

Abstract
The advent of scanning two-photon microscopy (2PM) has created a fertile new avenue for noninvasive investigation of brain activity in depth. One principal weakness of this method, however, lies with the limit of scanning speed, which makes optical interrogation of action potential-like activity in a neuronal network problematic. Encoded multisite two-photon microscopy (eMS2PM), a scanless method that allows simultaneous imaging of multiple targets in depth with high temporal resolution, addresses this drawback. eMS2PM uses a liquid crystal spatial light modulator to split a high-power femto-laser beam into multiple subbeams. To distinguish them, a digital micromirror device encodes each subbeam with a specific binary amplitude modulation sequence. Fluorescence signals from all independently targeted sites are then collected simultaneously onto a single photodetector and site-specifically decoded. We demonstrate that eMS2PM can be used to image spike-like voltage transients in cultured cells and fluorescence transients (calcium signals in neurons and red blood cells in capillaries from the cortex) in depth in vivo. These results establish eMS2PM as a unique method for simultaneous acquisition of neuronal network activity.
Keywords: multipoint, multiplexing, voltage-sensitive dyes, scanless two-photon microscopy
A persistent and challenging demand in neuroscience is the ability to monitor activity from defined populations of cellular targets in depth in the brain. Imaging with two-photon microscopy (2PM) of cells labeled with calcium-sensitive reporters (1, 2) has become the most popular approach to indirectly report spikes and yields micrometer-scale spatial resolution in vivo (3) to depths up to 1,000 μm (4). Conventional 2PM uses relatively slow scanning mechanisms, however, which do not permit the simultaneous acquisition of multiple millisecond-range signals emitted by cellular ensembles. In the last several years various attempts have been made to overcome this drawback. For example, using a mirror-based targeted path scanning technique that drastically reduced the background scanning time, Lillis et al. monitored the dynamics of spatially extended neuronal networks (5). Another approach consisted of using acousto-optic deflectors to steer the laser beam in less than 10 μs between cells or subcellular sites of interest (6–9). Both of these sequential scanning methods, however, suffer from the fundamental trade-off between signal-to-noise ratio (SNR) and fast temporal resolution (which should be maximized for detection of fast events such as spikes). Increasing the SNR typically requires integrating more photons per pixel, which can be achieved by increasing either the excitation laser intensity or the pixel dwell time. Regarding the former, there is a ceiling beyond which the average laser power cannot be increased (2.5–10 mW), typically referred to as the photo-damage limit (10, 11). Regarding the latter, decreasing the scanning rate will increase photon count but with the cost of lowering temporal resolution, ultimately reducing precise acquisition of fast events and missed spikes. Most recently, intracellularly loaded organic dyes have been successfully used to follow the detection of voltage transients. One method used a CCD camera for simultaneous detection of multipoints (12). A second used single-voxel recording with 2P excitation (13). Although the wide-field CCD method yields good temporal and spatial resolution, it is not feasible for acquisition of signals in depth. Single-voxel 2PM signals, on the other hand, can be acquired in depth but are not simultaneous.
To overcome the above-described SNR limitation, without sacrificing time resolution, ideally one illuminates and collects from all sites simultaneously. With average powers of more than 3 W, it is possible, using multiple reflection beam splitters (14) or spatial light modulators (SLM) (15), to split the excitation beam of currently available femtosecond Ti:Sapphire lasers into subbeams of significant milliwatt power. Besides creating multisite excitation points, SLMs can also be used to generate defined patterns of excitation (16). The limitation of these multisite-imaging methods comes from the detection, specifically when using array detectors such as CCD cameras, because fluorescence signals originating from different sites within the sample are blurred by scattering and spatially overlap onto the camera, thus limiting the imaging depth (17).
Here, we present encoded multisite two-photon microscopy (eMS2PM), as an alternative method. With eMS2PM, a femtosecond laser beam is split with a liquid crystal SLM and focused onto chosen targets (16, 18). Target registration is accomplished by repeated amplitude modulation of each subbeam with a short specific binary encoding sequence, using a digital micromirror device (DMD). Fluorescence signals emitted from all sites are collected onto a single photodetector. Individual fluorescence signals originating from each target are then recovered by multiplication of the photodetector signal by a site-specific decoding sequence.
We first tested the principle of eMS2PM on a slide of fluorescent beads moving below a scattering medium. We then used this approach to measure voltage transients in HEK cells, using a FRET-based DiO-C16/dipicrylamine quenching signal and calcium transients reported by Oregon Green Bapta 1 (OGB1) in cortical neurons in vitro in brain slice up to a depth of 220 µm and down to 300 µm in vivo in mouse neocortex. Finally we fluorescently imaged red blood cell passages from up to 12 simultaneous points in plasma-labeled capillaries at a depth of 350 µm in mouse neocortex.
Results
Principle of Encoded Multisite Two-Photon Microscopy.
An eMS2PM setup (Fig. 1, red beams) was added to a conventional 2PM (Fig. 1, yellow beam). Implementation begins with a reference image being scanned and acquired with the 2PM. Next, the user draws regions of interest (ROIs), consisting of single points (near diffraction-limited excitation points) or clusters of points, onto the reference image. The beam from the eMS2PM is then expanded (5×) to illuminate a reflective liquid crystal SLM. A SLM phase profile, required to create the user-defined ROI intensity profile at the focal plane of the objective (OB), is then computed by an iterative Fourier-transform algorithm (18). With a DMD (Methods), having mirrors that can be switched between ON and OFF every 60 μs and placed at the focal plane of lens L2, ensembles of micromirrors, corresponding to each ROI, are repeatedly updated according to orthogonal binary coding sequences or S codes (for the principle of S codes see Methods, Fig. S1, and SI Text 1). S codes are a succession of 60-μs-long ON or OFF illumination bins with the number of bins varying from 4, 8, 12, and 16 for 3, 7, 11, and 15 ROIs, respectively. This creates fast intensity modulation patterns specific to each ROI. The size of the field of view (100 μm × 100 μm) is set by the magnification of the telescope L3-OB. Because fluorescence emission is quasi-instantaneous compared with the duration of each illumination bin, the fluorescence emitted by each ROI is modulated with exactly the same pattern of binary S codes, creating slow envelope modulations on top of the fast binary S codes (Fig. 1, green signals between L4 and L5). All ROI signals are summed onto a single photomultiplier tube (PMT), amplified, stored on computer RAM, and demultiplexed using a custom-made decoding algorithm. The time resolution of eMS2PM is set by the duration of S codes (0.24 ms for 2–3 ROIs, 0.48 ms for 4–7 ROIs, 0.72 ms for 8–11 ROIs, and 0.96 ms for 12–15 ROIs). The targeting precision, which depends on the submicrometric coalignment of the 2PM and the eMS2PM, is tuned with a semiautomatic alignment software (SI Methods). Thus, ROIs drawn onto 2PM images are targeted to less than 0.5 μm in 3D.
Fig. 1.

The encoded multisite two-photon microscope (eMS2PM) setup. A reference image, used for choosing and targeting regions of interest (ROIs), is first acquired with a conventional galvanometric mirror-based (GM) two-photon laser-scanning microscope (2PM), illustrated by the yellow beam. A spatial light modulator (SLM) splits light from a second laser (red beam) into multiple subbeams that target user-specified sites inside sample S, e.g., neurons n1, … , n3. Subbeams are first focused onto a digital micromirror device (DMD) by lens L2 and repeatedly modulated with a different binary submillisecond S code (Methods). The DMD plane is imaged into the sample by a dichroic mirror (DIC1), the tube lens L3, and the microscope objective (OB). Fluorescent transients, e.g., generated by action potentials (black traces from n1, … , n3), induce an envelope variation of the fast binary fluorescence signals. The collected light is then reflected by a dichroic mirror DIC2 and projected onto the photomultiplier tube (PMT) by lenses L4 and L5. All signals are simultaneously detected and then recorded onto a computer. Individual signals from all cells (Sn1, Sn2, and Sn3) are decoded by a postprocessing algorithm. (Inset) Main steps of eMS2PM recording: 1, image acquisition with 2PM; 2, targeting ROIs; 3, SLM hologram computation; 4, programming DMD sequences; 5, detection and acquisition of all fluorescence signals; 6, decoding individual signals.
eMS2PM Proof of Concept: Detection of Moving Fluorescent Samples.
To test targeting of micrometric size structures and recording of dynamic events, we mounted 4-μm-diameter fluorescent beads (500-nm emission wavelength) on a glass slide, which could be translated laterally with respect to the objective (Fig. 2). Transient fluorescence signals occurred as the beads crossed static eMS2PM foci. In Fig. 2 (Upper), nine points were placed on an image acquired first with the 2PM to produce synchronous points (1, 4, 6–8), asynchronous points (3, 5, 9), or a neutral point (2). Signals were recorded and decoded as beads passed at 0.26 mm⋅s−1 under targeted points, with a time resolution of 0.72 ms. Excitation power was around 2 mW per point. As expected, point 3 was the first to detect a bead passage and points 1, 4, 6, 7, and 8 responded simultaneously, followed by points 5 and 9. Point 2 did not record any signal. This experiment demonstrated that the eMS2PM setup was able to arbitrarily target micrometric-scale structures in 2D and to simultaneously detect and separate multiple fluorescence signals. We repeated this experiment after placing a 500-μm-thick layer of scattering medium (0.5-μm diameter beads, 263 μm scattering mean free path at 860 nm emission and g = 0.73 from Mie theory) above the sample to mimic the effect of scattering in brain tissue (19, 20). Excitation power was increased to ∼30 mW per point. Despite the scattering barrier, fluorescent signals were clearly detected and similar to those in the absence of the scattering layer (3.8 ± 0.3 μm FWHM and 4.0 ± 0.2 μm FWHM, respectively). The average SNR of bead signals was 12.8, instead of 19.3 without a scattering layer (signal and noise measured around the peak of each bead signal). Importantly we did not detect crosstalk between points separated by 5 μm (i.e., points 3, 4, and 5), demonstrating that even with emission through a scattering medium, eMS2PM maintains precise spatial representation. A slight increase of noise, however, was recorded when one or more ROIs crossed a bead (i.e., trace of ROI 3 when ROIs 1, 4, 6, 7, and 8 detect a bead) and noise level tended to increase when more beads were illuminated. Theoretical (SI Text 2) and experimental demonstrations (Fig. S2) indicate that the noise measured in decoded traces is proportional to the square root of the number of illuminated beads, e.g., to the total number of photons received at the detector. This means that the noise measured in decoded eMS2PM traces is not correlated to the fluorescence of the specific target (as typically observed in shot-noise–limited laser scanning methods) but rather to the total detected signal. Here we demonstrated the ability to follow millisecond timescale synchronized or asynchronized fluorescent signals in nine user-defined targets. Furthermore, in a similar experiment where a fluorescent standard sample was translated at 3 mm⋅s−1, we could record submillisecond ON/OFF transients in 15 sites at 0.96 ms time resolution (Fig. S3).
Fig. 2.

Simultaneous detection of moving fluorescent samples with eMS2PM. (A) (Upper) Five fluorescent beads (4 μm) mounted on a glass slide were translated laterally (0.26 mm⋅s−1) from left to right. Transient fluorescence signals were detected and decoded as the beads crossed nine targeted but static eMS2PM foci. Note that no signal was detected (target 2) in the absence of bead. (B) Similar fluorescence signals were detected when the beads moved below a 500-μm-thick layer of scattering medium (0.5-μm diameter beads, 260-μm scattering mean free path at 860 nm).
eMS2PM Imaging of Spike-Like Voltage Transients in Cultured HEK Cells.
We next applied eMS2PM to record voltage-dependent fluorescence transients at several sites, using a FRET-based DiO-C16/dipicrylamine (DiO/DPA) method, where fluorescence emission of DiO is quenched by 5 µM DPA upon membrane depolarization (∼50% per 100 mV) with a response time of ∼0.1 ms (21). HEK-293 cells were labeled with DiO-C16, and their fluorescence was scanned by standard 2PM. Next, we voltage clamped the cells in whole-cell patch-clamp mode at a potential of −60 mV. Then two ROIs, composed of five points, were drawn: one ROI on the membrane of the patched cell and a second ROI on a neighboring uncoupled cell (Fig. 3A). Finally, while the bath was perfused with 5 μM DPA, five spike-like voltage commands were periodically applied at 90 Hz and decoded quenched fluorescence signals revealed spike-like transients from the patched cell and not the control cell. The 100-mV transient voltage commands induced fluorescence changes of −33.7 ± 4.1% (n = 20) with a half-width of 1.94 ± 0.21 ms. These data indicate that eMS2PM can follow quenched fluorescent bursts of spike-like events up to 90 Hz.
Fig. 3.

Recording spike-like waveforms with eMS2PM in HEK cells. (A) Two HEK-293 cells were labeled with DiO-C16 and then perfused with DPA. The cell at the bottom was voltage-clamped at −60 mV in whole-cell patch clamp mode. In each cell, two regions of interest (ROIs) were generated by focusing five points per ROI on the cell membrane. (Scale bar: 15 µm.) (B) Fluorescence quenching responses to five spike-like waveforms used as command voltage, 0.48-ms time resolution. (Left) Single sweeps. (Right) Averages of six trials. (Inset) Detail of spike waveforms (solid) and single-trial fluorescence response in bottom cell (shaded).
eMS2PM Imaging of Calcium Signals in Acute Cortical Slices.
Having established the speed at which eMS2PM can acquire signals, we investigated how well this method would perform to record neuronal activity within scattering brain tissue. Using acute neocortical brain slices from young mice, we imaged simultaneously the activity of up to six pyramidal neurons labeled with OGB1 AM at a depth of 100–220 µm (22). Fig. 4A illustrates the responses of five cells to 100 µM glutamate perfused onto the slice (100 µm in depth). Two points were targeted per ROI. Fluorescence was simultaneously recorded from six ROIs (five somata and one neuropil) with a time resolution of 2 ms for periods of 4 min. We found glutamate evoked asynchronous activity in two neurons (Fig. 4A, asterisks in s1 and s3) before a large complex synchronous event that was detected with different amplitudes, shapes, and durations at all five ROIs, including the neuropil (Bg). Importantly, we did not observe any drift of baseline fluorescence over these long and permanent illumination periods, indicating that under these conditions eMS2PM phototoxicity is negligible. In a second set of experiments, we evoked synaptic responses using local electrical stimulation with a small bipolar electrode (theta borosilicate pipette). Stimulation (trains of 5-Hz stimuli, repeated every 25 s over 240 s) evoked calcium increases of varying amplitudes in all six ROIs targeted onto neuronal somata (Fig. 4B, 150 µm in depth). Note that synaptic responses varied only slightly and independently over the 4 min of recordings. Repeated stimulation was possible without detectable phototoxicity. At a greater depth (Fig. 4C, 220 µm), independent calcium responses were still easily distinguishable in nearby ROIs of cortical neurons electroporated with OGB1 (23). In response to local stimulation (four pulses, 2 ms, 5 Hz), the calcium rise time was faster in the dendrite (d2) emerging from the soma S2 (Fig. 4C, Inset). Taken together these results clearly indicate that eMS2PM can be used to acquire separate and simultaneous calcium-evoked signals from multiple ROIs without detectable phototoxicity.
Fig. 4.

Simultaneous imaging of calcium signals with eMS2PM. (A) Calcium signals simultaneously detected in an acute slice of the mouse neocortex. (Left) The slice was labeled with a pipette containing Oregon Green Bapta 1 AM (OGB-AM) at a depth of 100 µM. Two points per ROI were targeted in each of five neurons and in the neuropil (background, Bg). (Center) Perfusion of glutamate evoked spontaneous activity in some neuronal somata (asterisks in s1 and s3) and a synchronized burst in all cells and in the neuropil. Note the absence of crosstalk between cell signals. (Right) Enlargement of the traces near the synchronized burst. (B) Continuous single-point excitation with eMS2PM does not alter synaptic responses. Electrical stimulations (5 Hz, four pulse trains) were repetitively applied through a glass theta pipette, placed at 100 µm in depth. Calcium responses of all recorded somata, labeled with OGB, varied independently (arrows) and did not show alteration with time (4 min of continuous eMS2PM). (C) Different calcium signal dynamics recorded at 220 µm with eMS2PM. (Left) Cells were labeled with OGB1 by electroporation in an acute cortical slice (P9 mouse). Calcium responses to electrical stimulation (5 Hz, four pulses, applied through a glass theta pipette) were recorded in three somata (s1, s2, and s4) and one dendrite of s2 (d2). (Center) Single trials (t1 and t2). (Right) (Inset) Note the faster signal kinetics in d2. (Scale bars, 15 µm.)
eMS2PM Imaging of Fluorescence Transients in the Cortex in Vivo.
We first investigated the potential of eMS2PM to image neuronal activity in young adult mice (postnatal days 20–40) in vivo. Pyramidal neurons labeled with electroporation of OGB1 (23) showed calcium increases in response to electrical stimulation. In Fig. 5A, the three somata (200 µm in depth) and the dendrite emerging from S1 simultaneously responded to electrical stimulation (four pulses, 2 ms, 5 Hz). These responses did not involve optical crosstalk between the sites, as revealed by the presence of large asynchronous spontaneous signals observed only in some ROIs (Fig. 5A, Right). eMS2PM could also be used to image calcium signals along the dendritic arbor of a single-layer II/III pyramid (Fig. 5B). Electrical stimulation (two pulses, 5 Hz) from a theta pipette placed at 200 µm in depth elicited a slow calcium signal in the soma (ROI 7, 300 µm) and faster ones in the apical dendrite (ROI 5, 275 µm; and ROI 1, 250 µm). Calcium responses were much weaker in nearby cellular processes (ROIs 2 and 4) and were not detected in the neuropil (ROIs 3 and 6). To image at 250, 275, and 300 µm, the laser power was tuned at 50, 65, and 75 mW per point, respectively (surface measurements). We then used eMS2PM to monitor transient fluorescence in cortical vessels. In capillaries labeled with fluorescein dextran, single red blood cells can be observed as longitudinal dark shadows in the fluorescent plasma (24). At a depth of 350 µm (Fig. 5C), we targeted 12 ROIs in three vessels (temporal resolution 0.96 ms): 10 points along a single capillary (ROIs 1–10) and 2 in neighboring larger vessels (ROIs 11 and 12). Traces 1–10 show that transient fluorescence drops (Fig. 5C, arrowhead in trace 1), indicating red blood cell passage, were correlated in all traces. The time shift (Fig. 5C, slanted dashed lines) allowed us to determine an average red blood cell velocity of 0.4 mm⋅s−1. As expected, individual red blood cells could not be extracted in larger vessels (ROIs 11 and 12) (Fig. 5C, Lower, gray traces). Taken together these results demonstrate that eMS2PM can detect simultaneously and without crosstalk fluorescent signals reporting physiological events in the brain at depth down to 300–350 µm with submillisecond temporal resolution.
Fig. 5.

In vivo imaging of fluorescence signals with eMS2PM. (A and B) Calcium signals simultaneously detected in neurons of the mouse neocortex (P38). (A) (Left) Bulk injection of OGB1 AM revealed labeled somata (s1–3) and dendrites (d1, emitted by s1) at a depth of 200 µm. (Center) Electrical stimulation (four pulses at 5 Hz) evoked calcium increases of different amplitudes. (Right) Spontaneous activity at the same four sites demonstrates the independence of fluorescence collection. (B) Calcium signals at several sites of a layer 2/3 pyramidal cell. (Left) OGB1 electroporation labeled several pyramids and processes (diagram and Insets). (Right) Electrical stimulation (two pulses at 5 Hz) evoked calcium increases in a single-pyramid soma (ROI 7) and its processes (ROIs 5 and 1). (Inset) Note the different time course of responses in the soma and the dendritic process (average of two trials). (C) (Upper) Three vessels labeled with fluorescein dextran were imaged at a depth of 350 µm. Individual red blood cells, flowing from left to right (arrow), were observed as longitudinal shadows in the capillary. Simultaneous measurements at 12 sites revealed correlated fluorescence drops in the capillary (ROIs 1–10), indicating red blood cells passage (open arrowhead) and allowing measurement of blood velocity (∼0.4 mm⋅s−1). Red blood cells are not detected in larger vessels (ROIs 11 and 12). (Scale bars, 10 µm.)
Discussion
The novelty of eMS2PM is the ability to simultaneously record submillisecond fluorescence changes at multiple sites in a scattering medium. Indeed the decoded eMS2PM signals are proportional to the fluorescence emitted at each targeted ROI averaged simultaneously for every S-code period (e.g., 0.48 ms for three ROIs). Such a capability is crucial for monitoring neuronal network electrical activity, either in vitro or in vivo. Current 2PM techniques capable of recording fluorescence signals simultaneously from multiple points use multiple pixel detectors. Such configurations often use an array of foci that are scanned across the sample and a CCD camera to record an image for every position of the foci array until the whole field of view is probed. Compared with single-focus scanning, image acquisition speed is multiplied by the number of foci and can reach 600 frames per second, using resonance scanners (25). Scanless 2P imaging, which implements a SLM to target the cells of interest (15), wide field excitation with temporal focusing (26, 27), light sheet illumination (28, 29), or exciting with an array of very closely spaced foci (30), can in principle further improve temporal resolution. In all these cases, however, the imaging speed is limited by the camera frame rate. Moreover, in all of the techniques to date using multiple-pixel detectors and imaging through a few tens of micrometers in the brain, light scattering reduces the spatial discrimination between adjacent cells. It has been shown in cortical slices, for example, that at depths greater than 50–100 μm poor contrast is due to an increasing contribution of scattered nonballistic photons (14, 30, 31). Imaging depths of these microscopes can be slightly increased by either restricting the detection to ballistic photons with confocal filtering (14) or reducing the optical crosstalk between adjacent pixels, using arrays of large photodetectors such as multianode photomultiplier tubes (31). In any case the imaging depths reported with these improved microscopes still do not reach the standard of a single-point scanning setup. Comparing eMS2PM, which uses a single detector and encoding for spatial discrimination, to all multipixel-based methods indicates only eMS2PM can resolve fluorescence signals that are spatially within a few micrometers apart within a scattering sample. We show this with fluorescent beads separated by 5 µm and decoded through approximately two mean free paths (Fig. 2) and with neurons recorded in vitro at depths up to 220 µm in neocortical acute slices (Fig. 4) and 300 µm in vivo (Fig. 5 A and B), where we acquired uncorrelated fluorescent calcium signals of neurons less than 10 µm apart. Finally, we detected individual RBC displacement in a cortical brain capillary by recording changes in plasma fluorescence transients at 10 points separated by ∼7 µm each at 350 µm deep.
eMS2PM and random access multiphoton microscopy (RAMP) offer comparable temporal resolutions. In RAMP, acousto-optic deflectors (AOD) rapidly scan the laser beam between points of interest with transition times down to ∼4 μs (7). Cellular dwell times for SNR > 2 are typically between 50 and 100 μs, depending on the cell type and the fluorescent indicator (6, 9). For example, action potentials in five purkinje cells labeled with FM 4–64 and recorded at ∼2 kHz yielded a SNR ∼2 (9). In vivo spiking activity was recorded at ∼500 Hz in 30–40 cortical neurons loaded with OGB1 AM in 2D (6) and 3D (32). In contrast, with eMS2PM where fluorescence variations are detected simultaneously, the total number of photons detected when recording from N cells is potentially N/2 times greater than with RAMP with the same time resolution (N/2 because S codes have a duty cycle of 1/2). However, the noise of eMS2PM-decoded traces also increases with the number of recorded sites (Fig. 2 and Fig. S2). We demonstrated theoretically (SI Text 2) and measured experimentally in standard fluorescent sample that, when using single-point ROIs, the SNR of eMS2PM traces is smaller by a factor 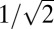 than the SNR of RAMP traces acquired with the same time resolution. However, in eMS2PM, one ROI can be probed with multiple excitation foci simultaneously (Figs. 3 and 4 A and C). In this case then, provided the laser power is sufficient (which is reasonable with average laser output powers of ∼3 W and recording up to ∼10 cells at depth <200 µm), the SNR of the decoded signal is multiplied by the square root of the number of foci. Also, targeting cells with multiple lower-intensity foci instead of single high-intensity spots as in RAMP could significantly reduce photobleaching rates [proportional to the third or fourth power of the intensity (33, 34)] without reducing SNR (SI Text 2). Under such conditions, we believe that eMS2PM could be more efficient than RAMP in following voltage transients in neuronal networks in vivo. On the other hand, our current eMS2PM is limited to simultaneous acquisition in the XY plane, whereas RAMP has recently been adapted for rapid 3D measurements over cubic-millimeter volumes (32).
than the SNR of RAMP traces acquired with the same time resolution. However, in eMS2PM, one ROI can be probed with multiple excitation foci simultaneously (Figs. 3 and 4 A and C). In this case then, provided the laser power is sufficient (which is reasonable with average laser output powers of ∼3 W and recording up to ∼10 cells at depth <200 µm), the SNR of the decoded signal is multiplied by the square root of the number of foci. Also, targeting cells with multiple lower-intensity foci instead of single high-intensity spots as in RAMP could significantly reduce photobleaching rates [proportional to the third or fourth power of the intensity (33, 34)] without reducing SNR (SI Text 2). Under such conditions, we believe that eMS2PM could be more efficient than RAMP in following voltage transients in neuronal networks in vivo. On the other hand, our current eMS2PM is limited to simultaneous acquisition in the XY plane, whereas RAMP has recently been adapted for rapid 3D measurements over cubic-millimeter volumes (32).
What about possible phototoxic effects of eMS2PM? Targeting sites of interest has the advantage over wide-field two-photon imaging (35) in avoiding phototoxic effects outside targeted sites. However, we were concerned that eMS2PM could irreversibly damage cells because fluorescence signal is integrated from parked diffraction-limited foci. We could record calcium responses to repeated electrical stimulations of cortical neurons (Fig. 4B) without any reduction in the ΔF/F amplitude for 4 min, indicating that targeted cells remained viable. That said, the absence of phototoxicity of eMS2PM should be confirmed in other tissue preparations and recording conditions (depth, laser power, and temporal resolution).
Can eMS2PM be further improved? In terms of imaging depth we could record uncorrelated calcium signals in multiple cells down to 220 µm in cortical slices (Fig. 4C) and up to 300 µm in in vivo neocortex of an adult mouse (Fig. 5B). RBC shadows in plasma-labeled capillaries could be imaged up to 350 µm (Fig. 5C). Currently deeper observations with eMS2PM are difficult because of limited laser power at the sample (∼500 mW total, i.e., ∼100 mW per site for five sites). Using DMD with greater diffraction efficiency should raise power at the sample to ∼1 W. Higher-power lasers are available but care will have to be taken to stay below the damage threshold of the DMD matrix, which is currently not specified for high-intensity femtosecond pulses. In terms of time resolution, with the current DMD, we could acquire up to 15 fluorescent ROIs with a submillisecond resolution (Fig. S3). Because temporal resolution is the product of the number of bins in the S code by the DMD frame duration, a faster DMD (although not currently existing on the market) would permit higher temporal resolution or recording from more cells with submillisecond time resolution. A new DMD with fewer pixels, or arbitrarily updating subgroups of pixels at high rates, could possibly achieve such a task. Both are conceivably features that could appear in the near future. Finally, we used only single- or multiple-point targets for excitation. Alternatively, arbitrary shapes could be formed by the SLM (16, 18). This is of particular interest to target membranes of cells loaded with voltage-sensitive dyes or photoactivatable channels. To create arbitrary shapes with micrometric axial resolution and homogeneous intensity profiles, temporal focusing and generalized phase-contrast methods (36) could be combined into our eMS2PM setup. In the future the SLM can also be used to correct for optical aberration and intensity loss in scattering brain tissue, thus improving the spatial resolution and SNR in depth (37–39). The particular combination SLM and DMD developed for the eMS2PM setup offers the unique possibility to shape arbitrary illumination patterns with micrometric precision and to apply them in any order with submillisecond temporal precision. Thus, the eMS2PM setup could also be used to photostimulate targeted sites with subcellular spatial resolution and with any predefined millisecond-range temporal patterns. Furthermore, this could be achieved in conjunction with neuronal activity recording.
Methods
Encoding/Decoding Principle.
The principle of multiplexing/demultiplexing with orthogonal functions has been used in many fields such as telecommunications and spectroscopy (40). Initially we considered using frequency multiplexing, an approach that was successfully used in biomedical imaging to encode magnetic resonance images and more recently applied to fluorescence (41) and phosphorescence lifetime (42) imaging with a single detector. With this type of encoding, demultiplexing is easily achieved by analyzing the detector signal in the frequency domain (e.g., with Fourier transform) and extracting the signal at the corresponding frequencies. This method fails when applied to monitoring neuronal activity, however, which requires a temporal resolution <1 ms and excitation subbeams being modulated at frequencies >10 kHz. To our knowledge, liquid crystal SLMs cannot be updated at this rate. In addition, if DMD frames can be updated up to 20 kHz, all pixels are actuated synchronously and it is not possible to address subgroups of pixels at different frequencies in the range 10–20 kHz. Therefore, instead of frequency multiplexing we considered an encoding scheme that used short uncorrelated binary sequences. S codes were chosen as they have the advantage of being unipolar (0 and 1) rather than bipolar (−1, +1) Walsh functions or Golay sequences. S-codes are the rows of S matrices, which are derived from Hadamard matrices (43) (SI Text 1). S matrices are symmetrical square matrices of dimension N with an associated decoding matrix S* composed of “+1” and “−1” such that
 |
For example,
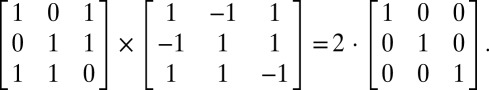 |
Eq. 1 can be also written
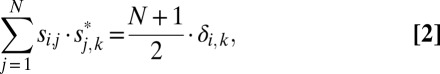 |
where δi,k = 1 for I = k and δi,k = 0 for i ≠ k.
We used S matrices with dimensions n = 3, 7, 11, 15 (higher dimensions exist but were not used in the present work). The codes used in eMS2PM for multiplexing and demultiplexing were the rows of S and S* matrices, respectively. “+1” and “0” corresponded to DMD pixels ON and OFF, respectively. If the signals emitted by M ROIs were a1, a2, … , aM, then the signal detected at the detector during one S code was a time sequence D:{d1, d2, …, dN},
 |
for j = 1…N (j being the time index). Here we assume that ROI signals ai do not vary over the duration of an S code.
The dot product of sequence D by the kth rows of S* yielded a value proportional to ak (Eqs. 2 and 3):
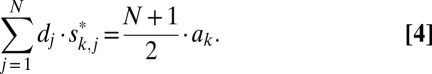 |
Thus, individual ai signals from each ROI were computed by simple multiplication of the detected sequence (D) by a decoding sequence that was a row of the S* matrix. The derivation of S and S* matrices from Hadamard matrices is explained in SI Text 1. The principle of coding with S codes is illustrated in Fig. S1. The S matrices that were used for up to 15 sites are presented in Table S1. S codes were played repeatedly by the DMD for the entire duration of data acquisition and synchronized with detector signal acquisition.
2PM and eMS2PM Setups.
For 2PM imaging, the Ti:Sapphire femtosecond laser (Tsunami; Spectra Physics) emitted ∼300 mW at 925 nm for in vitro experiments (Figs. 2–4) and 600 mW at 860 nm for in vivo experiments (Fig. 5). Laser power was attenuated by an electro-optics Pockels cell (350-80C; Conoptics) driven by a voltage amplifier (PZD700; Treck), itself computer controlled via a D/A card (USB6008; National Instruments). For reference image acquisition galvanometric scanner (GS) mirrors (VM500; GSI Lumonics) were raster-scanned over a 100-μm × 100-μm field of view with a pixel size 0.2 μm × 0.2 μm or smaller. Targeted ROIs were drawn in this reference frame. Fluorescence light collected by the objective was sent to a pair of lenses (Fig. 1, L4 and L5), coupled into a 2-mm diameter core polymethyl methacrylate optical fiber (not drawn in Fig. 1), and projected onto a PMT (R6357-SELECT; Hamamatsu) as previously described in ref. 44. A near infrared blocking filter (E700 sp 2ph; Chroma Technologies) suppressed scattered laser light. For eMS2PM recording, the second femtosecond laser (Mai Tai eHP; SpectraPhysics) with a dispersion compensation module (Deepsee; Spectraphysics) emitted ∼70-fs pulses at 80 MHz with an average power of 2.6 W. Laser wavelength, shutter, and dispersion compensation were controlled via RS232 interface by LabVIEW software integrated into the main setup user interface program. In vitro and in vivo experiments were performed at 860 nm and 905 nm, respectively. These wavelengths were close to the peak of two-photon excitation spectra of the fluorophores, within the SLM operation range (800–910 nm), and minimized reflection/transmission losses through the setup. Power sent to the SLM was controlled by a liquid crystal attenuator (Newport) and a computer-operated AD card (USB6008; National Instruments). The beam was enlarged to ∼10 mm diameter by a 5× beam expander (BE05M-B; Thorlabs) to fill the SLM aperture. The phase profile sent to the reflective SLM (LCOS-SLM X10468-02; Hamamatsu) was updated by the video card of the computer, using software written in LabVIEW. The DMD (0.7-in XGA DMD controlled by the DLP Discovery 4100 fast chipset; Texas Instruments) was controlled by a custom-made LabVIEW program, using dll routines (ALP-4; VIALUX). Frame rates up to 22 kHz can be reached, but we did not operate the DMD above 16 kHz to maintain sufficient average reflectivity (an ∼10-μs settling period occurs at each frame update, during which light is not fully reflected into the setup). The eMS2PM illumination patterns were coupled into the conventional 2PM with a dichroic mirror (Fig. 1, DM1): long-pass 900DCXXR (Chroma Technologies) for in vitro experiments (Figs. 2–4) and a short-pass T883SPXT (Chroma Technologies) for in vivo work (Fig. 5). The sizes of the two-photon excitation spots created by the eMS2PM were 0.73 µm laterally and 1.43 µm axially (FWHM) as measured on a thin (∼1 µm) sheet of rhodamine B in plastic.
Setup Light Transmission.
See SI Methods.
SLM and DMD Damage Threshold.
See SI Methods.
2PM and eMS2PM Optical Setups Coalignment.
See SI Methods.
Aberration Correction for Fluorescence Intensity Optimization.
See SI Methods.
Data Processing.
See SI Methods.
In Vivo Experimental Procedures.
See SI Methods.
Supplementary Material
Acknowledgments
The authors thank Karine Hérault for help with HEK cell cultures, Isabelle Arnoux who prepared acute mouse cortical slices, Andréa Virolle for help in animal surgeries, Brandon Stell for advice with DiO/DPA labeling methods, Eirini Papagiakoumou for guidance with the SLM, Cathie Ventalon for technical and scientific discussions, and Laurent Moreaux and Valentina Emiliani for discussions at the beginning of the project. Support was provided by the Institut National de la Santé et de la Recherche Médicale, the Leducq Foundation, the Human Frontier Science Program Organization, and the Fondation pour la Recherche Médicale. The team of Serge Charpak is part of the École des Neurosciences de Paris Ile-de-France network.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1307818110/-/DCSupplemental.
References
- 1.Piston DW, Kirby MS, Cheng H, Lederer WJ, Webb WW. Two-photon-excitation fluorescence imaging of three-dimensional calcium-ion activity. Appl Opt. 1994;33(4):662–669. doi: 10.1364/AO.33.000662. [DOI] [PubMed] [Google Scholar]
- 2.Yuste R, Denk W. Dendritic spines as basic functional units of neuronal integration. Nature. 1995;375(6533):682–684. doi: 10.1038/375682a0. [DOI] [PubMed] [Google Scholar]
- 3.Helmchen F, Svoboda K, Denk W, Tank DW. In vivo dendritic calcium dynamics in deep-layer cortical pyramidal neurons. Nat Neurosci. 1999;2(11):989–996. doi: 10.1038/14788. [DOI] [PubMed] [Google Scholar]
- 4.Theer P, Hasan MT, Denk W. Two-photon imaging to a depth of 1000 microm in living brains by use of a Ti:Al2O3 regenerative amplifier. Opt Lett. 2003;28(12):1022–1024. doi: 10.1364/ol.28.001022. [DOI] [PubMed] [Google Scholar]
- 5.Lillis KP, Eng A, White JA, Mertz J. Two-photon imaging of spatially extended neuronal network dynamics with high temporal resolution. J Neurosci Methods. 2008;172(2):178–184. doi: 10.1016/j.jneumeth.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grewe BF, Langer D, Kasper H, Kampa BM, Helmchen F. High-speed in vivo calcium imaging reveals neuronal network activity with near-millisecond precision. Nat Methods. 2010;7(5):399–405. doi: 10.1038/nmeth.1453. [DOI] [PubMed] [Google Scholar]
- 7.Otsu Y, et al. Optical monitoring of neuronal activity at high frame rate with a digital random-access multiphoton (RAMP) microscope. J Neurosci Methods. 2008;173(2):259–270. doi: 10.1016/j.jneumeth.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 8.Reddy GD, Saggau P. Fast three-dimensional laser scanning scheme using acousto-optic deflectors. J Biomed Opt. 2005;10(6):064038. doi: 10.1117/1.2141504. [DOI] [PubMed] [Google Scholar]
- 9.Sacconi L, et al. Optical recording of electrical activity in intact neuronal networks with random access second-harmonic generation microscopy. Opt Express. 2008;16(19):14910–14921. [PubMed] [Google Scholar]
- 10.König K, So PT, Mantulin WW, Gratton E. Cellular response to near-infrared femtosecond laser pulses in two-photon microscopes. Opt Lett. 1997;22(2):135–136. doi: 10.1364/ol.22.000135. [DOI] [PubMed] [Google Scholar]
- 11.Hopt A, Neher E. Highly nonlinear photodamage in two-photon fluorescence microscopy. Biophys J. 2001;80(4):2029–2036. doi: 10.1016/S0006-3495(01)76173-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holthoff K, Zecevic D, Konnerth A. Rapid time course of action potentials in spines and remote dendrites of mouse visual cortex neurons. J Physiol. 2010;588(Pt 7):1085–1096. doi: 10.1113/jphysiol.2009.184960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Acker CD, Yan P, Loew LM. Single-voxel recording of voltage transients in dendritic spines. Biophys J. 2011;101(2):L11–L13. doi: 10.1016/j.bpj.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Niesner R, Andresen V, Neumann J, Spiecker H, Gunzer M. The power of single and multibeam two-photon microscopy for high-resolution and high-speed deep tissue and intravital imaging. Biophys J. 2007;93(7):2519–2529. doi: 10.1529/biophysj.106.102459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nikolenko V, et al. SLM microscopy: Scanless two-photon imaging and photostimulation with spatial light modulators. Front Neural Circuits. 2008;2:5. doi: 10.3389/neuro.04.005.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Papagiakoumou E, de Sars V, Oron D, Emiliani V. Patterned two-photon illumination by spatiotemporal shaping of ultrashort pulses. Opt Express. 2008;16(26):22039–22047. doi: 10.1364/oe.16.022039. [DOI] [PubMed] [Google Scholar]
- 17.Kurtz R, Fricke M, Kalb J, Tinnefeld P, Sauer M. Application of multiline two-photon microscopy to functional in vivo imaging. J Neurosci Methods. 2006;151(2):276–286. doi: 10.1016/j.jneumeth.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Lutz C, et al. Holographic photolysis of caged neurotransmitters. Nat Methods. 2008;5(9):821–827. doi: 10.1038/nmeth.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheong W, Prahl S, Welch AJ. A review of the optical properties of biological tissues. IEEE J Quantum Electron. 1990;26(12):2166–2185. [Google Scholar]
- 20.Oheim M, Beaurepaire E, Chaigneau E, Mertz J, Charpak S. Two-photon microscopy in brain tissue: Parameters influencing the imaging depth. J Neurosci Methods. 2001;111(1):29–37. doi: 10.1016/s0165-0270(01)00438-1. [DOI] [PubMed] [Google Scholar]
- 21.Bradley J, Luo R, Otis TS, DiGregorio DA. Submillisecond optical reporting of membrane potential in situ using a neuronal tracer dye. J Neurosci. 2009;29(29):9197–9209. doi: 10.1523/JNEUROSCI.1240-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garaschuk O, Linn J, Eilers J, Konnerth A. Large-scale oscillatory calcium waves in the immature cortex. Nat Neurosci. 2000;3(5):452–459. doi: 10.1038/74823. [DOI] [PubMed] [Google Scholar]
- 23.Nagayama S, et al. In vivo simultaneous tracing and Ca(2+) imaging of local neuronal circuits. Neuron. 2007;53(6):789–803. doi: 10.1016/j.neuron.2007.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kleinfeld D, Mitra PP, Helmchen F, Denk W. Fluctuations and stimulus-induced changes in blood flow observed in individual capillaries in layers 2 through 4 of rat neocortex. Proc Natl Acad Sci USA. 1998;95(26):15741–15746. doi: 10.1073/pnas.95.26.15741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bahlmann K, et al. Multifocal multiphoton microscopy (MMM) at a frame rate beyond 600 Hz. Opt Express. 2007;15(17):10991–10998. doi: 10.1364/oe.15.010991. [DOI] [PubMed] [Google Scholar]
- 26.Yew EYS, Choi HJ, Kim D, So PTC. 2011. Wide-field two-photon microscopy with temporal focusing and HiLo background rejection. Proc SPIE 790310.
- 27.Therrien OD, Aubé B, Pagès S, Koninck PD, Côté D. Wide-field multiphoton imaging of cellular dynamics in thick tissue by temporal focusing and patterned illumination. Biomed Opt Express. 2011;2(3):696–704. doi: 10.1364/BOE.2.000696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Truong TV, Supatto W, Koos DS, Choi JM, Fraser SE. Deep and fast live imaging with two-photon scanned light-sheet microscopy. Nat Methods. 2011;8(9):757–760. doi: 10.1038/nmeth.1652. [DOI] [PubMed] [Google Scholar]
- 29.Zanacchi FC, et al. 2011. Two-photon fluorescence excitation within a light sheet based microscopy architecture. Proc SPIE 79032W.
- 30.Cheng A, Gonçalves JT, Golshani P, Arisaka K, Portera-Cailliau C. Simultaneous two-photon calcium imaging at different depths with spatiotemporal multiplexing. Nat Methods. 2011;8(2):139–142. doi: 10.1038/nmeth.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim KH, et al. Multifocal multiphoton microscopy based on multianode photomultiplier tubes. Opt Express. 2007;15(18):11658–11678. doi: 10.1364/oe.15.011658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katona G, et al. Fast two-photon in vivo imaging with three-dimensional random-access scanning in large tissue volumes. Nat Methods. 2012;9(2):201–208. doi: 10.1038/nmeth.1851. [DOI] [PubMed] [Google Scholar]
- 33.Patterson GH, Piston DW. Photobleaching in two-photon excitation microscopy. Biophys J. 2000;78(4):2159–2162. doi: 10.1016/S0006-3495(00)76762-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen TS, Zeng SQ, Luo QM, Zhang ZH, Zhou W. High-order photobleaching of green fluorescent protein inside live cells in two-photon excitation microscopy. Biochem Biophys Res Commun. 2002;291(5):1272–1275. doi: 10.1006/bbrc.2002.6587. [DOI] [PubMed] [Google Scholar]
- 35.Choi H, et al. 3D-resolved fluorescence and phosphorescence lifetime imaging using temporal focusing wide-field two-photon excitation. Opt Express. 2012;20(24):26219–26235. doi: 10.1364/OE.20.026219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Papagiakoumou E, et al. Scanless two-photon excitation of channelrhodopsin-2. Nat Methods. 2010;7(10):848–854. doi: 10.1038/nmeth.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ji N, Milkie DE, Betzig E. Adaptive optics via pupil segmentation for high-resolution imaging in biological tissues. Nat Methods. 2010;7(2):141–147. doi: 10.1038/nmeth.1411. [DOI] [PubMed] [Google Scholar]
- 38.Débarre D, et al. Image-based adaptive optics for two-photon microscopy. Opt Lett. 2009;34(16):2495–2497. doi: 10.1364/ol.34.002495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poland S, Fruhwirth G, Ng T, Ameer-beg S. 2011. Using adaptive optics for deep in vivo multiphoton FLIM. Proc SPIE 79032C.
- 40.Golay MJE. Multi-slit spectrometry. J Opt Soc Am. 1949;39(6):437–444. doi: 10.1364/josa.39.000437. [DOI] [PubMed] [Google Scholar]
- 41.Futia G, Schlup P, Winters DG, Bartels RA. Spatially-chirped modulation imaging of absorbtion and fluorescent objects on single-element optical detector. Opt Express. 2011;19(2):1626–1640. doi: 10.1364/OE.19.001626. [DOI] [PubMed] [Google Scholar]
- 42.Howard SS, Straub A, Horton N, Kobat D, Xu C. Frequency multiplexed in vivo multiphoton phosphorescence lifetime microscopy. Nat Photonics. 2013;7(1):33–37. doi: 10.1038/nphoton.2012.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harwit M, Sloane NJA. Hadamard Transform Optics. New York: Academic; 1979. [Google Scholar]
- 44.Ducros M, et al. Efficient large core fiber-based detection for multi-channel two-photon fluorescence microscopy and spectral unmixing. J Neurosci Methods. 2011;198(2):172–180. doi: 10.1016/j.jneumeth.2011.03.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.