

Abstract
Recently, we have demonstrated that 13-cis retinoic acid (13cRA) down-regulates rat angiotensin type 1A receptor (AT1AR) gene transcription through a MAP Kinase (ERK1/2) dependent mechanism in rat liver epithelial and aortic smooth muscle cells. However, the exact mechanism remained unknown. In this study, we determined the signaling intermediates activated by ERK1/2 involved in 13cRA mediated AT1AR down-regulation. Serially deleted rat AT1AR promoter CAT constructs indicate fragments containing a region −2541 and −1836 bp upstream of the 5 prime possess an Sp1 consensus sequence (5′-TGGGGCGGGGCGGGG-3′) have reduced CAT activity. Mobility shift analysis using untreated nuclear extracts in the presence of mithramycin A suggest the trans-acting factor binding to this cis-acting element is Sp1. 13cRA significantly reduced specific binding without any change in Sp1 protein expression. Studies showed 13cRA maximally phosphorylates and tranlocates to the nucleus ERK1/2 within 5–10 minutes, activating Egr-1 mRNA expression at 20 minutes followed by de novo protein synthesis, leading to an Egr-1/Sp1 interaction. siRNA silencing of Egr-1 restored AT1AR mRNA and protein expression in 13cRA treated cells, and Sp1 silencing results in complete loss of AT1AR expression. Our study suggests that 13cRA mediated activation of ERK1/2, through Egr-1, is capable of disrupting Sp1, the requisite transactivator for AT1AR expression, providing a novel paradigm in AT1AR gene transcription.
INTRODUCTION
Angiotensin II (AngII) is a crucial hormone in fluid volume control and vasoconstriction through its stimulation primarily of the angiotensin type 1 receptor (AT1R) (Burnier, 2001; Atlas, 2007). As a result, it has become a key factor in the realm of hypertension research. Moreover, a greater understanding of AngII stimulation of AT1R effects as a growth factor, stimulator of angiogenesis, and oxidative stressor has expanded the interest in AT1R research beyond hypertension (Braunwald, 2008; Pan et al., 2010; Abadir, 2011). AT1R is a ubiquitously expressed protein, with significant effects in renal, cardiovascular, hepatic, and neural physiology. However, AT1R protein expression is tissue and cell-specific primarily due to differential transcription of the AT1R gene (Hannan et al., 2004; Tower et al., 2010; Braga, 2011). Changes in AT1R transcription also occur under pathophysiological conditions, such as damaged cardiac tissue after myocardial infarction (Sun & Weber, 1994), atrial tissue and the rostral ventrolateral medulla in chronic heart failure (Kaprielian et al., 1997; Gao et al., 2008), and in cardiac, renal and neural tissue in cases of spontaneous hypertension (Esler, 1993; Raizada et al., 1993; Lenkey et al., 1997). The coordination of AT1R gene regulation is complex. Research has shown that the basal transcription of AT1R largely relies on the activity of the transcription factor specificity protein 1 (Sp1) (Zhao et al., 2001). Moreover, up-regulation of AT1R in the vascular wall is involved in the induction of oxidative stress and in enhancement of endothelial dysfunction and plaque instability (Nickenig & Harrison, 2002). The cellular stress resultant from generation of reactive oxygen species induces greater binding activity of activator protein 1 (Ap1) (Wu et al., 2005), leading to a cyclic pattern in which AT1R becomes deregulated, first up-regulated by oxidant stress then contributing to a further ROS burden in the vasculature. Our own studies have shown that hormonal signaling has diverse effects on AT1R transcription. Growth factors, such as growth hormone, insulin, platelet-derived growth factor, and epidermal growth factor have a direct up-regulatory effect on AT1R transcription, acting through cis-acting elements (Wyse et al., 2000). Alternatively, AT1R (AT1AR – in studies of rodent species) transcription may be down-regulated by treatment with tannic acid (Yesudas et al., 2012), the estrogen metabolite 2-methoxyestradiol (Koganti et al., 2012), or high glucose (Thomas & Thekkumkara, 2004), a study in which we isolated a novel glucose response element (GluRE), though characterization of cis-acting elements for the other agents remains undetermined. As this receptor is regulated in multiple ways, addressing the mechanism of gene transcription is essential developing better strategies to control the effects of AngII.
Retinoic acids, metabolites of preformed vitamin A, exert their function through the binding of nuclear receptors (RAR and RXR) and forming complexes with conserved sequences on gene promoters termed retinoic acid response elements (RARE) (Mark et al., 2006). 13-cis retinoic acid (13cRA) is a synthetic form that functions similarly to the other isoforms, but its exact mechanism of action is unclear. It may spontaneously isomerize to all-trans retinoic acid, or possess a novel signaling pathway of its own (Kim et al., 1994; Ganceviciene & Zouboulis, 2007). However, 13cRA has relatively weak transactivation activity for RAR/RXR compared with the endogenously formed isoforms all-trans and 9-cis retinoic acids (Mangelsdorf et al., 1994). Although 13cRA has multiple applications as a dermatological treatment (Landthaler et al., 1980) and as an effective therapeutic for numerous myelodysplastic and proliferative conditions (Piattellia et al., 1999; Zhang & Duvic, 2003; Siitotoen et al., 2007), the cellular mechanisms and molecular targets are not well understood. Studies have shown that 13cRA can act as a potent inhibitor of many of the retinoid and hydroxysteroid mediated pathways (Gamble et al., 1999). This may indicate how 13cRA mediates its antagonistic effects on sebum secretion and skin growth factors. However, many of these studies cannot provide compelling evidence of a distinct cellular mechanism. Interestingly, it has been proposed that 13cRA may have non-nuclear targets, based upon differential activity from all-trans and 9-cis retinoic acids (Blaner, 2001).
In a recently published study, we have demonstrated that in both rat liver epithelial and aortic smooth muscle cells, 13cRA is capable of down-regulating AT1AR by inhibiting AT1AR mRNA transcription (Snyder & Thekkumkara, 2012). Additionally, the study revealed that 13cRA induced AT1AR down-regulation is dependent on the activation of MAPK p42/p44 (ERK1/2). Importantly, this study suggested that the response in AT1R expression was effective in cells of hepatic origin, in which it has been shown that AT1R signaling blockade or interference provides substantial attenuation of inflammatory processes leading to fibrosis and steatosis (Ratziu and Zelber-Sagi, 2009). Angiotensin converting enzyme inhibition (ACEI) and angiotensin receptor blocker (ARB) use can significantly lower fibrosis index scores (Yoshiji et al. 2009; Kim et al. 2008). Thus, the findings of our preceding study were valuable in demonstrating a method of down-regulating AT1AR expression in hepatic and cardiac tissue, possibly providing an alternate approach to traditional ARB and ACEI pharmacology. However, the downstream mechanism(s) involved in 13cRA induction of ERK1/2 leading to AT1AR down-regulation remained undetermined, as the classical retinoic acid receptors (RAR/RXR) were not involved. The purpose of the present study was to identify the downstream effectors responsible for the inhibition of AT1AR transcription to further elucidate the mechanism of ERK1/2 mediated AT1AR repression.
METHODS
Materials
Continuously passaged rat liver epithelial cells (WB cells) were kindly provided by Dr. H. Shelton Earp, University of North Carolina at Chapel Hill (Chapel Hill, NC, USA). Richter’s improved minimal essential medium was obtained from Cellgro-Mediatech Inc. (Manassas, VA, USA). Fetal bovine serum (FBS) was from Equitech-Bio, Inc. (Kerrville, TX, USA). Oligonucleotide primers and EMSA nucleotide duplex probes were obtained from Integrated DNA technologies, Inc. (Coralville, IA). PCR master mix was from Roche (Indianapolis, IN, USA). Losartan was provided by Merck Sharp & Dohme Research Laboratories (Rahway, NJ, USA). 13-cis retinoic acid, insulin, mithramycin A, and gentamicin were from Sigma (St. Louis, MO, USA). DNA/RNA extraction reagents were from Ambion/ABI (Austin, TX, USA). [3H]AngII was from American Radiolabeled Chemicals (St. Louis, MO, USA). [α-32P]dGTP was from Perkin-Elmer (Waltham, MA, USA). PD98059 was from Calbiochem (La Jolla, CA, USA). Sp1 (SC-59) was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Egr-1 (cat no. 4153), phospho-ERK1/2 (cat no. 4370) and total ERK1/2 (cat no. 4695) antibodies were from Cell Signaling (Danvers, MA, USA). siRNA Silencer® Select pre-validated oligonucleotides, Opti-MEM®, and transfection reagents (Lipofectamine LTX® and RNAiMAX®) were from Life Technologies. Electrophoresis regents were from Bio-Rad (Hercules, CA, USA) and all other chemicals and molecular biology grade agents were purchased from Fisher Scientific (Fairlawn, NJ).
Construction of Expression Plasmids
All DNA manipulations were carried out according to previously published methods (Wyse et al., 2000). 3,372 bp of the 5-prime promoter sequence of the rat AT1AR, (ref# NC005116.2) was custom cloned into pUC57 carrier vector and sequence confirmed by GenScript® (Piscataway, NJ). By using the genomic clone as a template for polymerase chain reaction (PCR) and oligonucleotides corresponding with the published sequence of the rat AT1R promoter, fragments of varying length were amplified. The oligonucleotide corresponding to exon 1 (+23 to +45 bp) of the rat AT1AR (5′-ACAGATCTTCTCCAGCGGGACA-3′) was used as the antisense primer for each reaction. The varying sense primers (rP1, 5′-GCCTTATGCTAGCCTCCCTCCATC-3′; rP2, 5′-CCTTTGCTAGCCTTCCTTCCATC-3′; rP3, 5′-GGGCTAGCGCGAGAGAGCCA-3′; rP4, 5′-AAGGCTAGCGCAGAAACAGACTCT-3′; rP5, 5′-GTTGAGCTAGCATAATGAGGGGCA-3′; rP6, 5′-CAAATACTGAGGTCAAAGCTAGCAGCAA-3′; and rP7, 5′-GAATTCGAGCTCGGTACCTCGCGA-3′) yielded fragments of 310, 652, 1266, 1836, 2541, 2824, and 3321 bp, respectively. Each fragment was double digested with BglII and (for rP1-rP6) and NheI. For rP7, it was necessary to use an alternate double digestion with BglII and KpnI due to the presence of an NheI digestion site downstream to the 5-prime terminus of the rP7 fragment. Fragments rP1 – rP7 utilized primers that had incorporated restriction sites for 5-prime selective NheI/KpnI digestion and 3-prime selective BglII digestion. Double digested fragments were unidirectionally ligated into the multiple cloning site of the pCAT-Basic vector (Promega) digested with the same enzymes. The authenticity of clones was confirmed by automated bidirectional dideoxy sequencing. Mutated rP7X promoter construct was assembled using similar techniques, with additional modification by selective PCR amplification of sequences flanking the upstream Sp1 response element, and subsequent BamHI digestion of modified primer sequences followed by ligation to exclude said response element.
Cell Culture and Transfection of Reporter Gene Constructs
The WB cells were maintained in Richter’s improved MEM supplemented with 10% FBS, 10 mM glucose, 17.8 mM HEPES, 5.4 μg/ml insulin, and 44.6 μg/ml gentamicin (complete medium), at 37°C in 5% CO2 with 100% humidity. For the studies, cells were grown to 70–80% confluence and the medium was exchanged with treatment medium (Richter’s improved MEM supplemented with 5% FBS, 5 mM glucose, 17.8 mM HEPES, 5.4 μg/ml insulin, and 44.6 μg/ml gentamicin). After 24 h cells were exposed to treatment medium containing 13cRA for indicated concentrations and times. For transient transfection of DNA constructs, WB cells were seeded in 6-well plates and grown to 70–80% confluence in complete medium. Cells were transiently transfected with 3 μg of reporter plasmids and cotransfected with 1 μg of pSV-β-galactosidase expression construct (to act as an internal control for transfection efficiency) by using Lipofectamine LTX and PLUS reagent according to manufacturers instructions (Invitrogen, Carlsbad, CA) and grown for 24 h in complete medium. Following 24 h growth in complete medium, treatment medium and 13cRA were added consistent with all other studies.
Chloramphenicol Acetyl-Transferase Assay
The CAT assays were performed according to manufacturer’s instructions (Invitrogen FASTCAT® Protocol). Briefly, cells were rinsed with phosphate-buffered saline (PBS) three times and harvested in the same buffer. Cells were then centrifuged and the resultant pellet was resuspended in 100 μl of 0.25 M Tris-HCl, pH 7.8. Cellular extracts were prepared by freeze thaw cycling from the −80°C freezer to the 37°C water bath for 4 times. The cells were then centrifuged (17,530 X g at 4°C) for 10 minutes. 30 μl of the supernatant was removed and β-galactosidase activity was measured using a colorimetric assay according to the previously published method (Wyse et al., 2000). The remaining supernatant was heated at 70°C for 10 min to inactivate endogenous acetylases and centrifuged further to remove cell debris. The assay for CAT activity was performed as described in the FAST-CAT protocol. The green fluorescence was visualized under the VERSA-Doc® System at 504 nm absorbance and 510 nm emission. The acetylated chloramphenicol was then measured by fluorescent densitometry and normalized to arbitrary β-galactosidase activity/mg protein.
Electrophoretic Mobility Shift Assay
Nuclear extracts were prepared using NE-PER nuclear protein extraction kit (Thermo-Scientific). Protein concentration was determined using Bio-Rad® protein assay reagent based on the Bradford method (Bradford, 1976), and extracts used immediately after preparation. Mobility shift assays were performed as described previously (Wyse et al., 2000). Briefly, a double-stranded Sp1 consensus sequence with a 5-prime overhang was custom synthesized as duplex DNA by Integrated DNA Technologies® (sense primer, 5′-CCCCGCCCCGCCCCA-3′; antisense primer, 5′-TGGGGCGGGGCGG-3′). The double-stranded Sp1 response element (Sp1RE) was labeled by filling the overhang with DNA polymerase Klenow in the presence of [α-32P]dGTP. The labeled probe was purified using a Sephadex G-25 column. 10 μg of nuclear extracts were preincubated with 2 μg of poly(dIdC) in a total volume of 20 μl of binding buffer comprised of 0.1 M Tris-HCl, 50% glycerol, 0.2 M KCl, 0.5 M EDTA, and 1.0 M DTT at 22°C for 20 min. Where appropriate, the reaction mixture was supplemented with either proteinase K, 10 μM mithramycin, or varying concentrations (0–200 fold excess) of double-stranded, unlabeled Sp1RE. The labeled probe (300,000 cpm) was added and the reaction mixture was further incubated for 30 min at 22°C. Reactions were separated on a 6% native polyacrylamide gel, dried, and exposed to Kodak XR-film at −70°C with intensifying screens.
Angiotensin Receptor Binding Studies
Receptor binding studies were performed in triplicate on WB cells as described previously (Snyder & Thekkumkara, 2012). Briefly, cells were washed twice with PBS and incubated with [3H]AngII (20 pM) at 22°C in a binding buffer containing 50 mM Tris HCl (pH 7.5), 120 mM NaCl, 4 mM KCl, 5 mM MgCl2, 1 mM CaCl2, 10 μg/ml bacitracin, 2 mg/ml dextrose, and 2.5 mg/ml bovine serum albumin. After 60 min incubation cells were washed with ice cold PBS three times to remove free radioactivity. Cells were then lysed with 0.1% Triton X-100 in PBS. The cells were scraped and transferred to counting vials and radioactivity was determined using a Beckman liquid scintillation counter. Specific [3H]AngII binding was defined as that portion of the total binding displaced by 1 μM unlabelled AngII. At equilibrium, specific binding was more than 95% of the total binding. Protein concentrations were determined using Bio-Rad protein assay system based on the Bradford method (Bradford, 1976).
Reverse Transcriptase/Dual PCR and real-time PCR
Cells were grown to 70–80% confluency and treated with agents for indicated times. Total RNA was isolated using guanidinium thiocyanate-phenol-chloroform method as described previously (Snyder & Thekkumkara, 2012). 5 μg total RNA was processed for cDNA template conversion using MLV-RT. The reaction without reverse transcriptase served as control for DNA contamination. For real-time quantitative analysis of Egr-1 mRNA, two parallel PCR reactions each containing 5 μg total mRNA and either Egr-1 (Egr-1 sense-5′GTTGCCTCCCATCACCTA-3′: Egr-1 antisense – 5′-CAG AGGAAGACGATG-3′), AT1R (AT1R sense-5′-TGATTCAGCTGGG CGTCATCCA-3′; AT1R antisense-5′-TTTCGTAGA CAGGCTTGAGTGGG-3′) or β-actin specific primers (β-actin sense – 5′-CGGAACCGCTCATTGCC-3′; β-actin antisense-5′-ACCCACACTGTGCCCATCTA-3′) were performed using 2X SYBR Green Master Mix (Applied Biosystems). Following the reaction, threshold cycles (Ct) were calculated for each sample for Egr-1 and β-Actin mRNA reactions, and quantitative concentrations were calculated using ΔΔCt calculations described by Perkin Elmer Applied Biosystems.
Western Blot Analysis
Cells were lysed by sonification in lysis buffer containing 50 mM HEPES, 1% Triton X-100, 50 mM NaCl, 50 mM NaF, 10 mM sodium-pyrophosphate, 5 mM EDTA, and one tablet per 10 ml of Mini-Complete® Protease Inhibitor (Roche, Branford CT). Equal amounts of protein (25 μg/lane) were resolved by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred onto a nitrocellulose filter (Millipore, USA). The filters were blocked with tris-buffered saline (TBS) containing 5% nonfat milk and incubated with monoclonal antibody against Sp1, Egr-1, phospho-ERK1/2 or total ERK1/2 (1:1000 dil., Cell Signaling, USA) and monoclonal antibody against actin (1:3000 dil., Santa Cruz, USA). After washing three times with TBS containing 0.5% Tween 20, bound primary antibody was detected with anti-rabbit HRP conjugated goat-IgG (1:5000 dil., Santa Cruz, USA). Immunoreactive proteins were visualized using the chemiluminescent substrate kit from Pierce Biomedical (Thermo Scientific Pierce, USA).
Immunofluorescent Microscopy
Cells were seeded and grown in four-chamber microscope slides (Bio-Tek). After indicated treatment times the cells were washed once with cold PBS and fixed for 20 minutes in 4% paraformaldehyde. Cell nuclei were further perforated by incubation in 100% pure methanol at −20°C for 10 min. The cells were then blocked in 5% goat serum for one hour and incubated overnight at 4°C in rabbit primary antibody [1:1000 dil. Cell Signaling Technologies] directed against rat Egr-1, Sp1, or phospho-ERK1/2 proteins. Cells were washed five times with cold PBS and incubated at room temperature for two hours with FITC conjugated secondary goat anti-rabbit antibody [1:3000 dil. Santa Cruz]. Cells were washed an additional five times and stained with 10 nM 4′,6-diamidino-2-phenylindole (DAPI) for 5 min. After a final wash, coverslips were fixed to slides with ProLong Gold AntiFade [Invitrogen] for 48 h. Image capture was performed at 22°C using an Olympus® IX-81 microscope equipped with an Olympus® U-CMAD3 camera under a PlanApo® 60x/1,40 oil immersion objective. Images were analyzed using Slidebook® software.
Co-Immunoprecipitation
Nuclear extracts were prepared as mentioned above from 100 mm plates of 90–100% confluent culture plates using the NE-PER® nuclear extraction kit (Thermo Scientific). Protein content was determined by Bio-Rad DC protein assay system, and equivalent portions of 150 μg were aliquoted for each reaction group to 25 μl of nuclear extract buffer, the sample was further diluted to 150 ul in reaction buffer (150 mM NaCl, 10 mM HEPES, pH 7.5, 0.2% Nonidet P-40, 5 mM sodium fluoride, 5 mM sodium pyrophosphate, 2 mM sodium orthovanadate, 10 mg/L aprotinin, 10 mg/L leupeptin, and 1 mM phenylmethyl-sulfonyl fluoride). Protein A/G Sepharose [PAGS] (Cell Signaling, USA) was washed with reaction buffer and samples were precleared by a 1 h incubation with 50% PAGS. After centrifugation, the precleared lysate was suspended with 4 μg of capture antibody (either anti-Egr1 [Cell Signaling, USA] or anti-Sp1 [Santa Cruz, USA]) and incubated with gentle rocking for 90 min at 4°C. 50 μl of washed 50% PAGS was added to the reaction and incubated for a further 90 min at 4°C with gentle rocking. The PAGS was separated by brief centrifugation, and washed with 500 μl of cold wash buffer (150 mM NaCl, 10 mM HEPES, pH 7.5, and 0.2% Nonidet P-40) three times. A final was with 500 μl of 50 mM HEPES, pH 7.5 was performed, and the supernatant was removed as completely as possible from the PAGS beads. The proteins/antibody complexes bound to sepharose were eluted by addition of 25 μl of reducing 2X SDS sample buffer (120 mM Tris-HCl, pH 6.8, 3.3% SDS, 10% glycerol, 40 ug/mL bromphenol blue, and 200 mM dithiothreitol) and incubation at 100°C in a tightly capped tube for 5 min. The PAGS was pelleted by centrifugation (14,000 X g 5 min, RT) and 20 μl of the supernatant was transferred to a clean tube. The sample was then loaded into a SDS-PAGE and analyzed by Western blot, directed against Egr-1 or Sp1, as dictated by the capture antibody employed before.
siRNA Transfection
Cells were plated in 100 mm plates and grown to 60% confluence. Cells remained in growth media. In order to ensure effective gene knockdown, we employed a combination of 3 separate 21 mer Silencer Select® validated siRNA oligos at a concentration of 50 picomoles/10 mL of transfection media for each knockdown experiment. Provided successful transfection, each oligo is guaranteed to specifically knockdown target gene expression at least 80%. Transfection mixtures were prepared as follows for the 100 mm plates: 50 picomoles of each interfering RNA suspensions combined (a total of 150 picomoles for the three respective siRNA molecules provided (Egr1 KO oligos – Life Technologies Cat# s127689, s127690, s127691; Sp1 KO oligos – Life Technologies Cat # s128430, s128431, s128429). These siRNAs were diluted in Opti-MEM serum free media (150 picomoles/1 ml). In an additional tube, 30 μl of Lipofectamine RNAiMAX was diluted in 1 mL of Opti-MEM. The diluted lipofectamine was added to the RNA containing Opti-MEM solution. The 2 mL of reaction mixture was incubated for 20 min at 22°C. At the end of this incubation, the lipofectamine/RNA complex was added dropwise to the 100 mM plates containing 10 mL of growth media. The plates were allowed to grow an additional 24 hours, when the growth media was replaced with treatment media, and either AT1AR, Egr1, or Sp1 expression was assessed by previously mentioned methods.
Data Analysis
Sequence analyses and alignments were performed using MacVector 12.0 sequence analysis software. Results are presented as mean ± S.E.M and the value of P<0.05 was considered statistically significant. Statistical analysis included column statistics to test for normal distribution and parametric t-test or one-way ANOVA with post-hoc Bonferroni analysis as appropriate. Values are normalized to milligrams of protein determined by Bio-Rad DC protein assay system based on the Bradford method (Bradford, 1976). Data were analyzed using the GraphPad Prism® software.
RESULTS
AT1AR Promoter Activity
In our previous study, we have shown that exposure to 13cRA reduced the expression of rat AT1AR protein via transcriptional down-regulation (Snyder & Thekkumkara, 2012). To further determine whether there was promoter activity of AT1AR, and if so, whether down-regulation is involved with specific cis-acting elements, we investigated the involvement of the −3276 to +45-base pair promoter sequence in the regulation of the rat AT1AR gene transcription. Functional analysis of the rat AT1AR gene transcriptional regulator unit was performed using a reporter gene assay, which relies on the linkage of putative regulatory sequences to a reporter chloramphenicol acetyl transferase (CAT) gene, whose transcription is detected after transfection into WB cells. To determine specific regions responsible for 13cRA mediated AT1AR down-regulation, serial deletions were constructed and placed upstream of the CAT reporter gene [Fig. 1A]. These serially deleted promoter fragment containing reporter gene constructs were transiently transfected into cells and were then treated with 25 μM 13cRA for 24 hours. Each group was compared parallel to a control group transfected with the same fragment construct but not treated with 13cRA, thereby accounting for any variable activity not due to the 13cRA treatment, as the gene may be affected by any number of different factors when the promoter is truncated unrelated to treatment (Thomas & Thekkumkara, 2004) [Fig. 1B]. Activity relative to control was not significantly different in groups pCAT-rP1 (−310 bp—p>0.05) through pCAT-rP4 (−1836 bp—p>0.05). However, the CAT activity (and thus the CAT gene expression) was significantly suppressed in retinoic acid treated cells expressing pCAT-rP5 (−2541 bp, 61.1±5.641% reduced activity, p<0.001) through pCAT-rP7 (−3321 bp, 62.26±5.748% reduced activity, p<0.001), indicating the presence of a cis-acting element between −2541 and −1836 bp responsible for inhibition. Although this sequence is considerable in length, consensus binding sites to characteristic response elements are rare in this region. Sequence analysis revealed a single true match for an Sp1 putative response element (5′-TGGGGCGGGGCGGGG-3′) consensus site within this upstream region, which led to further investigation of Sp1’s involvement in 13cRA-induced down-regulation of AT1AR.
Figure 1.
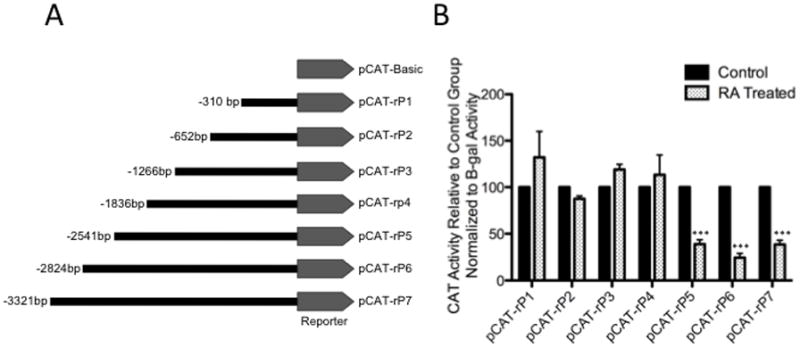
CAT activity of rat AT1R reporter gene constructs in 25 μM 13cRA. CAT activity for serially deleted promoter fragments in pCAT vector was determined. (A) Schematic representation of the pCAT reporter expression vector containing serially deleted 5′ promoter region of the rat AT1R gene. (B) Solid bars represent the CAT activity of control plasmids, the value set automatically at 100% activity relative to β-galactosidase co-transfection control, patterned bars represent the comparative CAT activity in 25 μM 13cRA treated cells. Data are expressed as mean ± SEM, **p<0.01, n=3.
Determination of the Role of Putative Sp1 Binding Site in 13cRA Mediated Down-Regulation of AT1AR
To determine whether the distal Sp1 response element is sensitive to 13cRA exposure, we performed a gel shift assay with nuclear extracts of control and 13cRA exposed WB cells using [32P] labeled Sp1 response element as a probe [Fig. 2A]. The result showed a distinct mobility shift of the labeled probe (indicated by arrow) with control nuclear extracts [Figure 2A, Lane 2] which was significantly reduced after pretreatment with 13cRA [Figure 2A, Lane 3]. Incubation of the extracts with proteinase K eliminated the signal, indicating the formation of a protein-DNA complex [Figure 2A, Lanes 4 & 5]. The protein-DNA binding was specific for nuclear factor(s) as evidenced by observed lack of mobility shift with the cytosolic extract [Figure 2A, Lane 6]. To determine the specificity of this reaction, nuclear extract was incubated with labeled probe and increasing concentrations (0–200X) of unlabeled Sp1 response element probe which progressively inhibited the appearance of labeled DNA-protein complex, demonstrating the specificity of the nuclear protein(s) binding to Sp1RE [Fig. 2B]. To further demonstrate the identity of the protein as Sp1, nuclear extracts were prepared after pre-treatment with mithramycin A, a nuclear factor inhibitor when used in conjunction with the consensus sequence allowed positive identification of Sp1 as the binding protein of this element [Fig. 2C]. When the nuclear extracts from mithramycin A treated cells were added to the probe, the binding of the protein to the labeled DNA was significantly reduced (85.31±4.44%, **p<0.01, n=3) [Figure 2C, Lane 3]. However, Western blot analysis revealed that Sp1 expression remained unaffected by the treatment with 13cRA in these cells [Fig. 2D]. Additionally, we performed radio-ligand binding studies to show the effect of mithramycin A, and thus Sp1 selective inhibition, on the expression of AT1R protein [Fig. 2E]. The results demonstrate that after treatment with 20 μM mithramycin A significantly reduced [3H]AngII binding (reduction of 52.7±9.91% compared to untreated control, p=0.006, n=3). Moreover, this reduction was not only similar to that resulting from 25 μM 13cRA treatment (p=0.6345, n=3), but combinatorial treatment of both mithramycin A and 13cRA resulted in no additive or synergistic effect (reduction of 57.9±9.14% compared to untreated control, p=0.0032; when compared to 13cRA treated group, p=0.6905; when compared to mithramycin A treated group, p=0.4166, n=3) suggesting that Sp1 inhibition by 13cRA was not enhanced by further addition of mithramycin A. Therefore, we propose that 13cRA may induce a trans-acting factor to interact with Sp1, thereby inhibiting Sp1’s ability to interact with the AT1AR promoter. However, in order to validate the distal Sp1 consensus sequence, a mutant promoter construct lacking the putuative response element (5′-TGGGGCGGGGCGGGG-3′) was created and designated as promoter construct “rP7X” [Fig. 2F]. CAT assay results indicate that following 13cRA treatment, there was a significant reduction in acetyl-transferase activity in rP7 non-mutated constructs as shown in Fig. 1B (reduction of 48.1±4.37% compared to untreated control, p=0.0027, n=3) [Fig. 2G]. However, after similar treatment with 25 μM 13cRA, rP7X mutated constructs showed no significant reduction of CAT activity (increased activity of 3.82±4.28% compared to untreated rP7X control, p=0.2629, n=3), indicating that loss of the putative Sp1 response element eliminates 13cRA’s ability to alter AT1R promoter activity.
Figure 2.
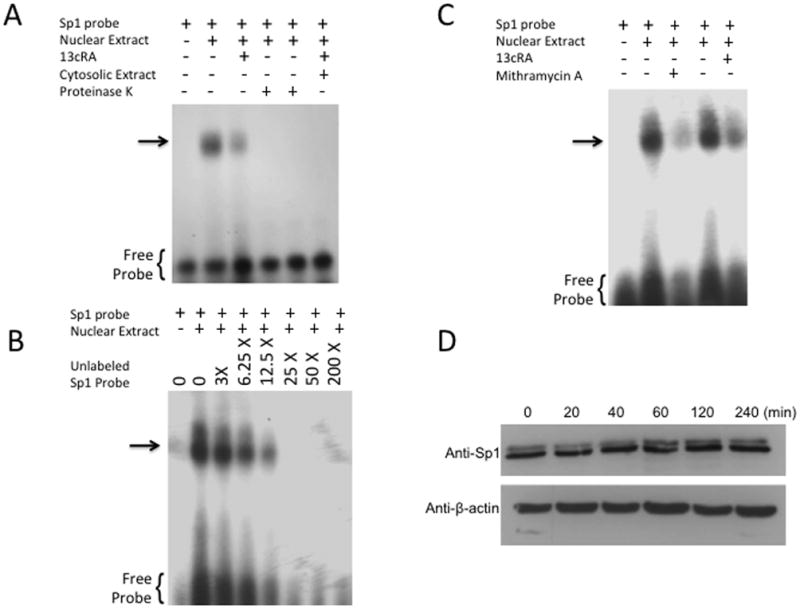
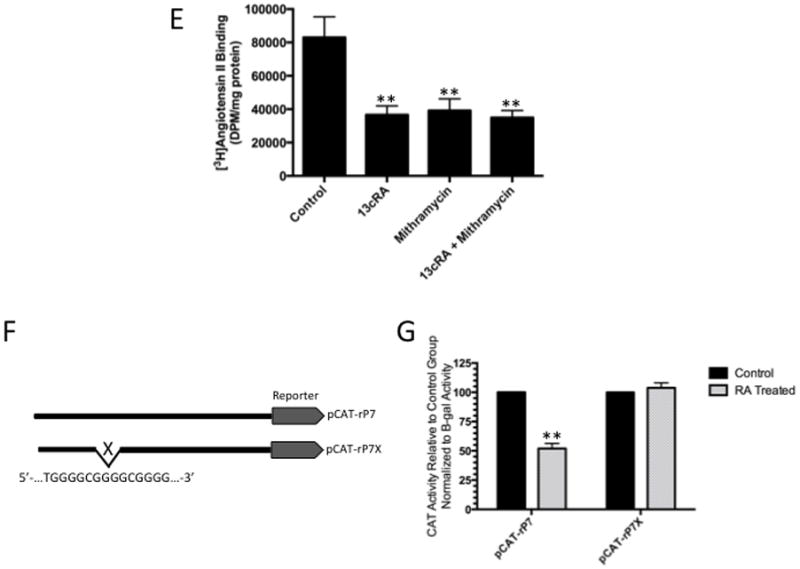
Identification of specific protein binding activity in WB nuclear extracts to Sp1RE. To identify protein binding to Sp1RE in 13cRA treated WB nuclear extracts, mobility shift assays were performed. 32P-labeled Sp1 probe was incubated with 10 μg of nuclear or cytosolic extracts. Samples were analyzed on 6% non-denaturing polyacrylamide gels and visualized by autoradiography. The position of the protein-DNA complex is indicated by arrow. (A) Labeled probe in the absence of nuclear or cytosolic extract (lane 1), in the presence of untreated nuclear extract (lane 2), in the presence of 13cRA exposed nuclear extract (lane 3), nuclear extracts of both untreated and treated with 13cRA in the presence of proteinase K (lanes 4 and 5), and in the presence of cytosolic extract (lane 6). (B) Mobility shift assay performed using labeled Sp1RE in the presence of increasing concentrations of unlabeled Sp1RE probe as indicated. The position of the protein-DNA complex is indicated by arrow. (C) Mobility shift assay of nuclear extracts treated with 13cRA or mithramycin A, both showing significant reduction in protein binding activity. (D) Time course 13cRA treatment Western blot analysis of Sp1 protein within the time-frame in which EMSA analysis was conducted reveals no significant change in Sp1 expression, p>0.05, n=3. (E) [3H]AngII binding assay demonstrating significant reduction in AngII binding in 13cRA, mithramycin A, and 13cRA + mithramycin A conditions (**p<0.01, n=3). (F) Schematic representation of the pCAT reporter expression vector unaltered rP7 portion of rat AT1R gene or modified mutant lacking indicated Sp1 response element. (G) Solid bars represent the CAT activity of control plasmids, the value set automatically at 100% activity relative to β-galactosidase co-transfection control, patterned bars represent the comparative CAT activity in 25 μM 13cRA treated cells. Data are expressed as mean ± SEM, **p<0.01, n=3.
13cRA Mediated de novo Synthesis of Egr-1
From previous studies, we confirmed that phosphorylation of ERK1/2 was essential for down-regulation of the AT1AR protein. In this study, when cells were exposed for different time periods, as indicated, we observed ERK1/2 was maximally phosphorylated within 5 minutes, followed by progressive dephosphorylation for the remainder of the 60-minute test period [Fig 3A]. Immunofluorescent microscopy studies revealed that phosphorylated ERK1/2 was primarily resident in the nucleus [Fig. 3B]. Upon further investigation, we observed increased Egr-1 expression in 13cRA exposed cells. We performed qPCR analysis using total RNA isolated from cells treated with 13cRA at different time points. qPCR results revealed that Egr-1 mRNA was up-regulated 403±57.8% (p<0.001, n=3) at 20 min, which indicated that 13cRA mediates increased Egr-1 gene transcription [Fig. 4A]. A 4-hour time-course Western blot analysis directed against Egr-1 showed a significant increase in Egr-1 expression in 13cRA treated cells, reaching maximum response at 40–60 minutes followed by a rapid decrease in expression for the remainder of the time points [Fig. 4B]. Egr-1 is a resident nuclear protein and early response gene, but in order to validate the Western blot data, we conducted a 120-minute time-course immunofluorescent microscopy study. The results of this study demonstrate that Egr-1 protein is up-regulated in 13cRA treated cells, consistent with the Western blot data, and the protein is localized primarily to the nucleus [Fig. 4C]. In order to determine whether Egr-1 up-regulation requires de novo synthesis, we performed a study in which cells were pretreated with the protein synthesis inhibitor cycloheximide. This analysis revealed that no Egr-1 protein up-regulation occurred if cells were pretreated with cycloheximide, and thus the Egr-1 up-regulation was due to de novo protein synthesis [Fig. 5A]. Egr-1 up-regulation was rapid and significant; however, we had not yet proven that this up-regulation was due to ERK1/2 phosphorylation. Western blot analysis in which cells were pretreated with the MEK inhibitor PD98059 revealed that if MEK and subsequent phosphorylated ERK1/2 were inhibited, no increase in Egr-1 production occurred [Fig. 5B]. After correlation with ERK1/2 activation, the next objective in our study was to establish Egr-1 interaction with Sp1 protein as described previously (Srivastava et al., 1998). We conducted a co-immunoprecipitation study in which antibodies targeting either Sp1 or Egr-1 in the nuclei of 13cRA exposed cells were immunoprecipitated and targeted with the corresponding detection antibody by Western blot analysis. The results of this study demonstrate that there is significant interaction between Egr-1 and Sp1 in 13cRA treated cells [Fig. 5C & 5D]. In Egr-1 Western blot analysis, some interaction is observed in untreated cells; however, this interaction is 518±67% stronger in 13cRA treated cells. The membranes were reprobed using the respective capture antibody to confirm that the target-precipitated protein was effectively transferred following protein electrophoresis [Fig. 5E & 5F]. Collectively these experiments suggest that there is a protein-protein interaction occurring between Egr-1 and Sp1.
Figure 3.
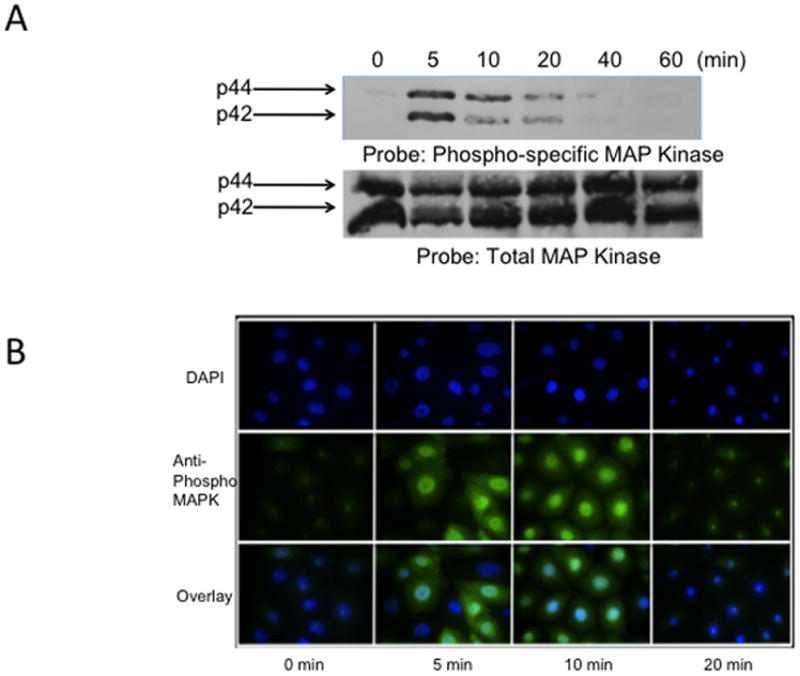
13cRA mediates early phosphorylation of ERK1/2. (A) Western blot analysis of phospho-MAPK relative to total-MAPK after treatment with 25 μM 13cRA at indicated times. (B) Immunofluorescent microscopy time-course directed against phospo-MAPK after treatment to 25 μM 13cRA at indicated times. Anti-phospho ERK1/2 labeled with FITC (green) and nuclei stained with DAPI.
Figure 4.
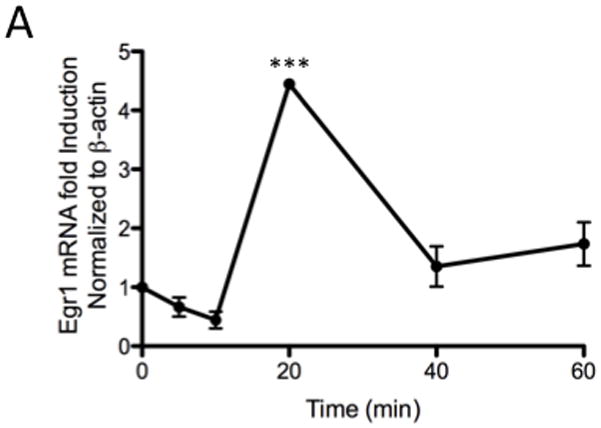
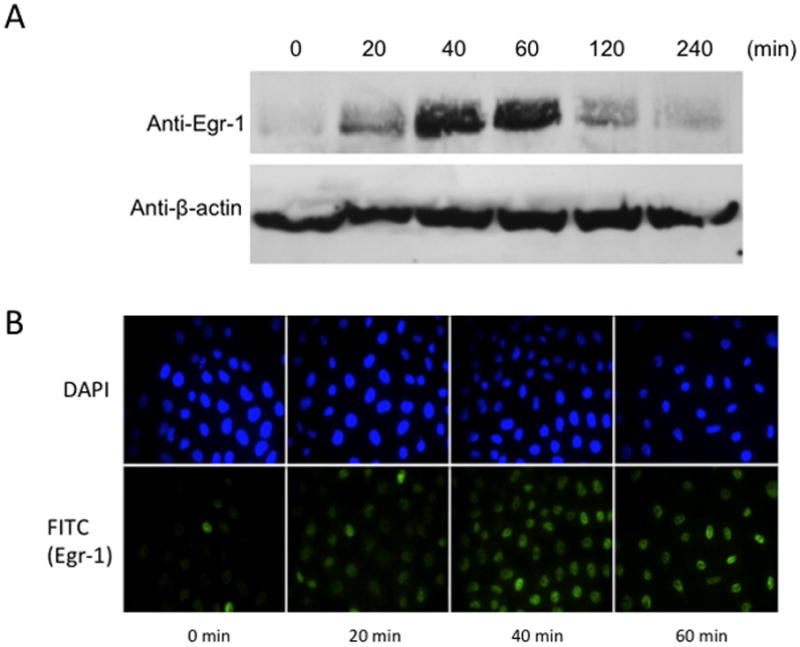
13cRA mediates activation of Egr-1 expression. (A) Real-time quantitative PCR detection of Egr-1 transcripts plotted as a time course, reaching maximum expression at 20 min (***p<0.001 compared to control, n=3). (B) Western blot analysis of Egr-1 after treatment with 25 μM 13cRA at indicated times. (C) Immunofluorescent microscopy time-course directed against Egr-1 after treatment to 25 μM 13cRA at indicated times. Anti-Egr-1 labeled with FITC (green) and nuclei stained with DAPI.
Figure 5.
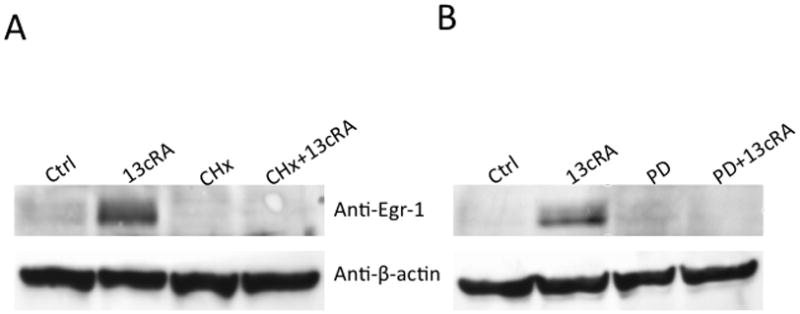
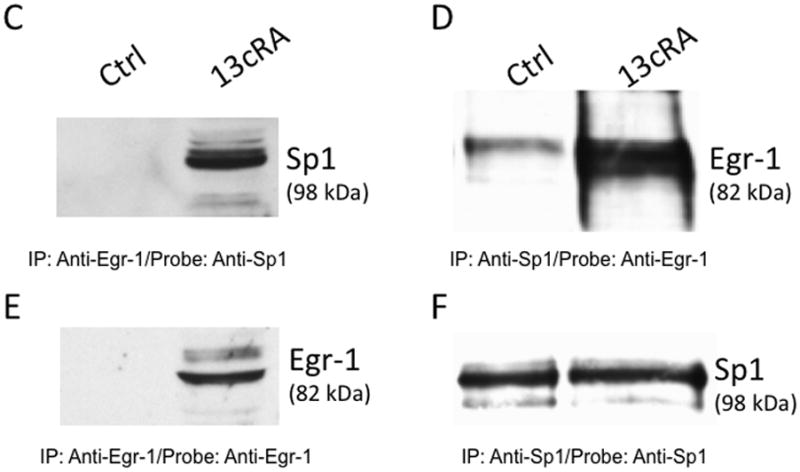
Egr-1 synthesis is dependent on ERK1/2 transcriptional activation and de novo synthesis. (A) Western blot analysis of Egr-1 after 1 h treatment with 25 μM 13cRA, with or without the MEK inhibitor (20 μM) PD98059. (B) Western blot analysis of Egr-1 after 1 h treatment with 25 μM 13cRA, with or without the translational inhibitor cycloheximide (CHx). (C) Western blot analysis of nuclear extracts treated with and without 13cRA, probed with anti-Sp1 primary antibody after immunoprecipitation with anti-Egr-1. (D) Western blot analysis of nuclear extracts treated with and without 13cRA, probed with anti-Egr-1 primary antibody after immunoprecipitation with anti-Sp1. (E & F) Corresponding reprobe controls with appropriate capture antibody to confirm efficient immunoprecipitation of target proteins.
Sp1 Dictates Basal Transcription of AT1R
In order to determine Sp1’s control of AT1AR expression, we selectively targeted Sp1 with siRNA oligonucleotides to silence gene expression. In order to ascertain the ability for the siRNA to silence Sp1 expression, Western blot analysis were performed. Western blot analysis using specific antibodies targeting Sp1 show significant knockdown of Sp1 expression in cells transfected with Sp1-specific siRNA compared to both scrambled negative control siRNA and untransfected cells (Sp1 KO control and Sp1 KO treated with 13cRA mean difference from control 90.80±2.963% and 92.54±2.502% respectively, p<0.0001, n=3) [Fig. 6A & 6B]. Consistently with our promoter analysis, AT1AR mRNA expression studies utilizing dual-PCR amplification indicate that upon knockdown of Sp1 expression, AT1AR mRNA expression down-regulates both in untreated control cells and cells exposed to 25 μM 13cRA for 24 hours (Sp1 KO control and Sp1 KO treated with 13cRA mean difference from control 84.53±2.348% and 84.92±3.113% respectively, p<0.0001, n=3) [Fig. 6C]. Furthermore, [3H]AngII binding studies indicate that receptor density on the plasma membrane is reduced 88.5±3.352% (p<0.0001, n=9) in cells transfected with Sp1-specific siRNA [Fig. 6D]. AngII binding among the untreated control and 13cRA exposed cells were not significantly different (p>0.05) among Sp1 knock-down groups. From the siRNA studies we concluded that Sp1 is a key regulator in the basal transcription of AT1AR.
Figure 6.
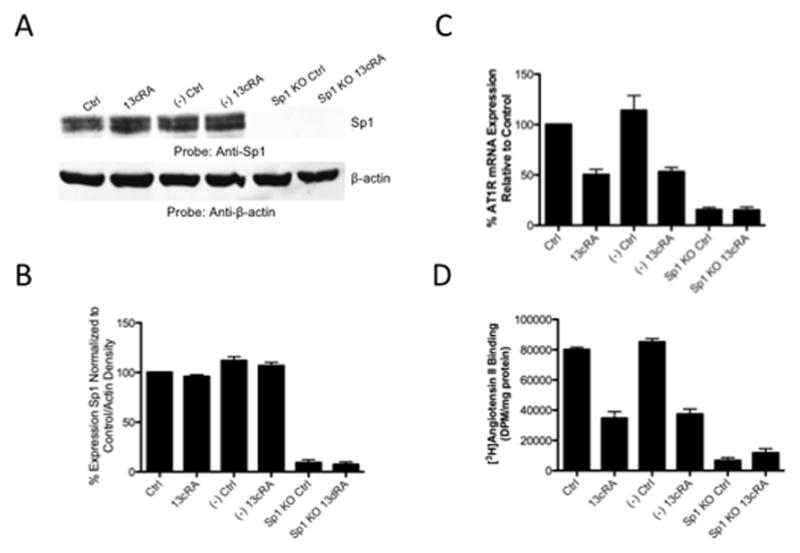
Knockdown studies reveal Sp1 expression is essential for AT1R basal transcription. (A) Western blot displaying sufficient Sp1 silencing by siRNA targeted for Sp1. Gel separation shows doublet of protein bands characteristic with polyclonal antibody SC59-X. (B) Densitometric analysis of Sp1 bands from Western blot shown in panel A. Displayed is a knockdown by Sp1 siRNA of 90–95% relative to control. 13cRA has no effect on level of expression of Sp1. (C) AT1AR mRNA expression eliminated after Sp1 knockdown (-out). mRNA transcription characteristically reduced after 13cRA treatment. (D) Radio-labeled ligand binding assay showing elimination of binding of AngII to membrane receptors after Sp1 knockout. All data are expressed as mean ± SEM. “(−)Ctrl” and “(−)13cRA” indicate cells that are transfected with scrambled negative control siRNA. All 13cRA results shown are treated with 13cRA for 20 h.
Establishment of Egr-1 as a Critical Mediator of 13cRA Induced AT1R Down-Regulation
Studies up to this point established that 13cRA mediated down-regulation of the AT1AR involves activation of ERK1/2; additionally, investigations demonstrated that Sp1 binding is affected on distal response elements, ERK1/2 activates the transcription and production of Egr-1, Egr-1 and Sp1 interact in a protein-protein complex detectable by co-immunoprecipitation, and Sp1 is essential for the basal transcription of the AT1AR gene. However, the studies did not demonstrate that Egr-1 is involved directly with the down-regulation of AT1AR. In order to determine whether Egr-1 is integral in ERK1/2 mediated AT1R down-regulation, we performed siRNA knockdown of Egr-1, similar to our method used for Sp1 knockdown, to determine Egr-1’s effect on AT1AR mRNA transcription and [3H]AngII binding in the presence of 13cRA. Western blot analysis indicates Egr-1 expression was significantly reduced compared both to untreated or scrambled siRNA when transfected with Egr-1 specific siRNA, and the siRNA transfected cells showed no significant increase in Egr-1 expression when exposed to 25 μM 13cRA for 24 hours [Fig. 7A]. Immunofluorescent microscopy targeting Egr-1 indicated that expression of the nuclear resident Egr-1 is significantly reduced in siRNA transfected cells, with no detectable increase in expression upon exposure with 13cRA [Fig. 7B]. Next, we examined the effect of Egr-1 siRNA transfection on 13cRA mediated AT1AR mRNA expression. AT1AR mRNA expression studies using dual-PCR amplification specific to β-actin and AT1AR mRNA display no significant reduction in siRNA transfected cells when treated with 13cRA (mean difference in 13cRA treated Egr-1 knock-down and Egr-1 knock-down control 2.663±8.842%, p=0.7783, n=3) [Fig. 7C]. Under similar conditions, we determined whether receptor-binding studies correlated with the detected restoration of mRNA expression. [3H]AngII binding displayed a complete restoration in AngII binding in Egr-1 siRNA transfected and 25 μM exposed cells (mean difference in 13cRA treated Egr-1 knock-down and Egr-1 knock-down control 9.055±7.224%, p=0.2281, n=6) [Fig. 7D]. Lastly, we performed additional EMSA analysis to correlate previous findings in mobility shift with the effect of selective knockdown of Sp1 and Egr-1 proteins on characteristic mobility shift patterns. Protein/DNA interaction was almost entirely eliminated after Sp1 knockdown in both untreated and 13cRA treated nuclear extracts [Fig. 7E, lanes 4 & 5 respectively], whereas robust protein/DNA interaction occurred in Egr-1 knockout nuclear extracts, regardless of 13cRA treatment [Fig. 7E, lanes 6 & 7]. Our results suggest that Egr-1 is essential for 13cRA mediated down-regulation of AT1AR, and we propose that its mechanism involves the disruption of Sp1 to initiate basal transcription of AT1AR mRNA by direct protein/protein interaction.
Figure 7.
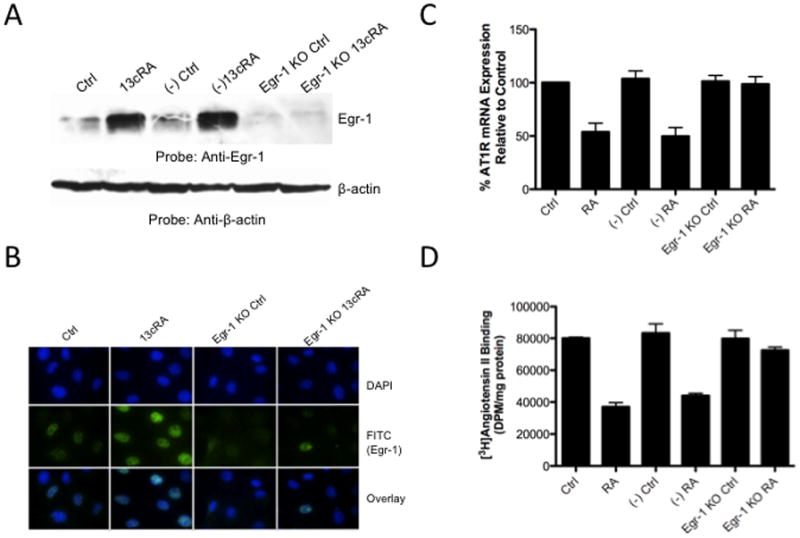
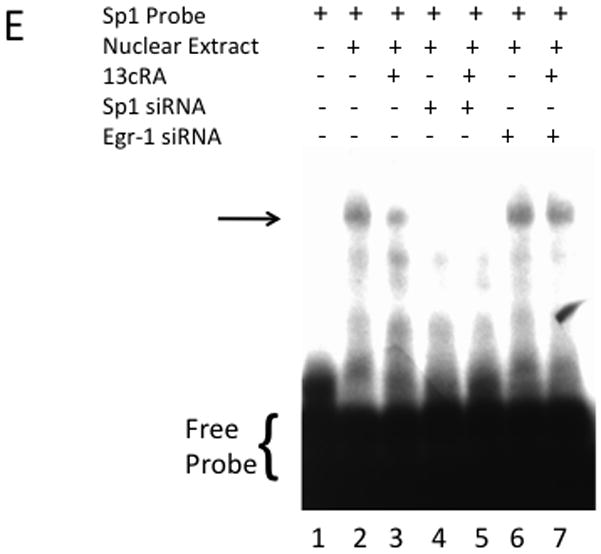
siRNA silencing of Egr-1 reverses 13cRA mediated down-regulation of AT1AR. (A) Western blot analysis of cells treated with or without 13cRA and transfected with negative control scrambled siRNA oligonucleotides or Egr-1 targeted siRNA oligonucleotides. (B) Immunofluorescent microscopy using anti-Egr-1 to confirm effective silencing of 13cRA induced Egr-1. Egr-1 antibody labeled with FITC (green) and nuclei are stained with DAPI (blue). (C) AT1AR mRNA expression restored after Egr-1 knockdown (-out) (D). Radio-labeled ligand binding assay showing restoration of binding of AngII to membrane receptors after Egr-1 knockout. All data are expressed as mean ± SEM. “(−)Ctrl” and “(−)RA” indicate cells that are transfected with scrambled negative control siRNA. Egr-1 imaging indicates 13cRA treatment for 60 min while AngII binding and mRNA studies reflect 20 hour 13cRA treatment. (E) Accompanying mobility shift in selective gene knockout studies included. Samples were analyzed on 6% non-denaturing polyacrylamide gels and visualized by autoradiography. The position of the protein-DNA complex is indicated by arrow. Labeled probe in the absence of nuclear extract (lane 1), in the presence of untreated nuclear extract (lane 2), in the presence of 13cRA exposed nuclear extract (lane 3), nuclear extracts of both untreated and treated with 13cRA after siRNA mediated silencing of Sp1 (lanes 4 and 5, respectively), and nuclear extracts of both untreated and treated with 13cRA after siRNA mediated silencing of Sp1 (lanes 6 and 7, respectively).
DISCUSSION
In our previous studies, we have shown that phosphorylation of ERK1/2 (MAPK p42/p44) results in the down-regulation of AT1AR mRNA expression (Snyder & Thekkumkara, 2012). However, signal transduction following the phosphorylation of ERK1/2 was not investigated. Therefore, we extended this study to elucidate the down-stream regulators responsible for AT1AR gene transcription. We observed that Sp1 binds to a distal cis-acting element on the AT1AR promoter for basal transcription, and upon 13cRA treatment, Egr-1, a trans-activator binds to Sp1 through protein-protein interaction, thereby down-regulating AT1AR expression. The observed effect requires ERK1/2 phosphoryation, which is unrelated to RAR/RXR activation, as retinoid receptor blockade has no effect on 13cRA-mediated down-regulation of AT1AR (Snyder & Thekkumkara, 2012). Sp1, along with Sp3, has been identified as an essential transcription factor for expression of human AT1R in H295-R cells (Zhao et al., 2001) and expression of rat AT1AR may be enhanced by Sp1 overexpression (Sugawara et al., 2001). Our study indicates that Sp1 is a constitutively active transcription factor. There is no current evidence of the involvement of distal Sp1 elements in the expression of human AT1R, particularly because most promoter studies examine much shorter promoter sequences. Therefore, it may be that any sequestration of Sp1 by Egr-1 in humans disrupts transcription by a similar mechanism, requiring additional studies; however, human studies are difficult, particularly because of the limited availability of cell lines with reliable expression of AT1R. However, to our knowledge this is the first instance in which disruption of a distal Sp1 binding element has resulted in a down-regulatory effect on AT1AR expression, suggesting that 13cRA’s effect is unique and is indicative of an entirely novel mechanism.
After determining the time course of early ERK1/2 phosphorylation, we had yet to establish the link between ERK1/2 and Sp1 binding disruption. The potential candidate was early growth response protein 1 (Egr-1). Egr-1 expression has been shown to be up-regulated upon ERK1/2 activation (Hoffman et al., 2008). In addition, Egr-1 has been associated with disruption of Sp1 binding ability with gene promoters, including response elements found in the AT1AR promoter (Hsu et al., 2009). Upon observing CXCR3 knockout mice have suppressed expression of Egr-1 and increased expression of AT1AR protein, the authors suggested a possible involvement of Egr-1 and Sp1 in the expression of AT1AR protein. These conclusions were based upon a previous study showing that Sp1 activity plays a major role in human AT1R gene transcription (Elton & Martin, 2006). Early growth response protein 1 (Egr-1), also known as zif268, NGFI-A, Krox24, and TIS8, is a zinc-finger transcription factor belonging to the same nuclear factor family as Sp1 and Sp3 (Lim et al., 1998). However, Egr-1 is distinct in its binding characteristics and is particularly active at 5′-GCGGGGGCG-3′ moieties within GC rich regions (Silverman & Collins, 1999). In fact, mutations of this characteristic element have shown that affinity for GC elements is lost if a single internal guanine residue or the 3-prime cytosine is exchanged for an alternate base (Zhang et al., 2007). Often, Egr-1 elements are integrated within regions sharing the particularly GC-rich motifs specific for Sp1 and Sp3 binding 5′-GGGCGG-3′ (Khachigian et al., 1995; Zhang et al., 2007). Egr-1, like many transcription factors, has either activation or repression function depending upon whether co-activators or co-repressors are recruited. Egr-1 can interact directly with CREB-binding protein (CBP) and p300, very similarly to Sp1, in order to activate gene transcription by the co-activators’ inherent histone acetyl transferase (HAT) activity. As far as Egr-1’s relevance in the current study, the distal Sp1 binding element used in our mobility shift does not possess a putative Egr-1 binding sequence. Sequence analysis revealed no characteristic Egr-1 binding elements located within the 3300 base pairs investigated in the rat AT1AR promoter. Therefore, Egr-1 binding may not be necessarily important as a direct trans-acting factor binding to cis-acting elements on the AT1AR promoter, but rather an indirect protein interference factor inhibiting binding of Sp1 by a protein-protein interaction as shown in this study.
Early growth response protein-1, as its name suggests, is an immediate response gene, though some sources characterize it as an intermediate response protein (Zhou et al., 2010) putting it in the same time-frame for activation of other kinase dependent effector transcription factors like c-fos and c-jun. Its steady state expression is relatively low, but upon activation by MAPKs, it is rapidly induced (Hoffman et al., 2008). In hypoxia-reoxygenation experiments, Zhou et al. found that Egr-1 expression was increased upon hypoxic conditions followed by reoxygenation (Zhou et al., 2010). This was accomplished by activation of cellular stress kinases (p38, p42/p44, JNK); the subsequent activation of Egr-1 led to transactivation of downstream targets such as TGF-β, ICAM-1, tissue factor, and PDGF (Khachigian et al., 1995; Silverman & Collins, 1999; Okada et al., 2001; Khachigian, 2006) that lead to vascular hyperpermeability, coagulation, and inflammation. Therefore, Egr-1 production is detrimental to the outcome of hypoxia-reoxygenation. However, in the present study’s findings coupled with what is known about exposure to 13cRA, Egr-1 is not itself an inflammatory mediator, and the transient up-regulation seen in these studies had no damaging effects in these cells. Egr-1 activation in the past has been accomplished by chronic cell stress leading to multiple MAPK activations and subsequent activation of negative pathways, but the data suggests that 13cRA is more selective as it is not associated with the activation of p38 or JNK (Jameel et al., 2009), and specific activation of ERK1/2 may give different results. The direct interaction between in Egr-1 and Sp1 has been recognized in previous studies. Jain et al. have suggested that Egr-1 and Sp1 can directly bind one another (Jain et al., 1996) and may limit their respective binding to gene promoters (Srivastava et al., 1998), but this interaction is dictated by Egr-1 phosphorylation by casein kinase II. When phosphorylated by casein kinase II, Egr-1 has lower binding affinity for Sp1; interestingly, casein kinase II has also been associated with the inactivation of Sp1 by phosphorylation (Harris et al., 2008). If Egr-1 first binds to Sp1, then dissociates by up-regulation of casein kinase II, there could be indirect inactivation of Sp1 by the same mechanism, which requires further investigation.
As we have shown in this study, Egr-1 up-regulation is an ERK1/2 dependent process. It has also been demonstrated that 17β-estradiol levels are associated with improved vascular condition (Kublickiene et al., 2008; Kitamura et al., 2009). Egr-1 is activated in MCF-7 cells upon treatment with 17β-estradiol (Chen et al., 2004). Moreover, this activation is through a non-genomic or “extra-nuclear” pathway. The extra-nuclear effect could have a strong parallel with the current study as we have shown that 13cRA mediates its effects independent of retinoic acid nuclear receptors (Snyder & Thekkumkara, 2012). In further support of the estrogen theory, the aforementioned study by Srivastava et al. controlled phosphorylation of Egr-1 and subsequent binding to Sp1 by estrogen replacement in ovariectomized animals (Srivastava et al., 1998). Estrogen replacement reduced the activity of casein kinase II and thus increased the formation of interactions between Egr-1 and Sp1. Especially with regard to our own research (Koganti et al., 2012) which has seen a down-regulatory effect by estrogen metabolites through extra-nuclear mechanisms, this comes as a link between disparate phenomena connected by ERK1/2. With regard to direct ERK1/2 activation by 13cRA, it may bind a number cytosolic binding proteins, such as CRABP-II or FABP5, leading to divergent effects through distinct signal transduction pathways (Schug et al., 2008) resulting in tissue-specific effects. These suggested upstream mechanisms of ERK1/2 phosphorylation in this model require further investigation. In summary, our results show for the first time that the mechanism of 13cRA mediated AT1AR down-regulation is reliant on both ERK1/2 phosphorylation and Egr-1 synthesis ultimately leading to the disruption in binding of Sp1 to the AT1AR promoter.
Acknowledgments
A portion of this work was presented at Experimental Biology 2011 in Washington, D.C.
FUNDING
This study was supported in part by a research grant from the National Institute of Health (#DK072140) and by a graduate fellowship from the Texas Tech University Health Sciences Center School of Pharmacy to R.S.
Footnotes
DECLARATION OF INTEREST
The authors wish to declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of this research.
References
- 1.Abadir PM. The frail renin-angiotensin system. Clin Geriatr Med. 2011;27(1):53–65. doi: 10.1016/j.cger.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atlas SA. The renin-angiotensin aldosterone system: pathophysiological role and pharmacologic inhibition. J Manag Care Pharm. 2007;13(8 Suppl B):9–20. doi: 10.18553/jmcp.2007.13.s8-b.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blaner WS. Cellular metabolism and actions of 13-cis-retinoic acid. J Am Acad Dermatol. 2001;45(5):S129–35. doi: 10.1067/mjd.2001.113714. [DOI] [PubMed] [Google Scholar]
- 4.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 5.Braga VA. Differential brain angiotensin-II type I receptor expression in hypertensive rats. J Vet Sci. 2011;12(3):291–3. doi: 10.4142/jvs.2011.12.3.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braunwald E. Biomarkers in Heart Failure. N Engl J Med. 2008;358:2148–2159. doi: 10.1056/NEJMra0800239. [DOI] [PubMed] [Google Scholar]
- 7.Burnier M. Novel angiotensin II inhibitors in cardiovascular medicine. Expert Opin Investig Drugs. 2001;10(11):1957–64. doi: 10.1517/13543784.10.11.1957. [DOI] [PubMed] [Google Scholar]
- 8.Chen CC, Lee WR, Safe S. Egr-1 is activated by 17beta-estradiol in MCF-7 cells by mitogen-activated protein kinase-dependent phosphorylation of ELK-1. J Cell Biochem. 2004;93(5):1063–74. doi: 10.1002/jcb.20257. [DOI] [PubMed] [Google Scholar]
- 9.Elton TS, Martin MM. Angiotensin II type 1 receptor gene regulation: transcriptional and posttranscriptional mechanisms. Hypertension. 2007;49(5):953–61. doi: 10.1161/HYPERTENSIONAHA.106.070565. [DOI] [PubMed] [Google Scholar]
- 10.Esler MD. Catecholamines and essential hypertension. Baillieres Clin Endocrinol Metab. 1993;7(2):415–38. doi: 10.1016/s0950-351x(05)80182-x. [DOI] [PubMed] [Google Scholar]
- 11.Gamble MV, Shang E, Piantedosi Zott R, Mertz JR, Wolgemuth DJ, Blaner WS. Biochemical properties, tissue expression and gene structure of a short chain dehydrogenase/reductase able to catalyze cis-retinol oxidation. J Lipid Res. 1999;40:2279–92. [PubMed] [Google Scholar]
- 12.Ganceviciene R, Zouboulis CC. Isotretinoin: state of the art treatment for acne vulgaris. Exp Rev Dermatol. 2007;2:693–706. doi: 10.1111/j.1610-0387.2009.07238.x. [DOI] [PubMed] [Google Scholar]
- 13.Gao L, Wang WZ, Wang W, Zucker IH. Imbalance of angiotensin type 1 receptor and angiotensin II type 2 receptor in the rostral ventrolateral medulla: potential mechanism for sympathetic overactivity in heart failure. Hypertension. 2008;52(4):708–14. doi: 10.1161/HYPERTENSIONAHA.108.116228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hannan RE, Gaspari TA, Davis EA, Widdop RE. Differential regulation by AT(1) and AT(2) receptors of angiotensin II-stimulated cyclic GMP production in rat uterine artery and aorta. Br J Pharmacol. 2004;141(6):1024–31. doi: 10.1038/sj.bjp.0705694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris SM, Harvey EJ, Hughes TR, Ramji DP. The interferon-gamma-mediated inhibition of lipoprotein lipase gene transcription in macrophages involves casein kinase 2- and phosphoinositide-3-kinase-mediated regulation of transcription factors Sp1 and Sp3. Cell Signal. 2008;20(12):2296–301. doi: 10.1016/j.cellsig.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoffmann E, Ashouri J, Wolter S, Doerrie A, Dittrich-Breiholz O, Schneider H, Wagner EF, Troppmair J, Mackman N, Kracht M. Transcriptional regulation of EGR-1 by the interleukin-1-JNK-MKK7-c-Jun pathway. J Biol Chem. 2008;283(18):12120–8. doi: 10.1074/jbc.M800583200. [DOI] [PubMed] [Google Scholar]
- 17.Hsu HH, Duning K, Meyer HH, Stölting M, Weide T, Kreusser S, van Le T, Gerard C, Telgmann R, Brand-Herrmann SM, Pavenstädt H, Bek MJ. Hypertension in mice lacking the CXCR3 chemokine receptor. Am J Physiol Renal Physiol. 2009;296(4):F780–9. doi: 10.1152/ajprenal.90444.2008. [DOI] [PubMed] [Google Scholar]
- 18.Jain N, Mahendran R, Philp R, Guy GR, Tan YH, Cao X. Casein kinase II associates with Egr-1 and acts as a negative modulator of its DNA binding and transcription activities in NIH 3T3 cells. J Biol Chem. 1996;271(23):13530–6. doi: 10.1074/jbc.271.23.13530. [DOI] [PubMed] [Google Scholar]
- 19.Jameel NM, Thirunavukkarasu C, Wu T, Watkins SC, Friedman SL, Gandhi CR. p38-MAPK- and caspase-3-mediated superoxide-induced apoptosis of rat hepatic stellate cells: reversal by retinoic acid. J Cell Physiol. 2009;218(1):157–66. doi: 10.1002/jcp.21581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaprielian RR, Dupont E, Hafizi S, Poole-Wilson PA, Khaghani A, Yacoub MH, Severs NJ. Angiotensin II receptor type 1 mRNA is upregulated in atria of patients with end-stage heart failure. J Mol Cell Cardiol. 1997;29(8):2299–304. doi: 10.1006/jmcc.1997.0458. [DOI] [PubMed] [Google Scholar]
- 21.Khachigian LM, Williams AJ, Coollins T. Interplay of Sp1 and Egr-1 in the proximal platelet-derived growth factor A-chain promoter in cultured vascular endothelial cells. J Biol Chem. 1995;270:27679–27686. doi: 10.1074/jbc.270.46.27679. [DOI] [PubMed] [Google Scholar]
- 22.Khachigian LM. Early growth response-1 in cardiovascular pathobiology. Circ Res. 2006;98(2):186–91. doi: 10.1161/01.RES.0000200177.53882.c3. [DOI] [PubMed] [Google Scholar]
- 23.Kim Y-M, Sharma RP, Li JK. Characterization of heterologously expressed recombinant retinoic acid receptors with natural or synthetic retinoids. J Biochem Toxicol. 1994;9:225–234. doi: 10.1002/jbt.2570090502. [DOI] [PubMed] [Google Scholar]
- 24.Kim MY, Baik SK, Park DH, Jang YO, Suk KT, Yea CJ, Lee IY, Kim JW, Kim HS, Kwon SO, Cho MY, Ko SB, Chang SJ, Um SH, Han KH. Angiotensin receptor blockers are superior to angiotensin-converting enzyme inhibitors in the suppression of hepatic fibrosis in a bile duct-ligated rat model. J Gastroenterol. 2008;43(11):889–96. doi: 10.1007/s00535-008-2239-9. [DOI] [PubMed] [Google Scholar]
- 25.Kitamura N, Araya R, Kudoh M, Kishida H, Kimura T, Murayama M, Takashima A, Sakamaki Y, Hashikawa T, Ito S, Ohtsuki S, Terasaki T, Wess J, Yamada M. Beneficial effects of estrogen in a mouse model of cerebrovascular insufficiency. PLoS One. 2009;4(4):e5159. doi: 10.1371/journal.pone.0005159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koganti S, Snyder R, Thekkumkara T. Pharmacologic effects of 2-methoxyestradiol on Angiotensin type 1 receptor down-regulation in rat liver epithelial and aortic smooth muscle cells. Gend Med. 2012;9(2):76–93. doi: 10.1016/j.genm.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kublickiene K, Fu XD, Svedas E, Landgren BM, Genazzani AR, Simoncini T. Effects in postmenopausal women of estradiol and medroxyprogesterone alone and combined on resistance artery function and endothelial morphology and movement. J Clin Endocrinol Metab. 2008;93(5):1874–83. doi: 10.1210/jc.2007-2651. [DOI] [PubMed] [Google Scholar]
- 28.Landthaler M, Kummermehr J, Wagner A, Plewig G. Inhibitory effects of 13-cis-retinoic acid on human sebaceous glands. Arch Dermatol Res. 1980;269:297–309. doi: 10.1007/BF00406424. [DOI] [PubMed] [Google Scholar]
- 29.Lenkey Z, Palkovits M, Corvol P, Llorens-Cortes C. Expression of angiotensin type-1 (AT1) and type-2 (AT2) receptor mRNAs in the adult rat brain: a functional neuroanatomical review. Frontiers in Neuroendocrinology. 1997;18:383–439. doi: 10.1006/frne.1997.0155. [DOI] [PubMed] [Google Scholar]
- 30.Lim C-P, Jain N, Cao X. Stress-induced immediate-early gene, egr-1, involves activation of p38/JNK1. Oncogene. 1998;16:2915–2926. doi: 10.1038/sj.onc.1201834. [DOI] [PubMed] [Google Scholar]
- 31.Mangelsdorf DJ, Umesono K, Evans RM. The retinoid receptors. In: Sporn MB, Roberts AB, Goodman DS, editors. The retinoids, biology, chemistry, and medicine. 2. New York: Raven Press; 1994. pp. 319–50. [Google Scholar]
- 32.Mark M, Ghyselinck NB, Chambon P. Function of retinoid nuclear receptors: lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis. Annu Rev Pharmacol Toxicol. 2006;46:451–80. doi: 10.1146/annurev.pharmtox.46.120604.141156. [DOI] [PubMed] [Google Scholar]
- 33.Nickenig G, Harrison DG. The AT(1)-type angiotensin receptor in oxidative stress and atherogenesis: Part II: AT(1) receptor regulation. Circulation. 2002;105(4):530–6. doi: 10.1161/hc0402.102619. [DOI] [PubMed] [Google Scholar]
- 34.Okada M, Fujita T, Sakaguchi T, Olson KE, Collins T, Stern DM, Yan SF, Pinsky DJ. Extinguishing Egr-1-dependent inflammatory and thrombotic cascades after lung transplantation. FASEB J. 2001;15(14):2757–9. doi: 10.1096/fj.01-0490fje. [DOI] [PubMed] [Google Scholar]
- 35.Pan P, Fu H, Zhang L, Huang H, Luo F, Wu W, Guo Y, Liu X. Angiotensin II upregulates the expression of placental growth factor in human vascular endothelial cells and smooth muscle cells. BMC Cell Biol. 2010;11:36. doi: 10.1186/1471-2121-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piattellia B, Fioronic M, Santinellid A, Rubinie C. Bcl-2 expression and apoptotic bodies in 13-cis-retinoic acid (isotretinoin)-topically treated oral leukoplakia: a pilot study. Oral Oncology. 1999;35(3):314–320. doi: 10.1016/s1368-8375(98)00095-5. [DOI] [PubMed] [Google Scholar]
- 37.Raizada MK, Sumners C, Lu D. Angiotensin II type 1 receptor mRNA levels in the brains of normotensive and spontaneously hypertensive rats. J Neurochem. 1993;60(5):1949–52. doi: 10.1111/j.1471-4159.1993.tb13426.x. [DOI] [PubMed] [Google Scholar]
- 38.Ratziu V, Zelber-Sagi S. Pharmacologic therapy of non-alcoholic steatohepatitis. Clin Liver Dis. 2009;13(4):667–88. doi: 10.1016/j.cld.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 39.Schug TT, Berry DC, Toshkov IA, Cheng L, Nikitin AY, Noy N. Overcoming retinoic acid-resistance of mammary carcinomas by diverting retinoic acid from PPARbeta/delta to RAR. Proc Natl Acad Sci U S A. 2008;105(21):7546–51. doi: 10.1073/pnas.0709981105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siitonen T, Timonen T, Juvonen E, Terävä V, Kutila A, Honkanen T, Mikkola M, Hallman H, Kauppila M, Nyländen P, Poikonen E, Rauhala A, Sinisalo M, Suominen M, Savolainen ER, Koistinen P, Koistinen Pirjo. Valproic acid combined with 13-cis retinoic acid and 1,25-dihydroxyvitamin D3 in the treatment of patients with myelodysplastic syndromes. Haematologica. 2007;92(8):1119–22. doi: 10.3324/haematol.11262. [DOI] [PubMed] [Google Scholar]
- 41.Silverman ES, Collins T. Pathways of Egr-1-mediated gene transcription in vascular biology. Am J Pathol. 1999;154(3):665–70. doi: 10.1016/S0002-9440(10)65312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Snyder R, Thekkumkara T. 13-cis-Retinoic acid specific down-regulation of angiotensin type 1 receptor in rat liver epithelial and aortic smooth muscle cells. J Mol Endocrinol. 2012;48(2):99–114. doi: 10.1530/JME-11-0095. [DOI] [PubMed] [Google Scholar]
- 43.Srivastava S, Weitzman MN, Kimble RB, Rizzo M, Zahner M, Milbrandt J, Ross FP, Pacifici R. Estrogen blocks M-CSF gene expression and osteoclast formation by regulating phosphorylation of Egr-1 and its interaction with Sp1. J Clin Invest. 1998;102:1850–1859. doi: 10.1172/JCI4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sugawara A, Takeuchi K, Uruno A, Ikeda Y, Arima S, Kudo M, Sato K, Taniyama Y, Ito S. Transcriptional suppression of type 1 angiotensin II receptor gene expression by peroxisome proliferator-activated receptor-gamma in vascular smooth muscle cells. Endocrinology. 2001;142(7):3125–34. doi: 10.1210/endo.142.7.8272. [DOI] [PubMed] [Google Scholar]
- 45.Sun Y, Weber KT. Angiotensin II receptor binding following myocardial infarction in the rat. Cardiovasc Res. 1994;28(11):1623–8. doi: 10.1093/cvr/28.11.1623. [DOI] [PubMed] [Google Scholar]
- 46.Thomas BE, Thekkumkara TJ. Glucose mediates transcriptional repression of the human angiotensin type-1 receptor gene: role for a novel cis-acting element. Mol Biol Cell. 2004;15(10):4347–55. doi: 10.1091/mbc.E04-03-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tower CL, Lui S, Charlesworth NR, Smith SD, Aplin JD, Jones RL. Differential expression of angiotensin II type 1 and type 2 receptors at the maternal-fetal interface: potential roles in early placental development. Reproduction. 2010;140(6):931–42. doi: 10.1530/REP-10-0307. [DOI] [PubMed] [Google Scholar]
- 48.Wu S, Gao J, Ohlemeyer C, Roos D, Niessen H, Köttgen E, Gessner R. Activation of AP-1 through reactive oxygen species by angiotensin II in rat cardiomyocytes. Free Radic Biol Med. 2005;39(12):1601–10. doi: 10.1016/j.freeradbiomed.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 49.Wyse BD, Linas SL, Thekkumkara TJ. Functional role of a novel cis-acting element (GAGA box) in human type-1 angiotensin II receptor gene transcription. J Mol Endocrinol. 2000;25:97–108. doi: 10.1677/jme.0.0250097. [DOI] [PubMed] [Google Scholar]
- 50.Yesudas R, Gumaste U, Snyder R, Thekkumkara T. Tannic acid down-regulates the angiotensin type 1 receptor through a MAPK-dependent mechanism. Mol Endocrinol. 2012;26(3):458–70. doi: 10.1210/me.2011-1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoshiji H, Noguchi R, Ikenaka Y, Namisaki T, Kitade M, Kaji K, Shirai Y, Yoshii J, Yanase K, Yamazaki M, Tsujimoto T, Kawaratani H, Akahane T, Aihara Y, Fukui H. Losartan, an angiotensin-II type 1 receptor blocker, attenuates the liver fibrosis development of non-alcoholic steatohepatitis in the rat. BMC Res Notes. 2009;2:70. doi: 10.1186/1756-0500-2-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang C, Duvic M. Retinoids: therapeutic applications and mechanisms of action in cutaneous T-cell lymphoma. Dermatol Ther. 2003;16(4):322–330. doi: 10.1111/j.1396-0296.2003.01644.x. [DOI] [PubMed] [Google Scholar]
- 53.Zhang P, Tchou-Wong KM, Costa M. Egr-1 mediates hypoxia-inducible transcription of the NDRG1 gene through an overlapping Egr-1/Sp1 binding site in the promoter. Cancer Res. 2007;67(19):9125–9133. doi: 10.1158/0008-5472.CAN-07-1525. [DOI] [PubMed] [Google Scholar]
- 54.Zhao X, Martin MM, Elton TS. The transcription factors Sp1 and Sp3 are required for human angiotensin II type 1 receptor gene expression in H295-R cells. Biochimica et Biophysica Acta (BBA) - Gene Structure and Expression. 2001;1522(3):195–206. doi: 10.1016/s0167-4781(01)00341-4. [DOI] [PubMed] [Google Scholar]
- 55.Zhou Y, Shi G, Zheng J, Huang Z, Gao F, Zhang Y, Guo F, Jia Q, Zheng Y. The protective effects of Egr-1 antisense oligodeoxyribonucleotide on cardiac microvascular endothelial injury induced by hypoxia-reoxygenation. Biochem Cell Biol. 2010;88(4):687–95. doi: 10.1139/O10-021. [DOI] [PubMed] [Google Scholar]