

Significance
Mitochondria, the powerhouses of the cell, constantly change their shape by fusing and dividing. How these two opposite processes are controlled remains unclear. In our study, we identified the Caenorhabditis elegans homolog of the human mitochondrial protein LRPPRC (leucine-rich pentatricopeptide repeat containing), which has been previously associated with the neurodegenerative French Canadian Leigh Syndrome. Analysis of this protein revealed an evolutionary conserved pathway that regulates mitochondrial shape. Specifically, we show that mitochondria transiently form a highly connected network to compensate for a decrease of the activity of the complex IV of the electron transport chain.
Keywords: mitochondrial dynamics, cytochrome c oxidase deficiency neurodegeneration
Abstract
Mitochondrial morphology changes in response to various stimuli but the significance of this is unclear. In a screen for mutants with abnormal mitochondrial morphology, we identified MMA-1, the Caenorhabditis elegans homolog of the French Canadian Leigh Syndrome protein LRPPRC (leucine-rich pentatricopeptide repeat containing). We demonstrate that reducing mma-1 or LRPPRC function causes mitochondrial hyperfusion. Reducing mma-1/LRPPRC function also decreases the activity of complex IV of the electron transport chain, however without affecting cellular ATP levels. Preventing mitochondrial hyperfusion in mma-1 animals causes larval arrest and embryonic lethality. Furthermore, prolonged LRPPRC knock-down in mammalian cells leads to mitochondrial fragmentation and decreased levels of ATP. These findings indicate that in a mma-1/LRPPRC–deficient background, hyperfusion allows mitochondria to maintain their functions despite a reduction in complex IV activity. Our data reveal an evolutionary conserved mechanism that is triggered by reduced complex IV function and that induces mitochondrial hyperfusion to transiently compensate for a drop in the activity of the electron transport chain.
Mitochondria are dynamic organelles that frequently fuse and divide, and their steady-state morphology is determined by the relative rates of fusion and fission (1–5). Mitochondrial fusion and fission are under the control of a conserved family of GTPases, the dynamin-related proteins (DRPs). For example, mammalian Drp1 or Caenorhabditis elegans DRP-1 are required for mitochondrial fission (6, 7). Conversely, mammalian Mfn1/2 and Opa1 or C. elegans FZO-1 and EAT-3 are required for the fusion of the outer and inner mitochondrial membrane, respectively (8–13).
Mitochondrial morphology can change in response to various cellular stimuli, including metabolic signals. Electron microscopic studies revealed that the ultrastructure of mitochondria changes depending on their rate of oxidative phosphorylation. For example, inner mitochondrial membrane cristae change from an “orthodox” to a “condensed” conformation in response to increased oxidative phosphorylation [i.e., increased activity of the electron transport chain (ETC)] (14). Furthermore, there is evidence for a bidirectional, functional link between the activity of the ETC and mitochondrial dynamics; that is, the ability of mitochondria to fuse and divide and, hence, change their steady-state morphology. On the one hand, mutations that reduce the activities of specific complexes of the ETC affect mitochondrial morphology and cause mitochondrial fragmentation. For example, fibroblasts from patients with defects in complexes I, III, or IV have fragmented mitochondria (15–17). Similarly, pharmacological inhibition of any of the ETC complexes in primary human fibroblasts and rat cortical neurons leads to mitochondrial fragmentation (18, 19). On the other hand, mutations that impair mitochondrial dynamics lead to defects in oxidative phosphorylation. For example, inactivation in HeLa cells of Drp1, which is required for mitochondrial fission, causes a decrease in mitochondrial membrane potential, respiration, and cellular ATP (20). Similarly, primary skin cells derived from patients with Optic Dominant Atrophy, who carry mutations in the mitochondrial fusion protein Opa1, exhibit reduced levels of complex IV subunits, resulting in decreased complex IV activity (21). Furthermore, the inactivation of the mitochondrial fusion protein Mfn2 in muscle cells grown in culture causes a reduction in the levels of subunits of several ETC complexes, as well as a decrease in mitochondrial membrane potential and respiration (22).
However, rather than fragmenting, mitochondria have recently also been shown to undergo a process referred to as “hyperfusion.” Specifically, in response to different forms of stress, such as the inhibition of cytosolic protein synthesis or starvation, mitochondria in cultured mammalian cells form large, hyperfused mitochondria (23–26). This hyperfusion appears to be mediated through the activation of the mitochondrial fusion proteins Mfn and Opa1, or the inactivation of the mitochondrial fission protein Drp1. In the case of mitochondrial hyperfusion in response to the inhibition of cytosolic protein synthesis, cells with hyperfused mitochondria have higher levels of cellular ATP than control cells (23). In the case of mitochondrial hyperfusion in response to starvation, cells with hyperfused mitochondria maintain normal levels of cellular ATP despite starvation conditions (24). Therefore, hyperfused mitochondria are likely to produce ATP more efficiently. It has been proposed that mitochondrial hyperfusion is a prosurvival response to various forms of stress because cells defective in mitochondrial hyperfusion are more sensitive to stress and undergo apoptosis (23, 24).
The mammalian LRPPRC gene is mutated in patients that suffer from French Canadian Leigh Syndrome, a neurodegenerative disease associated with complex IV deficiency (27). LRPPRC encodes a leucine-rich pentatricopeptide repeat containing protein that is targeted to the mitochondrial matrix (28). In the mitochondrial matrix, LRPPRC forms a ribonucleoprotein complex with the stem-loop RNA binding protein SLIRP and mitochondrial mRNAs (17, 29). The LRPPRC/SLIRP complex activates the mitochondrial poly(A) polymerase MTPAP, and thereby promotes the polyadenylation of mitochondrial mRNAs (29, 30). LRPPRC/SLIRP/MTPAP-dependent polyadenylation stabilizes mitochondrial mRNAs, such as COXI and COXII, which encode two subunits of complex IV (29, 30). In addition, LRPPRC plays an important role in the control of mitochondrial translation because its inactivation leads to abnormal patterns of mitochondrial translation, specifically resulting in complex IV deficiency (29).
Whether a homolog of mammalian LRPPRC exists in C. elegans has been unclear (28). We now report that C. elegans mma-1 encodes a structural and functional homolog of LRPPRC. Furthermore, we show that reducing the function of mma-1 in C. elegans or LRPPRC in mammalian cells causes a reduction in the level of complex IV of the ETC but not in the level of cellular ATP. In addition, we provide evidence that essential mitochondrial functions are maintained through mitochondrial hyperfusion, which is triggered by reducing complex IV activity. Our results reveal an evolutionary conserved compensatory mechanism that is induced by a deficiency in a specific complex of the ETC, complex IV, and whose role is to trigger mitochondrial hyperfusion to maintain mitochondrial function. Finally, we show that this compensatory mechanism is effective only transiently and that prolonged reduction of LRPPRC/mma-1 function causes a collapse of cellular functions.
Results
mma-1 Inactivation Causes Mitochondrial Hyperfusion.
To identify regulators of mitochondrial morphology in C. elegans, we performed a RNA-interference–mediated (RNAi) screen. To that end, we used a strain carrying a stable, integrated transgene expressing a mitochondrial matrix-targeted GFP (mitoGFP) in body wall muscle cells (Pmyo-3:mitoGFP). C14C10.4, one of the candidates identified in the screen, leads to an abnormal mitochondrial morphology phenotype when inactivated. We therefore named this gene “mma-1” (mitochondrial morphology abnormal-1). As shown in Fig. 1, in animals treated with mock double-strand RNA (dsRNA), mitochondria in body-wall muscle cells have a tubular morphology (96%) (Fig. 1A). In contrast, in mma-1(RNAi) animals, mitochondria in body-wall muscle cells have a hyperfused morphology (68%,) (Fig. 1A and Movie S1). [This phenotype was observed using three independent mma-1 RNAi clones (Fig. S1B)]. mma-1(RNAi) animals do not exhibit any other obvious defects and have normal broodsize and locomotion (Fig. S2).
Fig. 1.

mma-1(RNAi) causes mitochondrial hyperfusion. (A) L4 larvae carrying the transgene Pmyo-3:mitoGFP were treated with mock or mma-1 RNAi and mitochondrial morphology in body-wall muscle cells of L4 larvae of the F1 generation was analyzed by fluorescent microscopy. MitoGFP image and schematic of the representative mitochondrial morphology observed as well as a quantification are indicated (n = number of animals analyzed). (B) L4 larvae carrying the transgene Pmyo-3:PAmitogfp + Pmyo-3:mitodsred were treated with mock, drp-1, fzo-1, or mma-1 RNAi. PAmitoGFP was activated in body wall muscle of L4 larvae of the F1 generation by a 405-nm laser (white circle) and GFP signal was monitored during 25s. PAmitoGFP signals are shown in pseudocolor, where nonphoto-activated PAmitoGFP is in purple and photo-activated PAmitoGFP is in red. Intensity scale is indicated. For mma-1(RNAi), an image with a different intensity scale is also indicated. A quantification of the spread of the PAmitoGFP signal is also indicated. (n = 5–10 regions-of-interest in two to three animals; SD is indicated.) (C) N2 L4 larvae were grown on mock(RNAi) or mma-1(RNAi) plates for 24 h and transferred to TMRE plates for 15 h. Mitochondrial morphology in embryos was analyzed by Nomarski and fluorescent microscopy.
mma-1 is part of an operon (CEOP5360) composed of mma-1 and two other genes, C14C10.2 and C14C10.3 (Fig. S1A). All three genes are predicted to encode mitochondrial matrix-targeted proteins (99.4%, 88.9%, and 76.3% probability of mitochondrial targeting for MMA-1, C14C10.2, and C14C10.3, respectively) (31). Whereas the inactivation of C14C10.2 by RNAi does not cause an obvious mitochondrial morphology phenotype, the inactivation of C14C10.3 by RNAi causes a mitochondrial morphology phenotype similar to the phenotype observed upon mma-1 inactivation: that is, mitochondrial hyperfusion (77%) (Fig. S1B). Nuclear-localized polycistronic RNA can be silenced by RNAi. Therefore, RNAi treatment against one gene of an operon can cause the silencing of the entire polycistronic RNA. This silencing requires the Argonaute protein NRDE-3, which is necessary to transport siRNA from the cytoplasm to the nucleus (32). To determine whether the phenotype observed upon C14C10.3 RNAi treatment or mma-1 RNAi treatment is specific to C14C10.3 or mma-1, respectively, and not a result of inactivation of the entire operon, we repeated the experiment in animals carrying a nrde-3 loss-of-function mutation [nrde-3(gg66)]. Whereas the C14C10.3(RNAi)-induced mitochondrial hyperfusion phenotype was suppressed by nrde-3(gg66), the mma-1(RNAi)-induced mitochondrial hyperfusion phenotype was not [nrde-3(gg66); C14C10.3(RNAi): 22% and nrde-3(gg66);mma-1(RNAi): 88%] (Fig. S1B). This result demonstrates that it is the inactivation of mma-1 and not C14C10.3 that causes hyperfusion of the mitochondrial network.
To determine whether the mitochondrial matrix of animals treated with mma-1 dsRNA is interconnected, we used a transgene expressing mitochondrial matrix-targeted photoactivatable GFP (PAmitoGFP) in body-wall muscle cells (Pmyo-3:PAmitoGFP) (33). Multiple regions of interest were activated in transgenic animals using a 405-nm laser and the dispersal of PAmitoGFP fluorescence was monitored over time. In animals treated with mock dsRNA, the mitochondrial network shows a limited dispersal of the PAmitoGFP fluorescence, indicating a low level of interconnectivity (Fig. 1B). For comparison and consistent with previous studies, a higher level of interconnectivity is observed in animals treated with drp-1 dsRNA, whereas a lower level of interconnectivity is observed in animals treated with fzo-1 dsRNA (7, 10–13) (Fig. 1B). In animals treated with mma-1 dsRNA, we observed a collapse of the mitochondrial network into large, hyperfused organelles with a continuous matrix (Fig. 1B). Based on these results, we conclude that the inactivation of mma-1 causes complete hyperfusion of the outer and inner mitochondrial membrane, generating large, highly interconnected organelles.
Finally, to determine whether mma-1(RNAi)-induced mitochondrial hyperfusion is restricted to body-wall muscle cells, we tested the effect of mma-1(RNAi) on mitochondrial morphology in embryos, in which mitochondria were stained with TMRE (tetramethylrhodamine ester). In wild-type embryos at the two-cell stage of embryonic development, mitochondria have a tubular morphology (100%, n = 10) (Fig. 1C). In contrast, in mma-1(RNAi) embryos, mitochondria have a “ring”-like hyperfused morphology (100%, n = 10) (Fig. 1C). Therefore, the mitochondrial morphology phenotype caused by mma-1 inactivation is not restricted to body-wall muscle cells but can also be observed in other cell types, such as undifferentiated, embryonic cells.
C. elegans C14C10.4/MMA-1 Is the Structural and Functional Homolog of Mammalian LRPPRC.
In its N-terminal region, MMA-1 shares 46% similarities with the mammalian protein LRPPRC. Like LRPPRC, MMA-1 contains a mitochondrial targeting sequence (MTS) and pentatricopeptide repeats (PPRs). In addition, the two proteins share 43% similarity in their C-terminal region (Fig. 2A). In MMA-1, this region shows similarity to the bacterial EzrA protein, which regulates bacterial division by controlling the bacterial tubulin-like protein FtzZ (34). Using polyclonal anti–MMA-1 peptide antibodies that specifically recognize the MMA-1 protein (Fig. S3), we found that MMA-1 colocalizes with the mitochondrial ATP synthase in early embryos (Fig. 2B). In addition, we performed cell-fractionation experiments and found that MMA-1 is predominantly present in the mitochondria-enriched fraction (Fig. 2C).
Fig. 2.
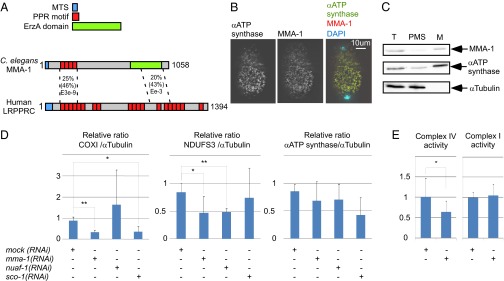
MMA-1 is the structural and functional homolog of mammalian LRPPRC. (A) Schematic of MMA-1 protein and its mammalian homolog LRPPRC with their different domains (MTS, PPR, EzrA domain). (B) Immunostaining of an early fertilized N2 egg with anti-ATP synthase and anti-MMA-1 antibodies. (C) Western analysis of different cell fractions (T, total lysate; PMS, postmitochondrial supernatant; M, mitochondria-enriched fraction). Equal volumes of each fraction were loaded on the gel. (D) Relative amount of the complex IV protein COXI, the complex I protein NDUFS3, and the α-subunit of the ATP synthase upon mock, mma-1, nuaf-1, or sco-1 RNAi (n = 12, three independent experiments). (E) Complex IV and complex I activities upon mock or mma-1 RNAi (n > 10). In D and E, SD are indicated. (*P < 0.05 and **P < 0.01, by Student’s t test.)
LRPPRC has been shown to control the stability and translation efficiency of mRNAs generated from several mitochondrial genes (17, 29). As shown in Fig. 2D, we found that mma-1(RNAi) significantly decreases the level of the mitochondria-encoded protein COXI (40% of control), a subunit of complex IV of the ETC. We also found that the level of NDUFS3, a subunit of complex I, was significantly decreased (50% of control). In contrast, the level of the α-subunit of the ATP synthase (complex V) was not affected. Because NDUFS3 is a nuclear-encoded protein, the effect of mma-1(RNAi) on NDUFS3 must be indirect. mma-1(RNAi) most likely induces a decrease in the level of mitochondria-encoded subunits of complex I, thereby leading to the disassembly of complex I and the degradation of its subunits, including NDUFS3. To determine whether mma-1(RNAi) also causes a reduction of complex I and IV activities, we measured these activities in a tissue-specific manner by enzymatic histochemistry (35). As shown in Fig. 2E, we detected a decrease of complex IV activity in body-wall muscle in response to mma-1(RNAi) (60% of control). In contrast, the activity of complex I was unaffected by mma-1(RNAi). Therefore, like mammalian LRPPRC, MMA-1 localizes to mitochondria and is required for complex IV activity. Together with the sequence homologies between MMA-1 and LRPPRC, these functional data support the notion that MMA-1 is the C. elegans homolog of mammalian LRPPRC.
Reducing Complex IV but Not Complex I Assembly Induces Mitochondrial Hyperfusion.
Because LRPPRC has been implicated in the coordination of mitochondrial translation (29), we tested whether blocking mitochondrial translation in C. elegans also affects mitochondrial morphology. As shown in Fig. 3, inactivation of the C. elegans homolog of mitochondrial elongation factor G1 (mefg-1) leads to a mitochondrial hyperfusion phenotype (Fig. 3). In contrast, we could not observe a mitochondrial morphology phenotype upon the inactivation by RNAi of genes encoding the mitochondrial RNA polymerase (mrpo-1) or the mitochondrial transcription factors (hmg-5 or mtfb-1) (Fig. S4).
Fig. 3.

Specific reduction of complex IV assembly leads to mitochondrial hyperfusion. L4 larvae carrying the Pmyo-3:mitoGFP transgene were treated with mock, mma-1, mefg-1, nuaf-1, or sco-1 RNAi and mitochondrial morphology in body-wall muscle cells of L4 larvae of the F1 generation was analyzed by fluorescent microscopy. (A) Schematic of the different mitochondrial morphologies observed. (B) MitoGFP image and schematic of the representative mitochondrial morphology observed as well as quantification are indicated. (n = number of animals analyzed.)
The inactivation of mma-1 leads to a decrease in both the levels of complex I as well as complex IV subunits. To determine whether complex I or complex IV destabilization is responsible for the induction of mitochondrial hyperfusion, we destabilized these two protein complexes individually by inactivating complex I- or complex IV-specific scaffold proteins. Inactivation of sco-1, which encodes the C. elegans ortholog of the mammalian complex IV scaffold protein SCO1, does not affect the level of NDUFS3, a component of complex I, but causes a specific reduction in the level of COXI protein (40% of control) (Fig. 2D). Inactivation of sco-1 causes a mitochondrial hyperfusion phenotype similar to the phenotype observed upon mma-1 inactivation (54%) (Fig. 3). Similarly, the inactivation of another complex IV scaffold protein, coaf-15, an ortholog of mammalian COX15, also causes a mitochondrial hyperfusion phenotype, although mitochondria do not appear as connected as upon mma-1(RNAi) or sco-1(RNAi) (72%) (Fig. S4). In contrast, the inactivation of nuaf-1, which encodes the C. elegans ortholog of the mammalian complex I scaffold protein NDUFAF1 (36), does not cause any obvious mitochondrial morphology phenotype (Fig. 3 and Fig. S4). Whereas nuaf-1(RNAi) did not affect the level of COXI protein, it did cause a specific reduction in the level of the complex I protein NDUFS3 (50% of control) (Fig. 2D). Based on these observations, we conclude that inhibiting mitochondrial translation or blocking the assembly of complex IV, but not complex I, causes mitochondrial hyperfusion.
Genetic Interactions Between mma-1 and fzo-1, eat-3, or drp-1.
In C. elegans, FZO-1 and EAT-3 are required for the fusion of the outer mitochondrial membrane and the inner mitochondrial membrane, respectively (7, 10–13). To determine whether mitochondrial hyperfusion induced by mma-1(RNAi) is dependent on these GTPases, we performed RNAi experiments in animals carrying a null-mutation in the fzo-1 gene [fzo-1(tm1133)] or the eat-3 gene [eat-3(tm1107)]. The loss of either fzo-1 or eat-3 function completely suppressed the ability of mma-1(RNAi) to induce mitochondrial hyperfusion (100%) (Fig. 4). Therefore, fzo-1 and eat-3 are epistatic to mma-1, which suggests that mma-1(RNAi)-induced mitochondrial hyperfusion is mediated through fzo-1 and eat-3.
Fig. 4.
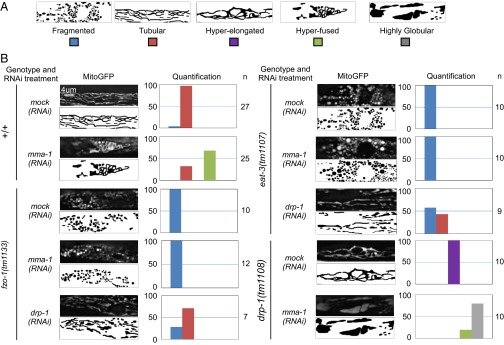
mma-1(RNAi) causes FZO-1- and EAT-3-dependent mitochondrial hyperfusion. Wild-type, fzo-1(tm1133), drp-1(tm1108), or eat-3(tm1107) L4 larvae carrying the Pmyo-3:mitoGFP transgene were treated with mock or mma-1 RNAi and mitochondrial morphology in body wall muscle cells of L4 larvae of the F1 generation was analyzed by fluorescent microscopy. [In the case of fzo-1(tm1133) or eat-3(tm1107) strains, P0 animals were treated for 24 h with RNAi and transferred to normal NGM plates] (A) Schematic of the different morphologies observed. (B) MitoGFP image and schematic of the representative mitochondrial morphology observed as well as a quantification are indicated (n = number of animals analyzed).
We also investigated the interaction between mma-1 and drp-1, which is required for mitochondrial fission (7). To that end, we inactivated mma-1 in animals carrying a null-mutation in the drp-1 gene [drp-1(tm1108)]. We found that mma-1(RNAi) in drp-1(tm1108) animals leads to a highly globular mitochondrial morphology phenotype that is different from both the mma-1(RNAi) phenotype and the drp-1(tm1108) phenotypes and that may represent a more severe hyperfusion phenotype (80%) (Fig. 4). This finding suggests that mma-1 and drp-1 may act in parallel. Therefore, mma-1(RNAi)-induced mitochondrial hyperfusion is most likely not caused by the inactivation of drp-1. This notion is further supported by the observation that mitochondrial fragmentation caused by the loss of either fzo-1or eat-3 is not suppressed by mma-1(RNAi) but is at least partially suppressed by drp-1(RNAi) (Fig. 4).
Mitochondrial Hyperfusion in mma-1(RNAi) Animals Is Critical for Viability.
The inactivation of mma-1 leads to a decrease in the levels of complex I and complex IV subunits (50% of controls) (Fig. 5A). Therefore, we expected to observe a decrease in the level of cellular ATP in mma-1(RNAi) animals. However, we found that, upon mma-1 inactivation, the level of cellular ATP remains unchanged (Fig. 5B). To test whether mitochondrial hyperfusion is required for the maintenance of cellular ATP levels in this mutant background, we inactivated mma-1 in animals carrying loss-of-function mutations in fzo-1 or eat-3 [fzo-1(tm1133) and eat-3(ad426)]. We found that the inactivation of mma-1 in the eat-3(ad426) mutant background leads to a highly penetrant synthetic larval arrest phenotype [eat-3(ad426);mma-1(RNAi): 88% compared with eat-3(ad426): 4%] (Fig. 5C). Furthermore, we found that the inactivation of mma-1 in the fzo-1(tm1133) mutant background results in a fully penetrant embryonic lethal/midlarval arrest phenotype [fzo-1(tm1133);mma-1(RNAi): 99% compared with fzo-1(tm1133): 22%] (Fig. 5C). In contrast, the inactivation of mma-1 in animals lacking drp-1 function [drp-1(tm1108)] does not increase the embryonic lethality caused by drp-1(tm1108). Because eat-3(ad426);mma-1(RNAi) animals and fzo-1(tm1133);mma-1(RNAi) animals are not viable, we were unable to measure the levels of cellular ATP in these mutant backgrounds. However, the fact that these animals are not viable indicates that preventing mitochondrial hyperfusion has severe consequences for viability and, hence, cellular functions in the mma-1 mutant background.
Fig. 5.

Complex IV-induced mitochondrial hyperfusion is a compensatory mechanism to maintain ATP level. (A) Level of NDUFS3 (complex I) and COXI (complex IV) upon mma-1 RNAi. (B) Level of ATP in the same animals shown in A. The difference was not statistically significant (P = 0.45). (C) mma-1(RNAi) and fzo-1(tm1133) or eat-3(ad426) loss-of-function mutations cause synthetic embryonic lethality and larval arrest (n > 50). (D) Average division time of the different cells indicated in the y axis in wild-type (+/+), fzo-1(tm1133), mma-1(RNAi)-treated wild-type embryos [mma-1(RNAi)], and mma-1(RNAi)-treated fzo-1(tm1133) embryos [fzo-1(tm1133);mma-1(RNAi)] [+/+: n = 5, fzo-1(tm1133): n = 4, mma-1(RNAi) in +/+: n = 3 and mma-1(RNAi) in fzo-1(tm1133): n = 3]. (E) ABarppppp lineage of wild-type and fzo-1(tm1133);mma-1(RNAi) embryo. (F) Recording images of mma-1(RNAi) in +/+ (left embryo) and mma-1(RNAi) in fzo-1(tm1133) (right embryo) at 16 min, 6 h, and 46 min, and 9 h and 3 min. In the Bottom panel, the mma-1(RNAi) in +/+ embryo on the left has already hatched and crawled away. In A, B, and D, SD are indicated. (*P < 0.05, **P < 0.01, and ***P < 0.001, by Student’s t test.)
To further investigate the embryonic lethality caused by mma-1(RNAi) in fzo-1(tm1133) animals, we analyzed their embryonic development using 4D lineage analysis (37). Specifically, using the SIMI BioCell software (Simi Reality Motion Systems GmbH, Unterschleissheim, Germany), we measured the cell-cycle length for the cells of the ABarppppp (V6R) lineage. As shown in Fig. 5D, the cell cycle lengths in wild-type embryos treated with mma-1 RNAi are very similar compared with those in wild-type embryos. Only in the later rounds of cell division (ABarppp, ABarpppp, and ABarppppp), minor differences are observed. Similar minor differences are observed in fzo-1(tm1133) embryos. Hence, the inactivation of fzo-1 or mma-1 minimally affects cell-cycle length during embryogenesis. In contrast, the simultaneous inactivation of fzo-1 and mma-1 results in a dramatic increase in cell-cycle length, especially in the later rounds of cell division. For example, on average, the ABarppppp cell took 82 min to divide in fzo-1(tm1133);mma-1(RNAi) embryos but only 48 min in wild-type embryos (Fig. 5 D and E). We followed the analyzed embryos until the completion of development. One of three fzo-1(tm1133);mma-1(RNAi) embryos hatched but arrested during larval development. The remaining two embryos arrested during embryogenesis and failed to hatch. As shown in Fig. 5F, fzo-1(tm1133);mma-1(RNAi) embryos develop much slower than wild-type embryos treated with mma-1 RNAi. After 6 h and 46 min, the mma-1(RNAi) embryo reached the twofold stage of embryogenesis, whereas the fzo-1(tm1133);mma-1(RNAi) embryo had only reached the bean stage of embryogenesis (Fig. 5F, Middle). [For comparison, mma-1(RNAi) embryos reached the bean stage after 4 h and 4 min.] Ultimately, the fzo-1(tm1133);mma-1(RNAi) embryos failed to hatch and died, whereas the mma-1(RNAi) embryo hatched and the emerging L1 larvae moved out of sight (Fig. 5F, Bottom). Based on these observations, we conclude that mitochondrial fusion in response to mma-1(RNAi) is essential for embryonic and larval development. In the absence of mitochondrial fusion, mma-1(RNAi) leads to mitochondrial dysfunction, which increases cell-cycle length and slows down development, ultimately leading to developmental arrest.
Knockdown of LRPPRC or SCO1 but Not the Mitochondrial Transcription Factor A (TFAM) Results in Mitochondrial Hyperfusion in Mammalian Cells.
Next, we addressed the question of whether the mitochondrial morphology alterations observed upon loss of mma-1 in C. elegans are a general phenomenon conserved between species. We therefore silenced expression of LRPPRC in human SH-SY5Y cells by RNAi (Fig. S5A). Whereas cells treated with control siRNA displayed predominantly a tubular mitochondrial network, a significant increase in hyperfused mitochondria was observed in LRPPRC knockdown cells (16.5% in control versus 48.5% in LRPPRC siRNA cells) (Fig. 6) (similar results were obtained using a different siRNA) (Fig. S5C). Consistent with published data, protein levels of COXI, COXII, and COXIII were decreased in LRPPRC-deficient human cells, indicating reduced levels of complex IV (Fig. S6 A and B). To test whether a decrease in complex IV activity is responsible for inducing mitochondrial hyperfusion, we transfected SH-SY5Y cells with SCO1 siRNA (Fig. S5B). Similarly to our results in C. elegans, down-regulation of SCO1 expression in SH-SY5Y cells induced mitochondrial hyperfusion (50%) (Fig. 6). [Similar results were obtained using a different siRNA (Fig. S5C).] In contrast, the tubular mitochondrial network was maintained in TFAM-deficient cells despite an efficient knock-down (67% in control versus 61% in TFAM siRNA cells) (Fig. S5 D–F). Mitochondrial hyperfusion induced by LRPPRC down-regulation in human cells was also dependent on a functional fusion machinery, because hyperfusion did not occur when Opa1 was simultaneously knocked down (Fig. S5 G–I). In conclusion, like in C. elegans, interfering with complex IV assembly or stability leads to mitochondrial hyperfusion in human cells.
Fig. 6.

Silencing of LRPPRC or SCO1 causes mitochondrial hyperfusion in mammalian cell culture. Human SH-SY5Y cells were treated with control, LRPPRC or SCO1 siRNAs. Three days after transfection, mitochondria were visualized using an anti-Tom20 antibody. Maximum projections of representative z-stacks as well as enlargements are shown for each condition. Data represent ±SEM of five independent experiments, each performed in duplicate. At least 150 cells were assessed per condition. SD are indicated. (*P < 0.05 and **P < 0.01, ANOVA and Bonferroni as a post hoc test.)
Mitochondrial Fusion Is a Transient Mechanism to Maintain ATP Levels in Response to Complex IV Reduction.
To test whether mitochondrial hyperfusion upon LRPPRC loss-of-function helps to maintain cellular ATP levels in human cells, we performed a time-course experiment. SH-SY5Y cells were transfected with control or LRPPRC siRNA, and both mitochondrial morphology and cellular ATP production was monitored on days 3–5 after transfection. To prevent compensation of decreased oxidative phosphorylation by increasing glycolysis, the cells were cultured in low glucose medium (38, 39). Remarkably, cellular ATP levels remained stable as long as mitochondrial hyperfusion occurred (days 3 and 4). However, at day 5 mitochondrial fragmentation prevailed, paralleled by a significant decrease in cellular ATP levels (Fig. 7) (LRRPRC knock-down is still efficient at day 5) (Fig. S6 C and D). These data support the model that upon complex IV deficiency, mitochondrial hyperfusion helps to temporarily maintain ATP production by oxidative phosphorylation.
Fig. 7.

LRPPRC siRNA-induced mitochondrial hyperfusion helps to maintain cellular ATP levels. (A) Cellular ATP levels of SH-SY5Y cells treated for 3, 4, or 5 d with control or LRPPRC siRNA. Data represent ±SEM of six independent experiments, each performed in duplicate. (B) Mitochondrial morphology of SH-SY5Y cells treated for 3, 4, or 5 d with control or LRPPRC siRNA. Mitochondria were visualized using an anti-Tom20 antibody. (C) Quantifications were based on three independent experiments, each performed in duplicate. At least 150 cells were assessed per condition. SD are indicated. (*P < 0.05, **P < 0.01, and ***P < 0.001, ANOVA and Bonferroni as a post hoc test.)
Discussion
We provide evidence that MMA-1 is the C. elegans homolog of LRPPRC. First, the MMA-1 protein shares significant sequence similarities with LRPPRC. Second, like LRPPRC (28), MMA-1 is targeted to mitochondria. Third, similar to what has been observed for LRPPRC, the inactivation of mma-1 leads to a decrease in COXI protein level and consequently complex IV activity. In contrast, despite a 50% reduction in the level of the complex I component NDUFS3, the activity of complex I remains unaffected in mma-1(RNAi) animals. This result could be because of a complex I-induced compensatory mechanism. Indeed, it has been previously shown that complex I activity is only partially decreased in mice knocked-out for another complex I subunit (NDUFS4) because of the formation of compensatory supercomplexes (40, 41). In conclusion, inactivation of mma-1 in C. elegans leads to a specific complex IV deficiency. This finding is significant because mutations in the LRPPRC gene have been associated with the French Canadian Leigh Syndrome, a neurodegenerative disease induced by complex IV deficiency (27). The conservation in structure and function between the MMA-1 and LRPPRC proteins establishes C. elegans mma-1 mutant animals as a model for the French Canadian Leigh Syndrome.
We also show that the inactivation of mma-1 in C. elegans or LRPPRC in mammalian cells leads to mitochondrial hyperfusion. Mitochondrial hyperfusion has so far been only observed in response to starvation signals (24, 25) or cell-cycle signals (42). We report that intramitochondrial signals, such as altered complex IV activity, can also control mitochondrial dynamics. Complex IV has been shown to be a key regulatory point of the ETC as its activity is controlled by the ratio of intramitochondrial ATP and ADP (ATP/ADP ratio) (43). Specifically, a high ATP/ADP ratio leads to the inhibition of complex IV activity and, consequently, a reduction in electron transport. Complex IV can therefore detect the level of ATP and adjust ATP synthesis to the requirement in cellular energy. Our work reveals an additional regulatory role of complex IV. As discussed in more detail below, our results indicate that a drop in complex IV activity induces mitochondrial hyperfusion, which is critical to maintain mitochondrial function.
Mitochondrial hyperfusion in response to complex IV inactivation could be the result of a decrease in mitochondrial fission or, alternatively, an increase in mitochondrial fusion. For the following reasons, we propose that mitochondrial hyperfusion in response to complex IV inactivation is the result of an increase in mitochondrial fusion. We show that in both C. elegans and mammalian cells, mitochondrial hyperfusion in response to complex IV inactivation is dependent on the mitochondrial fusion machinery. In contrast, in fission-defective C. elegans mutants [drp-1(tm1108)], the inactivation of complex IV results in a more severe mitochondrial hyperfusion phenotype. The notion that mitochondrial hyperfusion in response to complex IV inactivation is caused by the activation of mitochondrial fusion rather than a block in mitochondrial fission is furthermore supported by the finding that, although the inactivation of drp-1 can partially suppress the mitochondrial morphology phenotype caused by loss-of-function mutations of fzo-1 or eat-3, the inactivation of mma-1 is unable to do so. How does complex IV promote mitochondrial hyperfusion? Complex IV may indirectly affect Mfn1/2, which localize to the outer mitochondrial membrane and which have previously been implicated in stress-induced mitochondrial hyperfusion (23, 26). Alternatively, complex IV may directly affect Opa1, which localizes to the inner mitochondrial membrane. It has recently been shown that mammalian Opa1 can interact with some of the subunits of the ETC, including subunits of complex IV (21). Therefore, it is feasible that complex IV activates mitochondrial fusion by physically interacting with Opa1.
We show that mitochondrial hyperfusion is critical for the maintenance of cellular functions in cells in which complex IV activity is reduced. The inactivation of mma-1 in C. elegans or LRPPRC in mammalian cells results in a decrease in complex IV activity but, surprisingly, not in a decrease in the level of cellular ATP. Because mma-1(RNAi) animals do not exhibit any other obvious defects and have a normal broodsize and locomotion, this stable ATP level is likely not caused by a reduction in ATP usage. In contrast, mma-1(RNAi) animals in which mitochondrial hyperfusion is prevented (by a fzo-1 or eat-3 loss-of-function mutation) exhibit increased embryonic lethality associated with an increase in cell-cycle length during embryogenesis. Furthermore, animals that escape embryonic lethality do not develop further than the L3 larval stage. Inactivation by RNAi of the atp-2 gene, which encodes one of the subunits of the ATP synthase, causes a similar increase in cell-cycle length and embryonic lethality (44, 45). Furthermore, homozygous atp-2(ua2) mutants originating from heterozygous atp-2(ua2)/+ mothers develop normally through embryogenesis but arrest as L3 larvae. Hence, a maternal contribution of mRNA or protein supports the development of the homozygous offspring until the L3 stage, but not further. It has therefore been proposed that an energy-sensing developmental checkpoint exists at the L3-to-L4 transition (45). We speculate that some of the fzo-1(tm1133);mma-1(RNAi) progeny arrest during embryogenesis because of mitochondrial dysfunction similar to what is observed upon atp-2 RNAi. We also speculate that the progeny that escapes embryonic lethality undergoes larval arrest because they do not meet the energy-sensing developmental checkpoint described by Tsang et al. (45).
Based on these observations we propose that mitochondrial hyperfusion in the mma-1 mutant background helps to maintain mitochondrial function despite a reduction of complex IV activity. This protective effect may involve an increased ability to produce ATP because hyperfused mitochondria have been shown to have higher levels of ATP synthase dimers (24) and because increased oligomerization of ATP synthase has been shown to cause increased ATP production (46). Alternatively, mitochondrial hyperfusion may protect mitochondria by preventing mitophagy (24) or by affecting other essential mitochondrial functions, such as β-oxidation or calcium storage.
Even though mitochondrial hyperfusion in response to a reduction in complex IV activity is critical for the maintenance of mitochondrial and cellular functions, it only transiently compensates for a drop in complex IV activity. Reducing LRPPRC function for prolonged periods of time results in a progressive fragmentation of the mitochondrial network and, concomitantly, a drop in the level of cellular ATP. Why is this compensation only transient? Prolonged periods in a hyperfused state may result in the block of other important mitochondrial functions, which eventually leads to mitochondrial fragmentation and, consequently, a drop in ATP level. This model is consistent with the observation that fibroblasts from patients suffering from French Canadian Leigh Syndrome have fragmented mitochondria (17). Indeed, the complex IV deficiency that these patients suffer from may have initially led to a transient mitochondrial hyperfusion response. However, this response ultimately fails, resulting in a fragmented mitochondrial network. The transient nature of this compensatory mechanism also explains why mutations in complex IV have previously been associated with a mitochondrial fragmentation phenotype.
Materials and Methods
General C. elegans Methods and Strains.
C. elegans strains were cultured as previously described (47). Bristol N2 was used as the wild-type strain. Mutations used in this study were described by Riddle et al. (48), except: (LG II) fzo-1(tm1133), eat-3(tm1107) (National BioResource Project), (LG IV) drp-1(tm1108) (National BioResource Project), and (LG X) nrde-3(gg66) (32). A summary of the transgenic lines is shown in Table S1.
Cell Cultures and RNA Interference.
SH-SY5Y (DSMZ number ACC 209) cells were cultivated in DMEM (Lonza) supplemented with 15% FCS (vol/vol) (Sigma) and maintained at 37 °C, 5% CO2. For acute knock-down experiments, cells were reversely transfected with the following stealth siRNA oligos (Invitrogen) using Lipofectamine RNAiMAX (Invitrogen): human LRPPRC HSS115403 and HSS173413, human SCO-1 HSS109546 and HSS109547, human TFAM HSS144251, and human OPA-1 HSS107432. Cells were harvested at day 3 after transfection, unless otherwise stated.
RNA Interference.
RNAi by feeding was performed as previously described (49). The different RNAi strains are indicated as described in Table S2. Briefly, L4 larvae were grown at 20 °C on RNAi plates containing 6 mM isfopropyl-β-d-thiogalactopyranoside for 4–5 d. fzo-1(tm1133) or the eat-3(tm1107) mutants were treated for 24 h with RNAi and transferred to normal NGM plates. Mitochondrial morphology in L4 larvae of the F1 generation was analyzed by fluorescent microscopy, as described previously (13). For protein analyses, mixed-stage populations of worms harvested on the day of the microscopy were analyzed as described below. For 4D lineaging experiments, L4 larvae were treated by RNAi for 18 h prior analysis of F1 embryos. Plasmids used for RNAi by injection are described in Table S3. dsRNA was synthesized in vitro using the T3 and T7 polymerases (Ambion). Young adults were injected with dsRNA and mitochondrial morphology of L4 larvae of the F1 generation was analyzed as previously described (13).
Photoactivation Experiment.
PAmitoGFP (33) was cloned in pPD96.52 to generate pBC900 (Pmyo-3:PAmitoGFP). Photoactivation was performed on L4 larvae using an A1 confocal microscope (Nikon). Images were acquired with a Plan Apo VC 60× Oil DIC N2 (NA 1.4). Excitation of the PAmitoGFP was done with a 405-nm laser (2% power for 2 s). GFP signal was acquired with a 488-nm laser for excitation and a 525/50 filter for emission (pinhole size: 29 μm). Data were analyzed using Nikon Elements 3.2.
Protein Analysis.
Rabbit polyclonal anti–MMA-1 antibodies were generated using two antigenic peptides by Thermo Fisher Scientific. The antibodies were affinity-purified using a Sulfolink resin (Pierce) and used at 1:500 for Western analysis. Cell fractionation was performed as previously described (50). Worm protein extracts were resolved by SDS/PAGE and analyzed by Western analysis. To detect COXI, NDUFS3, αATP synthase, and α-Tubulin, we used mouse antibodies from Mitoscience MS404 (1:2,000), MS112 (1:2,000), MDS507 (1:2,000), and from Abcam ab7291 (1:2,000), respectively. Images were quantified using the ChemiDoc XRS+ System (Bio-Rad).
Mammalian cells samples were lysed in PBS 0.5% Triton X-100 0.5% Deoxycholate and resolved by SDS/PAGE. The following primary antibodies were used: anti-COXI 1:500 (Mitosciences, MS404), anti-COXII 1:1,000 (Epitomics, 2575-1), anti-COXIII 1:500 (Mitosciences, MS406), anti-COXIV 1:500 (Cell Signaling, 4844), anti-Hsp60 1:100 (Santa Cruz, sc-1052), anti–α-actin 1:5,000 (Sigma, A2228), anti-LRP130 1:1,000 (Santa Cruz, sc-166178), anti-SCO1 1:1,000 (Novus Biologicals, NBP1-87073), anti-TFAM 1:500 (Abnova, H00007019-B01P), and anti-OPA1 1:1,000 (BD, 612607). Detection was done using ECL or ECL Plus (GE Healthcare). Data were quantified using ImageJ.
Immunocytochemistry.
Embryos were collected on poly-l-lysine slides, frozen on dry ice for 20 min, and incubated in methanol/acetone (1:1) for 10 min. Embryos were permeabilized with PBS 1% Triton X-100 (vol/vol) two times for 10 min. After three washing steps of 10 min with PBST (PBS 0.1% Tween), slides were blocked with PBSTB [PBST 1% BSA (wt/vol)] for 20 min. Slides were then incubated with anti–MMA-1 (1:100) and anti-αATP synthase antibody (1:150) overnight at 4 °C. After three washing steps of 10 min with PBST, slides were incubated with goat anti-mouse FITC (1:200) and goat anti-rabbit Texas Red (1:200) for 2 h. After four washing steps of 10 min with PBST, slides were mounted in DAPI Vectashield (Vector Lab, H-1200). Images were acquired as previously described (13).
Tissue-Specific Histochemical Method.
Measurements were performed as previously described (35), with the following modifications. A synchronized population of 200 L1 larvae of N2 was inoculated on mock or mma-1(RNAi) plates. After 4–5 d at 20 °C, 1 d adult worms were embedded in OCT medium. Quantification was performed with ImageJ on at least 10 worms.
ATP Measurement.
A synchronized population of 200 N2 L1 larvae was inoculated onto RNAi plates. After 4–6 d at 20 °C, adults was harvested with 1 mL of M9 medium and washed three times with 10 mL of M9 medium. ATP measurements were performed using the ATP bio-luminescence Assay Kit HS II (Roche). Worms were resuspended in 250 μL of lysis buffer, incubated on ice for 10 min (with vortexing every 2 min) and for 5 min at 95 °C. After centrifugation at 13,000 × g for 1 min, 50 μL of the supernatant was used for the measurements using a LB940 Multimode Reader Mithras (Berthold Technologies). Protein concentration was determined by BCA to standardize the ATP level. In mammalian cells, ATP measurements were performed as previously described (39).
Analysis of Mitochondrial Morphology in Cultured Cells.
Cells grown on 15-mm glass coverslips were fixed with PBS 3.7% PFA (vol/vol) for 10 min, permeabilized with PBS 0.1% Triton X-100 for 5 min, blocked with PBS 5% BSA (wt/vol) for 30 min, and incubated with anti-TOM20 (1:1,000; Santa Cruz) in PBS 5% BSA (wt/vol) overnight at 4 °C. After extensive washing, fixed cells were incubated with Alexa555-goat anti-rabbit (1:1,000) for 2 h at 20 °C. Nuclei were counterstained with DAPI. Images were acquired with a Zeiss LSM710 confocal microscope with a 63× Oil objective (NA 1.4). Cells displaying an intact tubular mitochondrial network were classified as tubular; cells with globular or rod-like mitochondria were classified as fragmented; cells with hyperelongated or hyperconnected mitochondria were classified as hyperelongated. Classification was performed blind. Quantifications were based on duplicates of at least three independent experiments.
Supplementary Material
Acknowledgments
We thank E. Lambie, D. Mokranjac, T. Mikeladze-Dvali, J. Tatzelt, and members of the B.C. laboratory for comments on the manuscript; N. Lebedeva, L. Jocham, and D. Mayka for excellent technical support; the Dartmouth Department of Biological Sciences Light Microscopy Facility, and especially Ann Lavanway, for excellent technical support for confocal microsopy; A. Fire (Stanford School of Medicine) for plasmids; A. van der Bliek (University of California at Los Angeles) for plasmids; R. Youle (National Institutes of Neurological Disorder and Stroke, National Institutes of Health) for plasmids; S. Kennedy (University of Wisconsin) for sharing information about nrde-3(gg66); S. Mitani (National BioResource Project, Tokyo, Japan) for fzo-1(tm1133), drp-1(tm1108), and eat-3(tm1107); and the Caenorhabditis elegans Genetics Center (supported by the National Institutes of Health National Center for Research Resources) for strains. This work was supported by funding from American Cancer Society Research Scholar Grant RSG-06-110-1-CCG (to B.C.); National Institutes of Health Grants GM069950 and GM076651 (to B.C.); the Deutsche Forschungsgemeinschaft (K.F.W.); the Center for Integrated Protein Science Munich (B.C.); the Bundesministerium für Bildung und Forschung Nationalen Genomforschungsnetz plus “Functional Genomics of Parkinson’s Disease (to K.F.W.); and the Helmholtz Alliance “Mental Health in an Ageing Society” (to K.F.W.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1303872110/-/DCSupplemental.
References
- 1.Okamoto K, Shaw JM. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu Rev Genet. 2005;39:503–536. doi: 10.1146/annurev.genet.38.072902.093019. [DOI] [PubMed] [Google Scholar]
- 2.Hoppins S, Nunnari J. The molecular mechanism of mitochondrial fusion. Biochim Biophys Acta. 2009;1793(1):20–26. doi: 10.1016/j.bbamcr.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 3.Lackner LL, Nunnari JM. The molecular mechanism and cellular functions of mitochondrial division. Biochim Biophys Acta. 2009;1792(12):1138–1144. doi: 10.1016/j.bbadis.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11(12):872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- 5.Chan DC. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- 6.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12(8):2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Labrousse AM, Zappaterra MD, Rube DA, van der Bliek AM. C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell. 1999;4(5):815–826. doi: 10.1016/s1097-2765(00)80391-3. [DOI] [PubMed] [Google Scholar]
- 8.Chen H, et al. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160(2):189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Misaka T, Murate M, Fujimoto K, Kubo Y. The dynamin-related mouse mitochondrial GTPase OPA1 alters the structure of the mitochondrial inner membrane when exogenously introduced into COS-7 cells. Neurosci Res. 2006;55(2):123–133. doi: 10.1016/j.neures.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 10.Kanazawa T, et al. The C. elegans Opa1 homologue EAT-3 is essential for resistance to free radicals. PLoS Genet. 2008;4(2):e1000022. doi: 10.1371/journal.pgen.1000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Breckenridge DG, et al. Caenorhabditis elegans drp-1 and fis-2 regulate distinct cell-death execution pathways downstream of ced-3 and independent of ced-9. Mol Cell. 2008;31(4):586–597. doi: 10.1016/j.molcel.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tan FJ, et al. CED-9 and mitochondrial homeostasis in C. elegans muscle. J Cell Sci. 2008;121(Pt 20):3373–3382. doi: 10.1242/jcs.032904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rolland SG, Lu Y, David CN, Conradt B. The BCL-2-like protein CED-9 of C. elegans promotes FZO-1/Mfn1,2- and EAT-3/Opa1-dependent mitochondrial fusion. J Cell Biol. 2009;186(4):525–540. doi: 10.1083/jcb.200905070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hackenbrock CR, Rehn TG, Weinbach EC, Lemasters JJ. Oxidative phosphorylation and ultrastructural transformation in mitochondria in the intact ascites tumor cell. J Cell Biol. 1971;51(1):123–137. doi: 10.1083/jcb.51.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koopman WJ, et al. Mitochondrial network complexity and pathological decrease in complex I activity are tightly correlated in isolated human complex I deficiency. Am J Physiol Cell Physiol. 2005;289(4):C881–C890. doi: 10.1152/ajpcell.00104.2005. [DOI] [PubMed] [Google Scholar]
- 16.Morán M, et al. Cellular pathophysiological consequences of BCS1L mutations in mitochondrial complex III enzyme deficiency. Hum Mutat. 2010;31(8):930–941. doi: 10.1002/humu.21294. [DOI] [PubMed] [Google Scholar]
- 17.Sasarman F, Brunel-Guitton C, Antonicka H, Wai T, Shoubridge EA. LSFC Consortium LRPPRC and SLIRP interact in a ribonucleoprotein complex that regulates posttranscriptional gene expression in mitochondria. Mol Biol Cell. 2010;21(8):1315–1323. doi: 10.1091/mbc.E10-01-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guillery O, et al. Modulation of mitochondrial morphology by bioenergetics defects in primary human fibroblasts. Neuromuscul Disord. 2008;18(4):319–330. doi: 10.1016/j.nmd.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 19.Liot G, et al. Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA- and ROS-dependent pathway. Cell Death Differ. 2009;16(6):899–909. doi: 10.1038/cdd.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parone PA, et al. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS ONE. 2008;3(9):e3257. doi: 10.1371/journal.pone.0003257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agier V, et al. Defective mitochondrial fusion, altered respiratory function, and distorted cristae structure in skin fibroblasts with heterozygous OPA1 mutations. Biochim Biophys Acta. 2012;1822(10):1570–1580. doi: 10.1016/j.bbadis.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Pich S, et al. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet. 2005;14(11):1405–1415. doi: 10.1093/hmg/ddi149. [DOI] [PubMed] [Google Scholar]
- 23.Tondera D, et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009;28(11):1589–1600. doi: 10.1038/emboj.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13(5):589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA. 2011;108(25):10190–10195. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shutt T, Geoffrion M, Milne R, McBride HM. The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep. 2012;13(10):909–915. doi: 10.1038/embor.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mootha VK, et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc Natl Acad Sci USA. 2003;100(2):605–610. doi: 10.1073/pnas.242716699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sterky FH, Ruzzenente B, Gustafsson CM, Samuelsson T, Larsson NG. LRPPRC is a mitochondrial matrix protein that is conserved in metazoans. Biochem Biophys Res Commun. 2010;398(4):759–764. doi: 10.1016/j.bbrc.2010.07.019. [DOI] [PubMed] [Google Scholar]
- 29.Ruzzenente B, et al. LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs. EMBO J. 2012;31(2):443–456. doi: 10.1038/emboj.2011.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chujo T, et al. LRPPRC/SLIRP suppresses PNPase-mediated mRNA decay and promotes polyadenylation in human mitochondria. Nucleic Acids Res. 2012;40(16):8033–8047. doi: 10.1093/nar/gks506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Claros MG, Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem. 1996;241(3):779–786. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- 32.Guang S, et al. An Argonaute transports siRNAs from the cytoplasm to the nucleus. Science. 2008;321(5888):537–541. doi: 10.1126/science.1157647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karbowski M, et al. Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J Cell Biol. 2004;164(4):493–499. doi: 10.1083/jcb.200309082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harry E, Monahan L, Thompson L. Bacterial cell division: The mechanism and its precison. Int Rev Cytol. 2006;253:27–94. doi: 10.1016/S0074-7696(06)53002-5. [DOI] [PubMed] [Google Scholar]
- 35.Hench J, et al. A tissue-specific approach to the analysis of metabolic changes in Caenorhabditis elegans. PLoS ONE. 2011;6(12):e28417. doi: 10.1371/journal.pone.0028417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van den Ecker D, et al. Identification and functional analysis of mitochondrial complex I assembly factor homologues in C. elegans. Mitochondrion. 2012;12(3):399–405. doi: 10.1016/j.mito.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 37.Schnabel R, Hutter H, Moerman D, Schnabel H. Assessing normal embryogenesis in Caenorhabditis elegans using a 4D microscope: Variability of development and regional specification. Dev Biol. 1997;184(2):234–265. doi: 10.1006/dbio.1997.8509. [DOI] [PubMed] [Google Scholar]
- 38.Rossignol R, et al. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004;64(3):985–993. doi: 10.1158/0008-5472.can-03-1101. [DOI] [PubMed] [Google Scholar]
- 39.Bouman L, et al. Parkin is transcriptionally regulated by ATF4: Evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ. 2011;18(5):769–782. doi: 10.1038/cdd.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calvaruso MA, et al. Mitochondrial complex III stabilizes complex I in the absence of NDUFS4 to provide partial activity. Hum Mol Genet. 2012;21(1):115–120. doi: 10.1093/hmg/ddr446. [DOI] [PubMed] [Google Scholar]
- 41.Sterky FH, et al. Altered dopamine metabolism and increased vulnerability to MPTP in mice with partial deficiency of mitochondrial complex I in dopamine neurons. Hum Mol Genet. 2012;21(5):1078–1089. doi: 10.1093/hmg/ddr537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci USA. 2009;106(29):11960–11965. doi: 10.1073/pnas.0904875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ludwig B, et al. Cytochrome C oxidase and the regulation of oxidative phosphorylation. ChemBioChem. 2001;2(6):392–403. doi: 10.1002/1439-7633(20010601)2:6<392::AID-CBIC392>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 44.Sönnichsen B, et al. Full-genome RNAi profiling of early embryogenesis in Caenorhabditis elegans. Nature. 2005;434(7032):462–469. doi: 10.1038/nature03353. [DOI] [PubMed] [Google Scholar]
- 45.Tsang WY, Sayles LC, Grad LI, Pilgrim DB, Lemire BD. Mitochondrial respiratory chain deficiency in Caenorhabditis elegans results in developmental arrest and increased life span. J Biol Chem. 2001;276(34):32240–32246. doi: 10.1074/jbc.M103999200. [DOI] [PubMed] [Google Scholar]
- 46.Strauss M, Hofhaus G, Schröder RR, Kühlbrandt W. Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 2008;27(7):1154–1160. doi: 10.1038/emboj.2008.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77(1):71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Riddle DL, Blumenthal T, Meyer BJ, Priess JR (1997) Introduction to C. elegans. C. elegans II, eds Riddle DL, Blumenthal T, Meyer BJ, Priess JR (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY), 2nd Ed.
- 49.Timmons L, Court DL, Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 2001;263(1-2):103–112. doi: 10.1016/s0378-1119(00)00579-5. [DOI] [PubMed] [Google Scholar]
- 50.Lu Y, Rolland SG, Conradt B. A molecular switch that governs mitochondrial fusion and fission mediated by the BCL2-like protein CED-9 of Caenorhabditis elegans. Proc Natl Acad Sci USA. 2011;108(41):E813–E822. doi: 10.1073/pnas.1103218108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.