive trial design workflow.
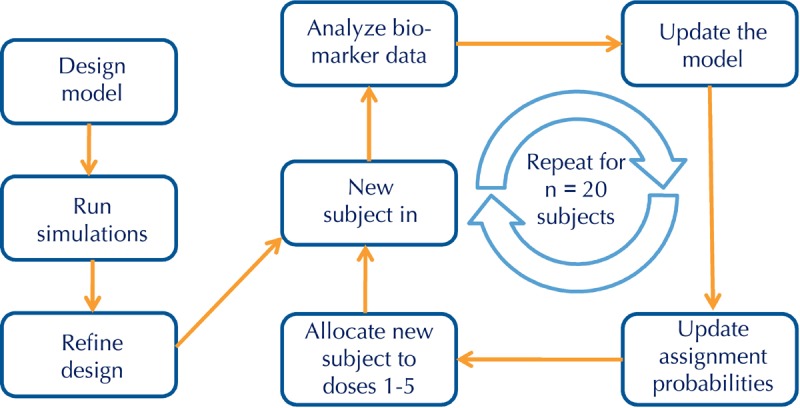
In the example shown, the adaptive trial is designed to determine an optimal dose using the continual reassessment method. All adaptive trials begin with a design model that is optimized with computer simulations before developing the clinical protocol. Adaptive trials allow for prospective modifications of one or more features of the protocol during the conduct of the study, in this case subject assignments to one of 5 dose groups, leading to significant efficiencies over conventional designs.
The IL-2/rapa trial demonstrated that robust changes in peripheral lymphocyte frequencies and function can be detected with state-of-the-art flow techniques in a small sample size. Thus, significant increases in Treg and NK cell frequencies, and significant changes in phosphorylated signal transducer and activator of transcription 5 (pSTAT5) levels, could be observed in only 9 subjects, and these changes were seen as early as 1 month after the start of therapy [16]. These rapid kinetics should enable the use of flow cytometry as a biomarker in an adaptive trial designed to determine a dose of IL-2 that enhances Treg without boosting undesired effector subsets. Based on preliminary results from a recently concluded trial of low-dose IL-2 (without rapamycin) in new-onset T1D patients, it appears that this goal may be attainable (David Klatzmann, personal communication), but the optimal dose and duration of IL-2 therapy remain to be determined. An adaptive trial would be well-suited to this task.
To address the use of flow cytometry as early biomarker, ITN is developing a trial design that is geared towards dose optimization using the continual reassessment method (Figure 3). In this design, subject assignment to one of five dose groups is continually updated based on flow data, such that most subjects are randomized to the effective doses, and randomization to ineffective doses is minimized (Figure 4). We have used trial simulations to model real-world scenarios (using assumptions about the shape of the dose response and the magnitude of Treg changes). We found that the design is remarkably efficient, yielding an optimal dose with ≤ 15 subjects, and in some scenarios with ≤ 10 subjects. This would represent a substantial saving in terms of trial duration and sample size compared with a conventional parallel-group dose-finding study, which we estimate would require 4-5 fold more subjects, and take 2-3 times longer to complete. Similar adaptive designs can be considered for the next phase of RETAIN (using gene expression changes as the biomarker) and, possibly, follow-up studies with ATG (based on early shifts in Treg/Teff ratios).
5. Conclusions
The current era of monotherapy trials has contributed an important chapter in the long-running search for effective interventions in T1D. The results have been sobering in highlighting how complex the autoimmune process is, and how refractory to single immune-modulating interventions. However, we have learnt important lessons from monotherapy trials, including:
1. The roles of the various components of the immune response
2. The treatment effects that are potentially linked to beneficial vs. adverse outcomes
3. The need to explore novel combination therapy approaches to interdict more than one pathway in the dysregulated innate and adaptive responses
4. A possible way forward with novel trial designs
Although much work still needs to be done, at ITN we believe that the stage is set for the next phase in tackling this hitherto intractable autoimmune disease, and we now have additional tools to make significant inroads in the search for disease-modifying interventions.
Disclosure: The authors report no conflict of interests.
Figure 4.

Proposed adaptive trial design for dose optimization of IL-2 in new-onset T1D
Acknowledgments
The research cited was performed with the support of the Immune Tolerance Network (NIH Contract #N01 AI15416), an international clinical research consortium headquartered at the University of California San Francisco, and supported by the National Institute of Allergy and Infectious Diseases (NIAID) and the Juvenile Diabetes Research Foundation (JDRF). We thank Kevan C. Herold, Carla J. Greenbaum, Steven E. Gitelman, Gordon C. Weir, Terry B. Strom, Mark R. Rigby, and the members of the AbATE, IL-2/Rapa, START, RETAIN, and T1DAL study teams for their numerous contributions to the design, conduct, and analysis of the ITN trials described in this review.
References
- 1.Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. 2010;464:1293–1300. doi: 10.1038/nature08933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waldron-Lynch F, Herold KC. Immunomodulatory therapy to preserve pancreatic beta-cell function in type 1 diabetes. Nat Rev Drug Disc. 2011;10:439–452. doi: 10.1038/nrd3402. [DOI] [PubMed] [Google Scholar]
- 3.Skyler JS, Ricordi C. Stopping type 1 diabetes: Attempts to prevent or cure type 1 diabetes in man. Diabetes. 2011;60:1–8. doi: 10.2337/db10-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mandrup-Poulsen T. Interleukin-1 antagonists and other cytokine blockade strategies for type 1 diabetes. Rev Diabet Stud. 2012;9(4):338–347. doi: 10.1900/RDS.2012.9.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steffes MW, Sibley S, Jackson M, Thomas W. Beta-Cell function and the development of diabetes-related complications in the diabetes control and complications trial. Diabetes Care. 2003;26:832–836. doi: 10.2337/diacare.26.3.832. [DOI] [PubMed] [Google Scholar]
- 6.Palmer JP, Fleming GA, Greenbaum CJ, Herold KC, Jansa LD, Kolb H, Lachin JM, Polonsky KS, Pozzilli P, Skyler JS, Steffes MW. C-peptide is the appropriate outcome measure for type 1 diabetes clinical trials to preserve beta-cell function. Diabetes. 2004;53:250–264. doi: 10.2337/diabetes.53.1.250. [DOI] [PubMed] [Google Scholar]
- 7.Lachin JM, McGee PL, Greenbaum CJ, Palmer J, Pescovitz MD, Gottlieb P, Skyler J Type 1 Diabetes Trial Network. Sample size requirements for studies of treatment effects on beta-cell function in newly diagnosed type diabetes. Plos One. 2011;6:e26471. doi: 10.1371/journal.pone.0026471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greenbaum CJ, Mandrup-Poulsen T, McGee PF, Battelino T, Haastert B, Ludvigsson J, Pozzilli P, Lachin JM, Kolb H Type 1 Diabetes TrialNet Research Group, European C-Peptide Trial Study Group. Mixed-meal tolerance test versus glucagon stimulation test for the assessment of beta-cell function in therapeutic trials in type 1 diabetes. Diabetes Care. 2008;31:1966–1971. doi: 10.2337/dc07-2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chatenoud L, Bluestone JA. CD3-specific antibodies: A portal to the treatment of autoimmunity. Nat Rev Immunol. 2007;7:622–632. doi: 10.1038/nri2134. [DOI] [PubMed] [Google Scholar]
- 10.Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, Gitelman SE, Harlan DM, Xu D, Zivin RA, Bluetsone JA. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346:1692–1698. doi: 10.1056/NEJMoa012864. [DOI] [PubMed] [Google Scholar]
- 11.Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, Gorus F, Goldman M, Walter M, Candon S. et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005;352:2598–2608. doi: 10.1056/NEJMoa043980. [DOI] [PubMed] [Google Scholar]
- 12.Herold KC, Gitelman SE, Willi SM, Gottlieb PA, Waldron-Lynch F, Devine L, Sherr J, Rosenthal SM, Adi S, Jalaludin MY. et al. Teplizumab treatment may improve C-peptide responses in participants with type 1 diabetes after the new-onset period: A randomized controlled trial. Diabetologia. 2012 doi: 10.1007/s00125-012-2753-4. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herold KC, Ehlers M, Boyle K, McNamara J, Gitelman S, Gottlieb P, Greenbaum C, Hagopian W, Bluestone JA. Metabolic parameters at baseline identify clinical responders to teplizumab 2 years after diagnosis of type 1 diabetes. Diabetologia. 2012;55(Suppl 1):S191. [Google Scholar]
- 14.Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, Sene D, Cacoub P, Klatzmann D. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med. 2011;365:2067–2077. doi: 10.1056/NEJMoa1105143. [DOI] [PubMed] [Google Scholar]
- 15.Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP, Armand P, Cutler C, Ho VT, Treister NS. et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med. 2011;365:2055–2066. doi: 10.1056/NEJMoa1108188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Long SA, Rieck M, Sanda S, Bollyky JB, Samuels PL, Goland R, Ahmann A, Rabinovitch A, Aggaewal S, Phippard D. et al. Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments Tregs yet transiently impairs beta-cell function. Diabetes. 2012;61:2340–2348. doi: 10.2337/db12-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang N, Su D, Qu S, Tse T, Bottino R, Balamurugan AN, Xu J, Bromberg JS, Dong HH. Sirolimus is associated with reduced islet engraftment and impaired beta-cell function. Diabetes. 2006;55:2429–2436. doi: 10.2337/db06-0173. [DOI] [PubMed] [Google Scholar]
- 18.Valle A, Jofra T, Stabilini A, Atkinson M, Roncarolo MG, Battaglia M. Rapamycin prevents and breaks the anti-CD3-induced tolerance in NOD mice. Diabetes. 2009;58:875–881. doi: 10.2337/db08-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piemonti L, Maffi P, Monti L, Lampasona V, Perseghin G, Magistretti P, Secchi A, Bonifacio E. Beta cell function during rapamycin monotherapy in long-term type 1 diabetes. Diabetologia. 2011;54:433–439. doi: 10.1007/s00125-010-1959-6. [DOI] [PubMed] [Google Scholar]
- 20.Eisenbarth GS, Srikanta S, Jackson R, Rabinowe S, Dolinar R, Aoki T, Morris MA. Anti-thymocyte globulin and prednisone immunotherapy of recent onset type 1 diabetes mellitus. Diabetes Res. 1985;2:271–276. [PubMed] [Google Scholar]
- 21.Saudek F, Havrdova T, Boucek P, Karasova L, Novota P, Skibova J. Polyclonal anti-T-cell therapy for type 1 diabetes mellitus of recent onset. Rev Diabet Stud. 2004;1:80–88. doi: 10.1900/RDS.2004.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Voltarelli JC, Couri CE, Stracieri AB, Oliveira MC, Moraes DA, Pieroni F, Coutinho M, Malmegrim KC, Foss-Freitas MC, Simoes BP. et al. Autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus. JAMA. 2007;297:1568–1576. doi: 10.1001/jama.297.14.1568. [DOI] [PubMed] [Google Scholar]
- 23.Couri CE, Oliveira MC, Stracieri AB, Moraes DA, Pieroni F, Barros GM, Madeira MI, Malmegrim KC, Foss-Freitas MC, Simoes BP. et al. C-peptide levels and insulin independence following autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus. JAMA. 2009;301:1573–1579. doi: 10.1001/jama.2009.470. [DOI] [PubMed] [Google Scholar]
- 24.Gitelman SE, Fisher LK, Gottlieb PA, Gottschalk M, Moore WV, Moran A, Rigby MR, Willi SM, Keyes-Elstein L, Pinckney A. et al. Effect of anti-thymocyte globulin (ATG) on preserving beta cell function in new-onset type 1 diabetes. Diabetologia. 2012;55(Suppl 1):S191. [Google Scholar]
- 25.Greenbaum CJ, Beam CA, Boulware D, Gitelman SE, Gottlieb PA, Herold KC, Lachin JM, McGee P, Palmer JP, Pescovitz MD. et al. Fall in C-peptide during first 2 years from diagnosis. Evidence of at least two distinct phases from composite TrialNet data. Diabetes. 2012;61:2066–2073. doi: 10.2337/db11-1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crystal RG. Alpha1-Antitrypsin deficiency, emphysema, and liver disease. Genetic basis and strategies for therapy. J Clin Invest. 1990;85:1343–1352. doi: 10.1172/JCI114578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tuder RM, Janciauskiene SM, Petrache I. Lung disease associated with alpha1-antitrypsin deficiency. Proc Am ThoracSoc. 2010;7:381–386. doi: 10.1513/pats.201002-020AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janciauskiene SM, Nita IM, Stevens T. Alpha1-Antitrypsin, old dog, new tricks. Alpha1-Antitrypsin exerts in vitro anti-inflammatory activity in human monocytes by elevating cAMP. J Biol Chem. 2007;282:8573–8582. doi: 10.1074/jbc.M607976200. [DOI] [PubMed] [Google Scholar]
- 29.Zhang B, Lu Y, Campbell-Thompson M, Spencer T, Wasserfall C, Atkinson M, Song S. Alpha1-antitrypsin protects beta-cells from apoptosis. Diabetes. 2007;56:1316–1323. doi: 10.2337/db06-1273. [DOI] [PubMed] [Google Scholar]
- 30.Kalis M, Kumar R, Janciauskiene S, Salehi A, Cilio CM. Alpha 1-antitrypsin enhances insulin secretion and prevents cytokine-mediated apoptosis in pancreatic beta-cells. Islets. 2010;2:185–189. doi: 10.4161/isl.2.3.11654. [DOI] [PubMed] [Google Scholar]
- 31.Nita I, Hollander C, Westin U, Janciauskiene SM. Prolastin, a pharmaceutical preparation of purified alpha1-antitrypsin, blocks endotoxin-mediated cytokine release. Resp Res. 2005;6:12. doi: 10.1186/1465-9921-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lewis EC, Mizrahi M, Toledano M, DeFelice N, Wright JL, Churg A, Shapiro L, Dinarello CA. Alpha1-Antitrypsin monotherapy induces immune tolerance during islet allograft transplantation in mice. Proc Natl Acad Sci USA. 2008;105:16236–16241. doi: 10.1073/pnas.0807627105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koulmanda M, Bhasin M, Hoffman L, Fan Z, Qipo A, Shi H, Bonner-Weir S, Putheti P, Degauque N, Libermann TA. et al. Curative and beta cell regenerative effects of alpha1-antitrypsin treatment in autoimmune diabetic NOD mice. Proc Natl Acad Sci USA. 2008;105:16242–16247. doi: 10.1073/pnas.0808031105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ehlers MR, Gottlieb PA, Herold K, Rigby MR, Willi SM, Aggarwal S, Phippard D, Gelmont DM, Jepson B, McNamara J. et al. Alpha-1 antitrypsin therapy in new-onset type 1 diabetes: Interim results from Part I of the RETAIN study. Diabetologia. 2012;55(Suppl 1):S67. [Google Scholar]
- 35.Krueger GG, Callis KP. Development and use of alefacept to treat psoriasis. J Am Acad Dermatol. 2003;49:S87–S97. doi: 10.1016/mjd.2003.552. [DOI] [PubMed] [Google Scholar]
- 36.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, Gottlieb PA, Marks JB, McGee PF, Moran AM. et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361:2143–2152. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, Gottlieb PA, Greenbaum CJ, Marks JB, Monzavi R. et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: A randomized, double-blind, placebo-controlled trial. Lancet. 2011;378:412–419. doi: 10.1016/S0140-6736(11)60886-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moran A. Canakinumab, an anti-IL-1 monoclonal antibody in recent-onset type 1 diabetes. Philadelphia. June 11, 2012; 72nd Scientific Sessions of the American Diabetes Association. [Google Scholar]
- 39.Mandrup-Poulsen T. IL-1 receptor antagonist in recent onset type 1 diabetes: A multicenter randomized, placebo-controlled trial. Philadelphia. June 11, 2012; 72nd Scientific Sessions of the American Diabetes Association. [Google Scholar]
- 40.Wherrett DK, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, Gottlieb PA, Greenbaum CJ, Herold KC, Marks JB. et al. Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: A randomized double-blind trial. Lancet. 2011;378:319–327. doi: 10.1016/S0140-6736(11)60895-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sherry N, Hagopian W, Ludvigsson J, Jain SM, Wahlen J, Ferry RJ, Bode B, Aronoff S, Holland C, Carlin D. et al. Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomized, placebo-controlled trial. Lancet. 2011;378:487–497. doi: 10.1016/S0140-6736(11)60931-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matthews JB, Staeva TP, Bernstein PL, Peakman M, von Herrath M ITN-JDRF Type 1 Diabetes Combination Therapy Assessment Group. Developing combination immunotherapies for type 1 diabetes: Recommendations from the ITN-JDRF Type 1 Diabetes Combination Therapy Assessment Group. Clin Exp Immunol. 2010;160:176–184. doi: 10.1111/j.1365-2249.2010.04153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ablamunits V, Henegariu O, Hansen JB, Opare-Addo L, Preston-Hurlburt P, Santamaria P, Mandrup-Poulsen T, Herold KC. Synergistic reversal of type 1 diabetes in NOD mice with anti-CD3 and interleukin-1 blockade. Diabetes. 2012;61:145–154. doi: 10.2337/db11-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bresson D, Togher L, Rodrigo E, Chen Y, Bluestone JA, Herold KC, von Herrath M. Anti-CD3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing Tregs. J Clin Invest. 2006;116:1371–1381. doi: 10.1172/JCI27191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parker MJ, Xue S, Alexander JJ, Wasserfall CH, Campbell-Thompson ML, Battaglia M, Gregori S, Mathews CE, Song S, Troutt M. et al. Immune depletion with cellular mobilization imparts immunoregulation and reverses autoimmune diabetes in nonobese diabetic mice. Diabetes. 2009;58:2277–2284. doi: 10.2337/db09-0557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gottlieb PA, Quinlan S, Krause-Steinrauf H, Greenbaum CJ, Wilson DM, Rodriguez H, Schatz DA, Moran AM, Lachin JM, Skyler JS. et al. Failure to preserve beta-cell function with mycophenolatemofetil and daclizumab combined therapy in patients with new-onset type 1 diabetes. Diabetes Care. 2010;33:826–832. doi: 10.2337/dc09-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sarikonda G, Sachithanantham S, Kupfer T, Posgai A, Wasserfall C, Bernstein P, Straub L, Pagni P, Schneider D, Calvo TR. et al. Transient B-cell depletion with anti-CD20 in combination with proinsulin DNA vaccine or oral insulin: Immunologic effects and efficacy in NOD mice. Plos One. 2013 doi: 10.1371/journal.pone.0054712. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brusko TM, Putnam AL, Bluestone JA. Human regulatory T cells: Role in autoimmune disease and therapeutic opportunities. Immunol Rev. 2008;223:371–390. doi: 10.1111/j.1600-065X.2008.00637.x. [DOI] [PubMed] [Google Scholar]
- 49.Putnam AL, Brusko TM, Lee MR, Liu W, Szot GL, Ghosh T, Atkinson MA, Bluestone JA. Expansion of human regulatory T-cells from patients with type 1 diabetes. Diabetes. 2009;58:652–662. doi: 10.2337/db08-1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Techmanska I, Juscinska J, Wujtewicz MA, Witkowski P, Mlynarski W, Balcerska A. et al. Administration of CD4+CD25highCD127- regulatory T cells preserves beta-cell function in type 1 diabetes in children. Diabetes Care. 2012;35:1817–1820. doi: 10.2337/dc12-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Horwitz DA, Zheng SG, Wang J, Gray JD. The real role of TGF-beta in regulatory T cells physiology. Critical role of IL-2 and TGF-beta in generation, function and stabilization of Foxp-3+CD4+ Treg. Eur J Immunol. 2008;38:912–915. doi: 10.1002/eji.200738109. [DOI] [PubMed] [Google Scholar]
- 52.FDA Guidance for Industry. Adaptive design clinical trials for drugs and biologics. U.S. Food and Drug Administration; 2010. Feb, [Google Scholar]
- 53.Jones D. Adaptive trials receive boost. Nat Rev Drug Disc. 2010;9:345–348. doi: 10.1038/nrd3174. [DOI] [PubMed] [Google Scholar]