

Abstract
More than three transcription factors often work together to enable cells to respond to various signals. The detection of combinatorial regulation by multiple transcription factors, however, is not only computationally nontrivial but also extremely unlikely because of multiple testing correction. The exponential growth in the number of tests forces us to set a strict limit on the maximum arity. Here, we propose an efficient branch-and-bound algorithm called the “limitless arity multiple-testing procedure” (LAMP) to count the exact number of testable combinations and calibrate the Bonferroni factor to the smallest possible value. LAMP lists significant combinations without any limit, whereas the family-wise error rate is rigorously controlled under the threshold. In the human breast cancer transcriptome, LAMP discovered statistically significant combinations of as many as eight binding motifs. This method may contribute to uncover pathways regulated in a coordinated fashion and find hidden associations in heterogeneous data.
Keywords: Bonferroni correction, gene expression
The regulation of gene expression by the binding of transcription factors (TFs) is a critical component of the cellular machinery. The joint activity of different TFs is essential for cells to respond to a wide spectrum of environmental and developmental signals (1, 2). A recent series of encyclopedia of DNA elements (ENCODE) project papers confirmed the importance of this combinatorial activity (3–6). Salient combinations of more than two TFs are commonly found in both mammals (7, 8) and plants (9, 10); however, existing computational methods are not sufficiently powerful to uncover these combinatorial regulations comprehensively. The earliest computational studies aimed to find a single TF-binding motif significantly associated with the expression levels of downstream genes, which is not appropriate for the detection of salient combinations because of their synergistic effects. The prioritization of motifs using machine learning methods such as support vector machine (SVM) and sparse regression (11–15) often falls short, as they mix up different mechanisms leading to the same phenomenon. Although a screening of all pairs of motifs (16–20) provides much better results than the single screening process, it will not discover higher-order effects. Using the current computational power, a comprehensive screening of combinations of three or more motifs may be possible. A crucial problem, however, is how to assess the statistical significance of discovered motifs.
When combinations of up to 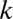 motifs are considered, the number of tested combinations increases exponentially to
motifs are considered, the number of tested combinations increases exponentially to 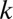 . It is well known that multiple tests may cause serious false positive problems. Hence, a multiple testing correction must be used (21). The family-wise error rate (FWER) indicates the probability that at least one false discovery happens in multiple tests. This rate increases at most linearly as the number of tests increases, which motivates the Bonferroni correction (22) that multiplies the raw P value by the number of tests. Because of the exponential growth in the number of tests, the discovery of combinations of higher-arity motif combinations (combinations of many motifs) is extremely unlikely, even with more sensitive corrections (23–28).
. It is well known that multiple tests may cause serious false positive problems. Hence, a multiple testing correction must be used (21). The family-wise error rate (FWER) indicates the probability that at least one false discovery happens in multiple tests. This rate increases at most linearly as the number of tests increases, which motivates the Bonferroni correction (22) that multiplies the raw P value by the number of tests. Because of the exponential growth in the number of tests, the discovery of combinations of higher-arity motif combinations (combinations of many motifs) is extremely unlikely, even with more sensitive corrections (23–28).
The problem with the Bonferroni correction is that the bound on FWER is proportional to the number of tests. However, as Tarone (26) showed in his pioneering paper, not every test increases the FWER. Thus, the Bonferroni factor may be improved by deliberately excluding such tests. Here, we propose an efficient branch-and-bound algorithm, called the “limitless arity multiple-testing procedure” (LAMP). LAMP counts the exact number of “testable” motif combinations and derives a tighter bound of FWER, which allows the calibration of the Bonferroni factor as the FWER is controlled rigorously under the threshold.
In comparison with existing methods that can find only two-motif combinations, our testing procedure may contribute to finding larger fractions of regulatory pathways and TF complexes, thus providing more concrete evidence for further investigation. In legacy yeast expression data (29), a four-motif combination corresponding to a known pathway was found using LAMP, whereas only two motifs in the combination had been predicted using the existing method. When applied to human breast cancer transcriptome data (30), combinations of up to eight motifs were found to be statistically significant.
Results
Method Overview.
To present our strategy for combinatorial regulation discovery, we assume the following simple scenario (Fig. 1). Only one expression level is available for each of the 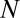 genes. If a conserved binding motif of a TF exists in the regulatory region of a gene, the motif is regarded to target the gene. For a given motif combination, the genes are partitioned in two ways. In one way, the genes are classified into
genes. If a conserved binding motif of a TF exists in the regulatory region of a gene, the motif is regarded to target the gene. For a given motif combination, the genes are partitioned in two ways. In one way, the genes are classified into 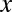 targeted and
targeted and 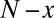 untargeted genes, depending on whether they are targeted by all members of the motif combination. In the other way, the genes are classified into
untargeted genes, depending on whether they are targeted by all members of the motif combination. In the other way, the genes are classified into 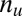 up-regulated genes and
up-regulated genes and 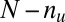 unregulated genes. If the P value of the division derived from Fisher’s exact test is below a threshold
unregulated genes. If the P value of the division derived from Fisher’s exact test is below a threshold 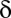 , the motif combination is considered regulatory.
, the motif combination is considered regulatory.
Fig. 1.

Salient three-motif combination. (A) Expressions of genes targeted by the motifs. In the first three boxes, the blue bars represent the expression levels of the genes targeted by each motif, whereas the bottom box represents those of the genes targeted by all three motifs. The genes in the red region are up-regulated, and those in the white region are unregulated. (B) P values of the motifs. Although any single motif is not significant, the three-motif combination is significant. (C) Flower diagram representation of the three-motif combination. Each petal corresponds to a single motif, and the center circle represents the motif combination. Color encodes the P values, and statistically significant ones are shown in red.
The principle behind the Bonferroni correction (22) is simple. Given 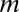 hypotheses, FWER is not higher than
hypotheses, FWER is not higher than 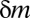 because of Boole’s inequality. In the Bonferroni correction,
because of Boole’s inequality. In the Bonferroni correction, 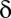 is determined as
is determined as 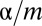 , which means that FWER ultimately is controlled under the significance level
, which means that FWER ultimately is controlled under the significance level 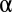 . If up to
. If up to 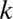 -motif combinations are considered, the number of hypotheses
-motif combinations are considered, the number of hypotheses 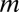 increases exponentially with
increases exponentially with 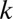 , which makes the threshold
, which makes the threshold 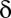 extremely small.
extremely small.
Interestingly, if the number of target genes 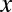 is sufficiently small, the motif combination may be ignored as nonregulatory without performing the test. Using Fisher’s exact test, the raw P value cannot be smaller than
is sufficiently small, the motif combination may be ignored as nonregulatory without performing the test. Using Fisher’s exact test, the raw P value cannot be smaller than
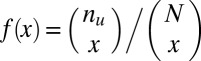 |
(Methods). Thus, if 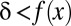 , this motif combination can never be significant, regardless of the expression levels targeted by the combination. This type of combination is called “untestable” (Fig. 2A). Tarone (26) showed that FWER does not increase with the addition of untestable combinations. Using this fact, the FWER bound can be tightened from
, this motif combination can never be significant, regardless of the expression levels targeted by the combination. This type of combination is called “untestable” (Fig. 2A). Tarone (26) showed that FWER does not increase with the addition of untestable combinations. Using this fact, the FWER bound can be tightened from 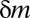 to
to 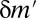 (Fig. 2B), where
(Fig. 2B), where 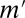 is the number of testable combinations that can be computed by an off-the-shelf frequent itemset mining algorithm (31–33) (see Methods for the full algorithm). This tightening leads to a dramatic reduction of the Bonferroni factor and enables us to discover statistically significant motif combinations without an arity limit.
is the number of testable combinations that can be computed by an off-the-shelf frequent itemset mining algorithm (31–33) (see Methods for the full algorithm). This tightening leads to a dramatic reduction of the Bonferroni factor and enables us to discover statistically significant motif combinations without an arity limit.
Fig. 2.

Relationships among threshold δ, the minimum P value f(x) of each motif combination, testable combinations, and the FWER bound. (A) Testable combinations. The bars represent the raw P values of motif combinations, and the red points on the bars indicate their minimum P values. The minimum P value of each combination is always below the raw P value. Testable motif combinations whose minimum P values are smaller than δ are illustrated in red. The testable combinations have the possibility of being considered regulatory. The number of all combinations of M motifs is 2M − 1, which is the Bonferroni factor without arity limit. (B) Comparison of FWER bounds between LAMP and Bonferroni correction. The FWER bound should be controlled under α. For a threshold δ, the FWER bound of LAMP is significantly smaller than that of the Bonferroni method, which indicates higher sensitivity of LAMP.
Most analyses involving multiple testing correction use the adjusted P value instead of the correction of 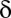 . Suppose
. Suppose 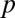 is the raw P value, and the adjusted P value of LAMP is calculated as
is the raw P value, and the adjusted P value of LAMP is calculated as 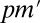 and is compared with
and is compared with  . In the following analysis, we use the adjusted P value.
. In the following analysis, we use the adjusted P value.
This method may be extended to other types of data, and the same principles may be applied to other types of statistical tests, including the Mann–Whitney U test for a single ranked series (see SI Appendix, Supporting Text 1 for details). Given a clustering result based on expression levels, the genes that belong to one of the clusters may be used as a substitute for the up-regulated genes.
Applications to Yeast Transcriptome.
Our analysis of yeast gene expression data yielded statistically significant but previously uncharacterized higher-arity motif combinations. The known or putative target genes of each known motif were collected from available ChIP–chip data (34). Microarray data from Gasch et al. (29), reflecting gene expression levels under 173 stress environments, also were used. A gene was considered up-regulated if its log2 ratio of expression level in target environment to that in control environment was at least 1.5. One-sided Fisher’s exact test was used, and the FWER was controlled under 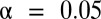 by the Bonferroni method and LAMP. The FWER is defined with respect to each environment separately. All motif combinations discovered are listed in SI Appendix, Table S1A.
by the Bonferroni method and LAMP. The FWER is defined with respect to each environment separately. All motif combinations discovered are listed in SI Appendix, Table S1A.
The number of testable combinations 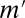 was optimized for each environment. The value of
was optimized for each environment. The value of  was in the range of 233–702 when maximum arity was not limited, whereas the original Bonferroni factor
was in the range of 233–702 when maximum arity was not limited, whereas the original Bonferroni factor 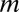 was 5,253 when maximum arity was two, which implies that LAMP has a higher sensitivity than the Bonferroni method. A total of 326 motif combinations were discovered, which is 1.70-fold higher than the number of motif combinations found using the Bonferroni correction (with a maximum arity of two) (SI Appendix, Supporting Text 2).
was 5,253 when maximum arity was two, which implies that LAMP has a higher sensitivity than the Bonferroni method. A total of 326 motif combinations were discovered, which is 1.70-fold higher than the number of motif combinations found using the Bonferroni correction (with a maximum arity of two) (SI Appendix, Supporting Text 2).
In the heat shock environment (shift from 25 °C to 37 °C), LAMP detected six motif combinations, including a four-motif combination, whereas the Bonferroni method found three singletons when the arity was limited to two and two singletons when the limit was set to three or four, failing to find any combinatorial regulations (Table 1). The members of the four-motif combination are bound by PHD1, SUT1, SOK2, and SKN7, respectively (34). Surprisingly, no single motif in the four-motif combination had a statistically significant P value (Fig. 3). According to the literature, PHD1, SUT1, and SKN7 are recruited by Tup1 (35, 36), a repressor under growth conditions, and the other gene, SOK2, is a candidate Tup1 recruiter (36), which suggests that all TFs bound to the motif jointly regulate genes within the Tup1-characterized pathway. Yu et al. (19) performed a comprehensive scanning of two-motif combinations with the expression data that includes our data. Indeed, they predicted a part of this four-motif combination (PHD1 and SKN7), but they failed to discover the whole combination. As shown in Fig. 3A, seven genes are found to have the four-motif combination in their regulatory regions, five of which, including a heat shock response TF (MGA1), stress response TF (MSN4), and heme activator protein (HAP4), are up-regulated. All proteins translated from the five genes are located in the nucleus, and three of them are related to RNA polymerase II, according to the Saccharomyces Genome Database (37), which suggests that this motif combination plays a role in the upstream regulation of the heat shock response.
Table 1.
Statistically significant motif combinations under a yeast heat shock condition
| Adjusted P value |
|||||
| Bonferroni |
|||||
| Motif combination | No. of target genes | LAMP | Max. arity ≤2 | Max. arity ≤3 | Max. arity ≤4 |
| HSF1 | 69 | 4.41E-24 | 7.64E-23 | 2.57E-21 | 6.44E-20 |
| MSN2 | 21 | 3.73E-11 | 6.46E-10 | 2.18E-08 | 5.45E-07 |
| MSN4 | 24 | 0.000532 | 0.00923 | 0.311 | >1 |
| SKO1 | 6 | 0.00839 | 0.145 | >1 | >1 |
| SNT2 | 18 | 0.0192 | 0.333 | >1 | >1 |
| PHD1, SUT1, SOK2, SKN7 | 7 | 0.0272 | — | — | >1 |
The bold numbers are significant after the correction. The optimal Bonferroni factor 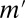 in LAMP is 167, whereas the Bonferroni factors are 5,253, 176,953, and 4,426,528 for maximum arities of 2, 3, and 4, respectively.
in LAMP is 167, whereas the Bonferroni factors are 5,253, 176,953, and 4,426,528 for maximum arities of 2, 3, and 4, respectively.
Fig. 3.

Statistical significance of the four-motif combination in yeast. (A) Expression ranks and P values of the genes targeted by the members of the combination. Adjusted P values are used. (B) Flower diagram of the combination. Although none of the singleton motifs was considered regulatory, the combination has a significant P value.
LAMP found a four-motif combination on raffinose medium; this combination consists of the binding motifs of the heme activators HAP1, HAP2, HAP3, and HAP5 (adjusted P value = 0.00754). Yu et al. (19) also detected a part of this combination (HAP3 and HAP5) but overlooked the higher-arity combination.
Applications to Breast Cancer Transcriptome.
Although both epidermal growth factor (EGF) and heregulin (HRG) belong to the ErbB family receptor ligands (38), EGF induces the proliferation of MCF-7 human breast cancer cells whereas HRG induces their differentiation (the accumulation of lipid droplets). A transcriptomics study (30) suggested the existence of unidentified mechanisms by which the same family ligands produce different patterns of early responsive gene expression. By using LAMP to reanalyze the gene expression profile of breast cancer cells treated with these two ligands, we identified statistically significant motif combinations related to proliferation- and differentiation-specific pathways. We applied LAMP with a one-sided Fisher exact test under the significance level 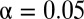 . The gene expression profiles were observed from 55 different conditions (30), which were stimulated with 0.1, 0.5, 1, and 10 nM of either EGF or HRG for 5, 10, 15, 30, 45, 60 (HRG only), and 90 min, and two controls. A gene was considered up-regulated if its expression level in the experimental sample was more than twice that in the control sample. We also used curated data from the Molecular Signatures Database (MSigDB) (39), including 397 types of binding sites, to determine the relationships between motifs and genes. Nomenclature of the motifs is due to MSigDB. The discovered motif combinations are listed in SI Appendix, Table S1 B and C. The value of
. The gene expression profiles were observed from 55 different conditions (30), which were stimulated with 0.1, 0.5, 1, and 10 nM of either EGF or HRG for 5, 10, 15, 30, 45, 60 (HRG only), and 90 min, and two controls. A gene was considered up-regulated if its expression level in the experimental sample was more than twice that in the control sample. We also used curated data from the Molecular Signatures Database (MSigDB) (39), including 397 types of binding sites, to determine the relationships between motifs and genes. Nomenclature of the motifs is due to MSigDB. The discovered motif combinations are listed in SI Appendix, Table S1 B and C. The value of 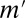 was ranged from 2,700,718 to 3,750,336 for EGF and from 1,174,108 to 2,874,627 for HRG. These values are 4,000 times higher than the value obtained in the yeast analysis, which reflects the greater complexity of the regulation of the human transcriptome. Our method found a total of 43 and 23 motif combinations in the EGF- and HRG-induced cells, respectively, including one eight-motif combination, whereas the Bonferroni correction (with a maximum arity of three) detected only 19 and 7 combinations, respectively, and found at most one three-motif combination.
was ranged from 2,700,718 to 3,750,336 for EGF and from 1,174,108 to 2,874,627 for HRG. These values are 4,000 times higher than the value obtained in the yeast analysis, which reflects the greater complexity of the regulation of the human transcriptome. Our method found a total of 43 and 23 motif combinations in the EGF- and HRG-induced cells, respectively, including one eight-motif combination, whereas the Bonferroni correction (with a maximum arity of three) detected only 19 and 7 combinations, respectively, and found at most one three-motif combination.
Fig. 4 shows the statistically significant high-arity combinations. The largest eight-motif combination was found after 15 min of treatment with 0.5 nM EGF (adjusted P value = 0.0137). Surprisingly, no single motif had an adjusted P value lower than 1 (Fig. 4A). Furthermore, none of the combinations with arity below six were statistically significant (Fig. 4B). According to existing knowledge, the eight-motif combination is highly relevant to the feature of MCF-7 cells and induced ligand type. A ChIP–seq study confirmed that some TFs bind to C/EBP, OCT1, and FOXO4 motifs in estrogen receptor (ER)-positive cells (40), and MCF-7 cells indeed are ER positive. Among the TFs known to bind to the motifs, CEBPB, GR, JUN, FOXA1, and FOXK2 are substantially expressed (SI Appendix, Supporting Text 3A). The transcription factor GR is highly regulated in MCF-7 cells by estrogen treatment (41). High expression of JUN increases biologic aggressiveness of MCF-7 cells (42).
Fig. 4.
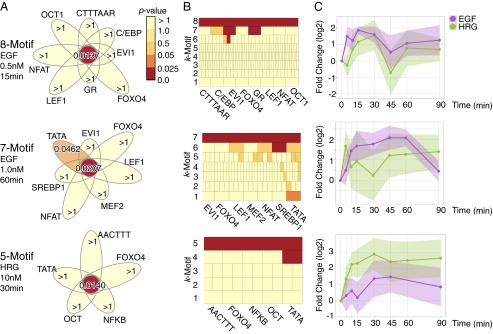
Statistically significant high-arity motif combinations in breast cancer cells. (A) Flower diagrams of motif combinations. (B) Adjusted P values of all subsets of the combinations. Each box represents one combination, and the color illustrates its P value. The k-th row consists of k-motif combinations. This figure shows that no subset with arity of two or three is significant. (C) Expression changes over time of the target genes of the k-motif combination. The averages and SDs of the expressions are represented as lines and color regions, respectively.
The second largest combination (adjusted P value = 0.0209) was a seven-motif combination identified in the cells treated with 1.0 nM EGF for 60 min. As shown in Fig. 4A, Middle, no single motif has a significant P value, except TATA (adjusted P value = 0.0462), which is known to be activated in the MCF-7 cells (43). Among the expressed TFs binding to the motifs (SI Appendix, Supporting Text 3B), MEF2 is of particular interest because it works with NFAT (44). The combination regulates 10 genes in total [brain-derived neurotrophic factor (BDNF), CHD2, EYA1, HOXA2, HAS2, interleukin 1 receptor accessory protein-like 1 (IL1RAPL1), PDZ domain containing ring finger 4 (PDZRN4), PHOX2B, PPARGC1A, and RBFOX1], five of which are related to differentiation (BDNF, EYA1, HOXA2, PHOX2B, and PPARGC1A) and two of which (HOXA2 and PHOX2B) are annotated as negative regulators of differentiation (Gene Ontology GO:0045665). In addition, Fig. 4C, Middle shows that expression levels of the 10 downstream genes in the EGF-induced cells are higher than those measured in the HRG-induced cells for the first 60 min. These results imply that the combination plays a negative regulatory role for differentiation in response to EGF.
A five-motif combination was found after 30 min of treatment with 10 nM HRG (Fig. 4 and SI Appendix, Supporting Text 3C). The combination targets 11 genes (BDNF, CYR61, EGR2, EHF, ETV1, HOXB3, HOXB6, ID2, IL1RAPL1, PDZRN4, and SOX5). Their average expression levels were higher than those observed in the EGF-induced cells, which suggests that the combination committed the cell toward an HRG-specific regulatory pathway.
LAMP identified several high-arity combinations that would have been missed by single-motif or motif-pair scans, providing a deeper understanding of the ligand-specific pathways. About half the genes targeted by the discovered high-arity combinations are related to the regulation of transcription; LAMP may contribute to capture key regulations that determine cell fate. Note that the biological views presented here are only hypothetical. Extensive biological investigation is necessary to confirm the hypotheses.
Discussion
To demonstrate that LAMP improves the detection of combinatorial regulations, we computed empirical FWERs with the 1,000 permuted sets of yeast data used to compute Table 1 and compared the FWERs with the Bonferroni correction. The significance level 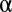 was set to 0.05. Although the Bonferroni correction never found significant combinations from the permutation data (with a maximum arity of three), the average FWER computed by LAMP was 0.013 (Fig. 5A), which demonstrates that the detection power of LAMP is potentially higher than that of the Bonferroni correction. We also computed the empirical FWER using human cancer cell data (Fig. 5B). As in the yeast data analysis, the results showed that the detection power of LAMP was higher than that of the Bonferroni correction.
was set to 0.05. Although the Bonferroni correction never found significant combinations from the permutation data (with a maximum arity of three), the average FWER computed by LAMP was 0.013 (Fig. 5A), which demonstrates that the detection power of LAMP is potentially higher than that of the Bonferroni correction. We also computed the empirical FWER using human cancer cell data (Fig. 5B). As in the yeast data analysis, the results showed that the detection power of LAMP was higher than that of the Bonferroni correction.
Fig. 5.

Comparison of the results of LAMP with those of Bonferroni corrections using randomly permuted datasets. (A and B) Comparisons of the empirical FWERs of LAMP with those of the Bonferroni correction with respect to the yeast and human data, respectively. (C and D) Computational time for the analysis of the yeast and human data, respectively. Note that the y-axis is in log scale.
The calculation speed of LAMP is sufficiently fast compared with other correction methods. As shown in Fig. 5C, LAMP required less than 1 s to analyze the yeast data (even without an arity limit), whereas the computing time of the Bonferroni correction increases exponentially with the number of tests. For the human data (Fig. 5D), LAMP required 2.3 h without an arity limit, which was faster than the Bonferroni method (maximum arity of three or greater). The estimated computing time for the analysis of all possible combinations of the yeast data using the Bonferroni method is more than 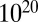 y. Thus, the branch-and-bound algorithm dramatically reduced the computing time. LAMP also is faster than other sensitive corrections, such as Šidák’s (23) and Holm’s (24) corrections. These corrections require listing and sorting an intractable number of tests when high-arity combinations are considered. LAMP avoids this calculation using a frequent itemset mining technique. Another approach for avoiding false discoveries in multiple testing is estimation of the error rate with random sampling-based methods (45–48). This approach would require an extraordinarily large amount of time because a large number of hypotheses must be investigated to discover high-arity combinations.
y. Thus, the branch-and-bound algorithm dramatically reduced the computing time. LAMP also is faster than other sensitive corrections, such as Šidák’s (23) and Holm’s (24) corrections. These corrections require listing and sorting an intractable number of tests when high-arity combinations are considered. LAMP avoids this calculation using a frequent itemset mining technique. Another approach for avoiding false discoveries in multiple testing is estimation of the error rate with random sampling-based methods (45–48). This approach would require an extraordinarily large amount of time because a large number of hypotheses must be investigated to discover high-arity combinations.
LAMP may be applied to any statistical test as long as the minimum P value is strictly positive. An important exception is the t test (45), whose minimum P value is always zero, because only one outlier can make the P value arbitrarily small. In this paper, we preferred Fisher’s exact test despite the need to set a threshold for expression levels, because up-regulation by external signals has to be discriminated from natural fluctuations. The Mann–Whitney U test may have been another choice, because it does not require dichotomization; however, the P value is likely to be affected by small expression changes not caused by the signal of interest.
We investigated how our results change with different thresholds (SI Appendix, Supporting Text 2) and observed that a lower threshold leads to a larger number of discoveries. Although it is common to determine the optimal threshold to maximize the number of discoveries (49), it leads to an unconventionally low threshold that cannot be justified from biological viewpoints. In our experiments, the threshold is selected so that it is large enough to rule out natural fluctuations and a large number of high-arity combinations are discovered.
The control of false discovery rate (FDR) instead of the FWER often is enough in biological problems (21, 45). Combining an FDR controlling method such as Benjamini–Hochberg (27) or Storey–Tibshirani (28) with the itemset mining algorithms we have done for the Bonferroni is a challenge for further research. In our yeast and human experiments, multiple-testing correction was performed separately for each environment. To obtain statistically stronger results, environment-wide correction would be necessary. LAMP possibly may be extended to this case by using an environment-wide mining algorithm. In this paper, we focused on finding significant joint effects only, but it would be interesting to decompose the effect to multilevel interaction effects. Also, LAMP possibly may be improved by incorporating the method by Roth (50), which uses a noninteger correction factor. On the application side, LAMP may be used to provide an integrated analysis of heterogeneous biological data. For example, recent experiments have shown the chromatin status of regulatory regions: the combination of these data with information on the binding motif sites may be used to uncover collaborative regulations between chromatin openings and the binding of TFs. The integration of microRNA (miRNA) targets and TF binding sites may show orchestral regulations by miRNAs and TFs.
Methods
Minimum P Value.
Suppose we have 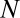 genes in total. We partition the genes in two ways: one is according to whether a gene is targeted by all members of a motif combination, the other according to whether a gene is up-regulated. Assume that
genes in total. We partition the genes in two ways: one is according to whether a gene is targeted by all members of a motif combination, the other according to whether a gene is up-regulated. Assume that 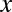 genes are targeted,
genes are targeted, 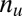 genes are up-regulated, and
genes are up-regulated, and  genes are both targeted and up-regulated. Because no value in the contingency table is negative,
genes are both targeted and up-regulated. Because no value in the contingency table is negative, 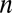 ranges from
ranges from 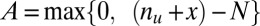 to
to 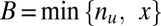 . Using one-sided Fisher’s exact test, the P value of the motif combination is computed as
. Using one-sided Fisher’s exact test, the P value of the motif combination is computed as
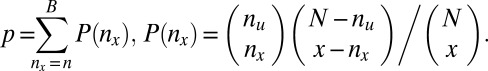 |
The P value 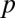 attains its minimum value when the contingency table is most biased; that is,
attains its minimum value when the contingency table is most biased; that is, 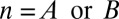 . Because
. Because 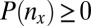 , the minimum value is attained at
, the minimum value is attained at 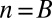 . If
. If 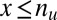 , the minimum is achieved at
, the minimum is achieved at 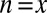 and described as
and described as 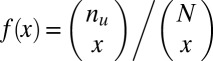 . If
. If 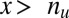 ,
,  is defined as
is defined as 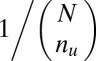 (SI Appendix, Supporting Text 4).
(SI Appendix, Supporting Text 4).
Algorithmic Details.
Fixing a P value threshold 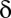 , the testable combinations are identified by computing the number of target genes
, the testable combinations are identified by computing the number of target genes 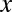 for all motif combinations that satisfy
for all motif combinations that satisfy 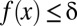 . Because
. Because 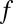 is a monotonic function (SI Appendix, Supporting Text 4), it is possible to determine
is a monotonic function (SI Appendix, Supporting Text 4), it is possible to determine 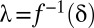 . Then, testable combinations may be collected by identifying the combinations that target
. Then, testable combinations may be collected by identifying the combinations that target 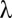 or more genes. Fortunately, this task may be performed efficiently by an itemset mining algorithm (SI Appendix, Supporting Text 5). As shown in Tarone (26), FWER is bounded by
or more genes. Fortunately, this task may be performed efficiently by an itemset mining algorithm (SI Appendix, Supporting Text 5). As shown in Tarone (26), FWER is bounded by 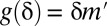 , where
, where 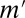 is the number of testable combinations.
is the number of testable combinations.
Because the domain of 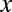 is discrete,
is discrete,  is defined as
is defined as 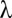 such that
such that 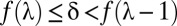 . Because of discontinuities of
. Because of discontinuities of 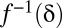 , the FWER bound is a stepwise linear function of
, the FWER bound is a stepwise linear function of 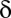 (SI Appendix, Fig. S1).
(SI Appendix, Fig. S1).
The P value threshold 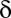 must be determined so that the FWER bound is calibrated to a predetermined level
must be determined so that the FWER bound is calibrated to a predetermined level 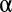 . To this aim, we construct the function
. To this aim, we construct the function 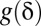 by drawing line segments in SI Appendix, Fig. S1 one by one from bottom to top. To draw the first segment, itemset mining is performed to find the combinations that target
by drawing line segments in SI Appendix, Fig. S1 one by one from bottom to top. To draw the first segment, itemset mining is performed to find the combinations that target 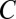 or more genes. If the number of such combinations is
or more genes. If the number of such combinations is 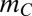 , the line segment is described as
, the line segment is described as
Itemset mining is repeated by decrementing the parameter one by one, and the procedure stops if one of the end points of the line segment goes beyond  (SI Appendix, Fig. S2). Finally, the crossing point is presented as the final result.
(SI Appendix, Fig. S2). Finally, the crossing point is presented as the final result.
Yeast Data.
To perform testing, gene expression data and TF binding site information must be combined. For gene expression data, we used microarray data observed over 173 different conditions on an average of 5,935.6 genes (29). Binding site positions are generated from Harbison et al. (34), and gene positions used are from the Saccharomyces Genome Database (37). We associated a motif with a gene when the TF binding site was located between 800 bp upstream and 50 bp downstream from the transcription start sites of the gene. The integrated data contained 102 types of motifs. The motifs are associated with an average of 30.1 genes.
Human Breast Cancer Data.
Human breast cancer data were generated from the MCF-7 breast cancer expression profile (accession no. GSE6462; normalized using the trimmed mean method with GCOS 1.2) (30) and relationships between motifs and genes provided by the MSigDB (39). The expression data contain samples induced by two different ErbB receptor ligands, EGF and HRG. We removed genes whose log2 expression levels are less than 4 over all conditions on each ligand. The numbers of genes in EGF- and HRG-induced samples were 12,773 and 12,851, respectively. To associate TF binding motifs with genes, we used the value “motif” in category C3 in the MSigDB (39). The data contained a few different quality scores for each motif. To remove the ambiguity, we merged associated genes over the different qualities for each motif. We associated the TF data with gene expression data through GenBank ID. The number of motifs is 397, and the motifs are associated with an average of 215.4 and 217.3 genes for EGF and HRG, respectively.
Calculation of Empirical FWER with Permutation Data.
We estimated empirical FWER and the average number of discoveries from randomly permuted data (Fig. 5). In permutation, gene expression levels were shuffled randomly. The yeast data are taken from the heat shock environment, and the human data are from EGF, 0.5 nM 15 min. For each organism, 1,000 different permuted datasets were generated. If at least one false discovery happened in a permuted dataset, the dataset was counted as a family-wise error. An estimate of empirical FWER is computed as the fraction of family-wise errors among the 1,000 datasets. In addition, the number of discoveries was recorded in each permuted dataset, and the average over all datasets was computed. The above experiment was repeated 10 times; Fig. 5 reports the average of estimated values and their SDs. All experiments were conducted on a machine with two AMD Opteron processors at 2.3 GHz with 32 gigabytes of random access memory running Red Hat Linux.
Software.
All programs to correct significance levels are available from http://seselab.org/lamp. The programs are written in Python except linear time closed itemset miner (LCM) (33) for frequent itemset mining.
Supplementary Material
Acknowledgments
The supercomputing resource was provided by the National Institute of Genetics, Research Organization of Information and Systems, Japan, and the Human Genome Center (The University of Tokyo). This work was made possible by the support of the Minato Discrete Structure Manipulation System Project, Exploratory Research for Advanced Technology, Japan Science and Technology Agency (to A.T. and K.T.); the Cell Innovation Project, Ministry of Education, Culture, Sports, Science, and Technology, Japan (to M.O.-H.); and Japan Society for the Promotion of Science (JSPS) KAKENHI Grants 23128504, 24240044, and 24680032 (to J.S.). A.T. is supported by JSPS Research Fellowships for Young Scientists. K.T. also is supported by the Funding Program for World-Leading Innovative R&D on Science and Technology.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1302233110/-/DCSupplemental.
References
- 1.Ravasi T, et al. An atlas of combinatorial transcriptional regulation in mouse and man. Cell. 2010;140(5):744–752. doi: 10.1016/j.cell.2010.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee B-K, et al. Cell-type specific and combinatorial usage of diverse transcription factors revealed by genome-wide binding studies in multiple human cells. Genome Res. 2012;22(1):9–24. doi: 10.1101/gr.127597.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dunham I, et al. ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gerstein MB, et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489(7414):91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charos AE, et al. A highly integrated and complex PPARGC1A transcription factor binding network in HepG2 cells. Genome Res. 2012;22(9):1668–1679. doi: 10.1101/gr.127761.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J, et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22(9):1798–1812. doi: 10.1101/gr.139105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miura H, et al. Identification of DNA regions and a set of transcriptional regulatory factors involved in transcriptional regulation of several human liver-enriched transcription factor genes. Nucleic Acids Res. 2009;37(3):778–792. doi: 10.1093/nar/gkn978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schlesinger J, et al. The cardiac transcription network modulated by Gata4, Mef2a, Nkx2.5, Srf, histone modifications, and microRNAs. PLoS Genet. 2011;7(2):e1001313. doi: 10.1371/journal.pgen.1001313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baudry A, et al. TT2, TT8, and TTG1 synergistically specify the expression of BANYULS and proanthocyanidin biosynthesis in Arabidopsis thaliana. Plant J. 2004;39(3):366–380. doi: 10.1111/j.1365-313X.2004.02138.x. [DOI] [PubMed] [Google Scholar]
- 10.Wang R, et al. Multiple regulatory elements in the Arabidopsis NIA1 promoter act synergistically to form a nitrate enhancer. Plant Physiol. 2010;154(1):423–432. doi: 10.1104/pp.110.162586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Middendorf M, Kundaje A, Wiggins C, Freund Y, Leslie C. Predicting genetic regulatory response using classification. Bioinformatics. 2004;20(Suppl 1):i232–i240. doi: 10.1093/bioinformatics/bth923. [DOI] [PubMed] [Google Scholar]
- 12.Schultheiss SJ, Busch W, Lohmann JU, Kohlbacher O, Rätsch G. KIRMES: Kernel-based identification of regulatory modules in euchromatic sequences. Bioinformatics. 2009;25(16):2126–2133. doi: 10.1093/bioinformatics/btp278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Narlikar L, et al. Genome-wide discovery of human heart enhancers. Genome Res. 2010;20(3):381–392. doi: 10.1101/gr.098657.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng C, et al. Understanding transcriptional regulation by integrative analysis of transcription factor binding data. Genome Res. 2012;22(9):1658–1667. doi: 10.1101/gr.136838.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arvey A, Agius P, Noble WS, Leslie C. Sequence and chromatin determinants of cell-type-specific transcription factor binding. Genome Res. 2012;22(9):1723–1734. doi: 10.1101/gr.127712.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pilpel Y, Sudarsanam P, Church GM. Identifying regulatory networks by combinatorial analysis of promoter elements. Nat Genet. 2001;29(2):153–159. doi: 10.1038/ng724. [DOI] [PubMed] [Google Scholar]
- 17.Banerjee N, Zhang MQ. Identifying cooperativity among transcription factors controlling the cell cycle in yeast. Nucleic Acids Res. 2003;31(23):7024–7031. doi: 10.1093/nar/gkg894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu Z, Shendure J, Church GM. Discovering functional transcription-factor combinations in the human cell cycle. Genome Res. 2005;15(6):848–855. doi: 10.1101/gr.3394405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu X, et al. Genome-wide prediction and characterization of interactions between transcription factors in Saccharomyces cerevisiae. Nucleic Acids Res. 2006;34(3):917–927. doi: 10.1093/nar/gkj487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shiraishi Y, Okada-Hatakeyama M, Miyano S. A rank-based statistical test for measuring synergistic effects between two gene sets. Bioinformatics. 2011;27(17):2399–2405. doi: 10.1093/bioinformatics/btr382. [DOI] [PubMed] [Google Scholar]
- 21.Noble WS. How does multiple testing correction work? Nat Biotechnol. 2009;27(12):1135–1137. doi: 10.1038/nbt1209-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bonferroni C (1936) Teoria Statistica Delle Classi e Calcolo Delle Probabilita, Pubblicazioni del R Istituto Superiore di Scienze Economiche e Commerciali di Firenze (Libreria Internazionale Seeber, Florence, Italy) Vol 8, pp 3–62.
- 23.Šidák Z. Rectangular confidence regions for the means of multivariate normal distributions. J Am Stat Assoc. 1967;62(318):626–633. [Google Scholar]
- 24.Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat. 1979;6(2):65–70. [Google Scholar]
- 25.Hochberg Y. A sharper Bonferroni procedure for multiple tests of significance. Biometrika. 1988;75(4):800–802. [Google Scholar]
- 26.Tarone RE. A modified Bonferroni method for discrete data. Biometrics. 1990;46(2):515–522. [PubMed] [Google Scholar]
- 27.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc, B. 1995;57(1):289–300. [Google Scholar]
- 28.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gasch AP, et al. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11(12):4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagashima T, et al. Quantitative transcriptional control of ErbB receptor signaling undergoes graded to biphasic response for cell differentiation. J Biol Chem. 2007;282(6):4045–4056. doi: 10.1074/jbc.M608653200. [DOI] [PubMed] [Google Scholar]
- 31. Agrawal R, Srikant R (1994) Fast algorithms for mining association rules. Proceedings of the 20th International Conference on Very Large Data Bases (Morgan Kaufmann, San Francisco), 487–499.
- 32. Han J, Pei J, Yin Y (2000) Mining frequent patterns without candidate generation. Proceedings of the 2000 ACM SIGMOD International Conference on Management of Data, eds Chen W, Naughton JF, Bernstein PA (Assoc Computing Machinery, New York), pp 1–12.
- 33. Uno T, Asai T, Uchida Y, Arimura H (2003) LCM: An efficient algorithm for enumerating frequent closed item sets. Proceedings of the ICDM 2003 Workshop on Frequent Itemset Mining Implementations, eds Goethals B, Zaki MJ. Available at http://ceur-ws.org/Vol-90. Accessed July 12, 2013.
- 34.Harbison CT, et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431(7004):99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ness F, et al. SUT1 is a putative Zn[II]2Cys6-transcription factor whose upregulation enhances both sterol uptake and synthesis in aerobically growing Saccharomyces cerevisiae cells. Eur J Biochem. 2001;268(6):1585–1595. [PubMed] [Google Scholar]
- 36.Hanlon SE, Rizzo JM, Tatomer DC, Lieb JD, Buck MJ. The stress response factors Yap6, Cin5, Phd1, and Skn7 direct targeting of the conserved co-repressor Tup1-Ssn6 in S. cerevisiae. PLoS ONE. 2011;6(4):e19060. doi: 10.1371/journal.pone.0019060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cherry JM, et al. SGD: Saccharomyces genome database. Nucleic Acids Res. 1998;26(1):73–79. doi: 10.1093/nar/26.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Citri A, Yarden Y. EGF-ERBB signalling: Towards the systems level. Nat Rev Mol Cell Biol. 2006;7(7):505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 39.Xie X, et al. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434(7031):338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carroll JS, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38(11):1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 41.Krishnan AV, Swami S, Feldman D. Estradiol inhibits glucocorticoid receptor expression and induces glucocorticoid resistance in MCF-7 human breast cancer cells. J Steroid Biochem Mol Biol. 2001;77(1):29–37. doi: 10.1016/s0960-0760(01)00030-9. [DOI] [PubMed] [Google Scholar]
- 42.Smith LM, et al. cJun overexpression in MCF-7 breast cancer cells produces a tumorigenic, invasive and hormone resistant phenotype. Oncogene. 1999;18(44):6063–6070. doi: 10.1038/sj.onc.1202989. [DOI] [PubMed] [Google Scholar]
- 43.Cavaillès V, Augereau P, Rochefort H. Cathepsin D gene is controlled by a mixed promoter, and estrogens stimulate only TATA-dependent transcription in breast cancer cells. Proc Natl Acad Sci USA. 1993;90(1):203–207. doi: 10.1073/pnas.90.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKinsey TA, Zhang CL, Olson EN. MEF2: A calcium-dependent regulator of cell division, differentiation and death. Trends Biochem Sci. 2002;27(1):40–47. doi: 10.1016/s0968-0004(01)02031-x. [DOI] [PubMed] [Google Scholar]
- 45.Dudoit S, Shaffer JP, Boldrick J. Multiple hypothesis testing in microarray experiments. Stat Sci. 2003;18(1):71–103. [Google Scholar]
- 46.Westfall P, Young S. Resampling-Based Multiple Testing: Examples and Methods for p-Value Adjustment. New York: Wiley; 1993. [Google Scholar]
- 47.Troendle JF. A permutational step-up method of testing multiple outcomes. Biometrics. 1996;52(3):846–859. [PubMed] [Google Scholar]
- 48.Korn E, Troendle J, McShane L, Simon R. Controlling the number of false discoveries: Application to high-dimensional genomic data. J Statist Plann Inference. 2004;124(2):379–398. [Google Scholar]
- 49.Budczies J, et al. Cutoff Finder: A comprehensive and straightforward Web application enabling rapid biomarker cutoff optimization. PLoS ONE. 2012;7(12):e51862. doi: 10.1371/journal.pone.0051862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roth AJ. Multiple comparison procedures for discrete test statistics. J Statist Plann Inference. 1999;82(1-2):101–117. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.