

Abstract
Cohesin’s Smc1 and Smc3 subunits form V-shaped heterodimers, the nucleotide binding domains (NBDs) of which bind the C- and N-terminal domains, respectively, of the α-kleisin subunit, forming a large tripartite ring within in which sister DNAs are entrapped, and thereby held together (sister chromatid cohesion). During replication, establishment of stable cohesion is dependent on Eco1-mediated acetylation of Smc3’s NBD, which is thought to prevent dissociation of α-kleisin from Smc3, thereby locking shut a “DNA exit gate.” How Scc3 and Pds5, regulatory subunits bound to α-kleisin, regulate cohesion establishment and maintenance is poorly understood. We show here that by binding to α-kleisin adjacent to its Smc3 nucleotide binding N-terminal domain, Pds5 not only promotes cohesin’s release from chromatin but also mediates de novo acetylation of Smc3 by Eco1 during S phase and subsequently prevents de-acetylation by the deacetylase Hos1/HDAC8. By first promoting cohesin’s release from chromosomes and subsequently creating and guarding the chemical modification responsible for blocking release, Pds5 enables chromosomal cohesin to switch during S phase from a state of high turnover to one capable of tenaciously holding sister chromatids together for extended periods of time, a duality that has hitherto complicated analysis of this versatile cohesin subunit.
Keywords: cell, gene
Sister chromatid cohesion is conferred by a protein complex called cohesin, the rod-shaped Smc1 and Smc3 subunits of which associate via a hinge dimerization domain to form V-shaped heterodimers. ABC-like nucleotide binding domains (NBDs) at the vertices of Smc dimers are bound by C- and N-terminal domains of α-kleisin subunits, respectively, thereby creating huge tripartite rings within which sister DNAs are thought be entrapped (1, 2). The ability of these rings to build, maintain, and dissolve cohesion depends on their association with a number of regulatory subunits, including Scc3, Pds5, Sororin, and Wapl (3–8).
How DNA molecules enter cohesin rings and either remain entrapped or subsequently escape is poorly understood. The molecules are thought to enter through a “gate” created by transient dissociation of the Smc-Smc hinge interface (9). This event, which leads to a potentially stable association with chromatin, depends on the activity of a separate complex called kollerin [composed of Sister chromatid cohesion protein 2 and 4 (Scc2/4)] (10, 11) as well as hydrolysis of ATP bound to both NBDs (12–15).
Cohesin’s release from its topological embrace with chromatin fibers occurs via two mechanisms. The cleavage of α-kleisin by separase, which opens the ring, destroys sister DNA cohesion and triggers sister chromatid disjunction at the metaphase to anaphase transition (16). A second mechanism, which is separase-independent but instead relies on cohesin’s Wapl subunit, releases cohesin throughout the cell cycle, enabling it to turn over on chromosomes (7, 17, 18). The rate of release is greatly increased as animal cells enter mitosis, a process known as the prophase pathway (19, 20) and responsible for removing most cohesin from chromosomes arms before activation of separase by the anaphase-promoting complex or cyclosome (APC/C).
Yeast cohesin complexes also possess a Wapl-dependent releasing activity but it is not noticeably increased as cells enter mitosis. As a result, most cohesin is removed from yeast chromosomes during mitosis by separase only upon APC/C activation. In addition to Wapl, releasing activity associated with yeast cohesin depends on a pair of lysine residues within Smc3’s NBD (K112 and K113), Scc3, and Pds5. Because fusion of Smc3’s C terminus to the N terminus of α-kleisin blocks cohesin’s turnover on chromatin (21), it is thought that release is mediated by the escape of DNAs through a gate created by dissociation of α-kleisin’s N-terminal domain from Smc3’s NBD.
Establishment of stable sister chromatid cohesion during DNA replication requires inactivation of Wapl-dependent “releasing” activity through acetylation of K112 and K113 by Eco1 (18, 22–26). In animal cells, acetylation facilitates recruitment of sororin, an additional event essential for creating stable cohesion (5, 18). In yeast, where no sororin ortholog has hitherto been detected, acetylation per se may inactivate releasing activity and lock shut the Smc3/α-kleisin DNA exit gate. Consistent with the notion that acetylation blocks dissociation of α-kleisin’s N-terminal domain from the Smc3 NBD, fusion of Smc3’s C terminus to the N terminus of α-kleisin mimics the effect of acetylation and enables yeast cells to create sister chromatid cohesion in the absence of Eco1 (21).
According to the hypothesis outlined above, acetylation blocks kleisin’s dissociation from Smc3’s NBD and must therefore be maintained throughout the G2 and M phases. De-acetylation eventually occurs at the hands of a class I deacetylase, called Hos1 in yeast (27–29) and HDAC8 in mammalian cells (30). In yeast, this process only takes place upon kleisin cleavage by separase at the metaphase-to-anaphase transition. Because artificial cleavage of kleisin by tobacco etch virus (TEV) protease is sufficient to trigger de-acetylation by Hos1 at other stages of the cell cycle, de-acetylation must be regulated by the state of the cohesin ring and not by cell cycle-regulated Hos1 activity (27). Kleisin cleavage is accompanied by cohesin’s dissociation from chromosomes, and it has therefore been suggested that cohesin’s association with chromatin inhibits Hos1 activity (27–29). Whatever the mechanism, protection must be highly robust as massive Hos1 overexpression during the G2/M phase causes only slow and partial de-acetylation (27).
In this article, we investigate the function of an important regulatory subunit associated with the cohesin ring, namely the HEAT repeat-containing protein Pds5 (3, 4). We show that by binding to a small domain immediately adjacent and C-terminal to the N-terminal helices that bind Smc3 NBDs, Pds5 promotes cohesin’s release from chromatin in cooperation with Wapl and Scc3. Remarkably, Pds5 has two other functions that together mitigate the potentially lethal consequences of its participation in releasing activity. Pds5 promotes acetylation of Smc3 by Eco1 during S phase and prevents de-acetylation by Hos1 during the G2 and M phases. Interestingly, Pds5 has yet another, as yet mysterious, function in stabilizing cohesion besides maintaining Smc3 acetylation. Finally, by showing that Smc3 de-acetylation is triggered by kleisin cleavage but not by that of Smc3’s coiled coil, we propose that Pds5’s ability to block Hos1-mediated de-acetylation is ended not by cohesin’s dissociation from chromatin, as previously thought (28), but by separase-mediated cleavage.
Results
Pds5 Is Recruited by Sequences Within α-Kleisin Adjacent to Its N-Terminal Smc3 Binding Domain.
To identify essential sequences within α-kleisin that might promote recruitment of regulatory subunits, such as Pds5 and Scc3, we created a series of deletions within its central domain, leaving undisturbed N- and C-terminal domains that bind the Smc NBDs. This process revealed that residues between 131 and 138 are essential for cell viability (SI Appendix, Fig. S1A). Deletions spanning 103–123 and 180–256 were also lethal, but these regions were ignored because smaller deletions subdividing these two intervals had little or no phenotype and the effect of the larger deletions could therefore be attributable to reductions in α-kleisin’s length.
Site-directed mutagenesis revealed that Scc1V137G L138G or Scc1V137K barely supported cell proliferation (Fig. 1A). These residues are highly conserved among related fungi and located in a predicted helix next to the N-terminal α-helical domain involved in binding the Smc3 NBD (SI Appendix, Fig. S1B). Coimmunoprecipitation showed both double and single mutations greatly reduced Scc1’s association with Pds5 in yeast extracts (Fig. 1B and SI Appendix, Fig. S1C). Importantly, Scc1L89K, which abolishes binding to the Smc3 NBD, had no such effect (Fig. 1B). Live-cell imaging showed that cohesin composed of Scc1V137K fused to GFP forms similar barrel shaped structures (in the presence of untagged wild-type Scc1) to those formed by wild-type Scc1-GFP (Fig. 1C). Chip-sequencing and Chip-qPCR confirmed that the distribution along chromosomes of Scc1V137K was broadly similar to that of wild-type, although the mutant complexes were slightly less abundant along chromosome arms (SI Appendix, Fig. S2 A and B). These results imply that V137K does not compromise loading of cohesin onto chromosomes, at least in the vicinity of centromeres.
Fig. 1.

Cohesin’s α-kleisin subunit recruits Pds5 via its distinct domain adjacent to the N-terminal α-helix. (A) Scc1(V137G L138G) and Scc1(V137K) mutations cause cell lethality. Different Scc1 mutants are expressed in cells whose endogenous Scc1 is under control of the GAL promoter (K9514, K16779, K16832, K17132, K17120, K17133, K17121, and K17119). Lethality was determined by growing cells on YEP+glucose plates at 30 °C. (B) Coimmunoprecipitation showing that Scc1V137K but not L89K abolishes Pds5 interaction (K8069, K16867, K17297, and K17289). (C) Live-cell imaging showing nuclear localization of ectopically expressed GFP-tagged wild-type Scc1 (Left, K18038) and Scc1V137K (Right, K17370) in G2/M cells. (D) Western blotting showing the ectopic wild-type Scc1 (K17356) and Scc1V137K (K17357) protein levels in Pds5-GFP diploid cells. (E) Distribution of Pds5-GFP in cells expressing ectopic wild-type Scc1 (K17356) or V137K (K17357) at the leu locus after switching off endogenous GAL1-SCC1. (F) Wild-type Scc1 (K18846), but not Scc1V137K (K18847), recruits pericentric Wapl-GFP after endogenous GAL1-SCC1 is switched off. (G) Scc1 with photocross-linking ρ-BPA substitution at positions M127, D130 and T133 cross-links to Pds5 (K17830, K17827, K17828, and K17829).
Live-cell imaging demonstrated that Scc1V137K is proficient in recruiting GFP-tagged Scc3 (SI Appendix, Fig. S3A) but not Pds5 to pericentric chromatin in vivo (Fig. 1 D and E). Thus, cohesin containing Scc1V137K loads onto chromosomes efficiently but fails to recruit Pds5; it also fails to recruit Wapl-GFP (Fig. 1F), which is consistent with the finding that mutations in the N-terminal part of Pds5 fail to recruit Wapl (21). The adverse effect of V137K on Scc1’s interaction with Pds5 cannot be attributed to the loss of Wapl, because wpl1Δ (which is not lethal) does not affect Pds5’s association with pericentric chromatin (SI Appendix, Fig. S3B). Importantly, cells expressing Scc1V137K in the absence of wild-type α-kleisin fail to establish sister-chromatid cohesion and arrest in mitosis with highly separated centromere clusters (Mtw1-RFP), even though core cohesin (Scc3-GFP) accumulates at pericentric regions (Figs. 1 E and F, and SI Appendix, Fig. S3A). We conclude that cohesin rings defective specifically in recruiting Pds5 fail to generate sister chromatid cohesion.
We used an in vivo site-specific cross-linking assay to address whether Pds5 actually binds the sequences surrounding V137. Residues within the N- and C-terminal domains of Scc1 were systematically mutated to amber stop codons (UAG) and ρ-benzoyl-phenylalanine (ρ-BPA) incorporated at these positions using an “orthogonal” aminoacyl-tRNA synthetase-tRNA pair (31). Proliferation of cells expressing amber codon containing SCC1 genes from a 2-μm plasmid is usually enhanced by addition of ρ-BPA (SI Appendix, Fig. S4A). Upon UV irradiation, ρ-BPA covalently links binding partners situated within ∼7 Å. We identified several residues—namely M127, D130, and T133—the substitution of which by ρ-BPA gave rise to efficient Pds5-Scc1 cross-link–shown different mobility (Fig. 1G). We speculate that these mobility differences in various ρ-BPA substitutions in Scc1 are probably because of different cross-linking sites in Pds5. Analysis of UV-induced high molecular weight species (SI Appendix, Fig. S4B, arrow) by mass spectrometry confirmed that they are composed of Scc1 and Pds5 sequences. In total, we identified five ρ-BPA substitutions between positions 127 and 133 capable of giving rise to Pds5 cross-links (SI Appendix, Fig. S4C). Importantly, no Pds5 cross-links were obtained with ρ-BPA substitutions within Scc1’s N-terminal or C-terminal domains.
Crucially, the Pds5-Scc1 cross-links generated by D130BPA were abolished by V137K (SI Appendix, Fig. S4D), implying that the interaction revealed by D130BPA is the same as that compromised by V137K. The residues surrounding M127, D130, and T133 appear to contribute to Pds5 binding because deletion of residues between 124 and 130 causes slow growth (SI Appendix, Fig. S1A). We conclude that V137G L138G and V137K abolish interaction with Pds5 because the latter actually binds to this region of α-kleisin.
Pds5 Is Required for Cohesin’s Release from Chromatin.
To address whether the cohesion defect caused by Scc1V137K is caused by a failure of cohesin to associate stably with chromatin, we used inverse fluorescence recovery after photobleaching (iFRAP) to compare the rate of mutant and wild-type Scc1-GFP turnover within pericentric chromatin in tetraploid cells. To do this, we measured the fluorescence intensity associated with the two sides of each barrel following selective photobleaching of one half. In contrast to previous reports that cohesin subunits are stably associated with pericentric barrels (32–34), we observed turnover (with a half life of ∼2 min) of one-half of pericentric Scc1-GFP. Identical results were obtained with cells expressing Scc1-GFP from endogenous genes (21) or from ectopic loci (SI Appendix, Fig. S5A). Far from increasing this turnover, V137K actually decreased it, an effect that was even more pronounced in diploid cells that had undergone replication in the absence of Eco1 activity (SI Appendix, Fig. S5B). These results imply that the defective cohesion caused by Scc1V137K is not because of cohesin’s unstable association with chromatin, a conclusion consistent with the finding that Pds5 is responsible for recruiting Wapl and with the identification of specific Pds5 alleles defective in cohesin’s turnover on chromosomes (21).
Pds5’s Association Is Required to Promote de Novo Smc3 Acetylation.
Cohesin that has been loaded onto chromosomes must subsequently be acetylated by Eco1 during S phase to build stable sister chromatid cohesion (22, 24–26, 35). To address whether Pds5 is required for this process, we measured the effect on Smc3 acetylation of depleting Pds5 using an auxin-dependent degron (AID) (36). Cells were first arrested in G1 by addition of pheromone, treated with auxin, which caused Pds5-AID depletion (Fig. 2A), and then released from pheromone to S phase in the presence of auxin. Strikingly, this process largely abolished acetylation, despite having only a minor effect on Scc1 protein levels (Fig. 2B and SI Appendix, Fig. S6). When this article was under the preparation, a similar observation was reported in fission yeast (37). Our findings indicate that promoting acetylation of cohesin by Eco1 may therefore be a highly conserved feature of Pds5.
Fig. 2.

The association of Pds5 and α-kleisin is required for Smc3 acetylation. (A) Depletion of Pds5 through auxin-dependent degradation pathway. Cells Pds5 tagged with auxin-degron (K18421) were pheromone arrested in G1 for 1.5 h (t = 0 min) and 3 mM Indole-3-acetic acid sodium salt (auxin) was added or not for another 1 h. Proteins were extracted to monitor the level of Pds5. (B) Pds5 is required for Smc3 acetylation. Cells (K18421) were arrested in G1 and Pds5 was depleted as described in A, and then released to S phase. (C) Pds5-binding mutant Scc1V137K is defective in Smc3 acetylation. Immunoprecipitation of Scc1 was performed on strains K10905, K17234, and K18326.
To address whether Pds5 promotes Smc3 acetylation through its association with cohesin rings, we compared the abundance of acetylated Smc3 in immunoprecipitates of wild-type or Scc1V137K protein. V137K greatly reduced the level of Smc3 acetylation (Fig. 2C), an effect that cannot be attributed to competition with wild-type protein (SI Appendix, Fig. S7). We conclude that Pds5 promotes Smc3 acetylation only when associated with cohesin’s kleisin subunit. This result is consistent with the finding that Schizosaccharomyces pombe’s Eco1 ortholog Eso1 interacts with Pds5 in a yeast two-hybrid assay (23).
There is a real possibility (see below) that the lack of Smc3 acetylation associated with V137K could be caused not by defective de novo acetylation but by a failure to block de-acetylation by Hos1 during the G2 and M phases (27, 28). To address this possibility, we measured Smc3 acetylation in strains whose Hos1 can be degraded in a conditional manner using an auxin-dependent degron (Hos1-AID) (36). Smc3 failed to be de-acetylated when Hos1-AID cells underwent anaphase following release from pheromone in the presence of auxin (SI Appendix, Fig. S8), demonstrating that this induces an effective depletion. Crucially, when auxin was first added to pheromone-arrested cells 40 min before release, we found lower levels of acetylated Smc3 in immunoprecipitates of HA-tagged Scc1V137K than those of HA-tagged wild-type Scc1 (SI Appendix, Fig. S9, Upper) as cells underwent S phase in the presence of auxin, which greatly reduced Hos1-AID levels (SI Appendix, Fig. S9, Lower). Strikingly, V137K caused a corresponding increase in the level of unacetylated Smc3 associated with Scc1 (see SI Appendix, Fig. S10 for unacetylated Smc3 antibody characterization). We conclude that the low levels of acetylated Smc3 associated with Scc1V137K are mainly a result of defective de novo acetylation, a conclusion consistent with the finding that the temperature-sensitive pds5-101 mutation causes a major decrease in the Smc3 acetylation as hos1Δ cells undergo S phase at the restrictive temperature (SI Appendix, Fig. S11).
During the course of these experiments, we observed a small reduction in Scc1 protein levels in pds5-ts cells at restrictive temperature (SI Appendix, Fig. S11C) after depletion of Pds5 (SI Appendix, Fig. S6), as well as a reduction in Scc1V137K protein levels arrested in G2/M because of activation of spindle checkpoint caused by cohesion defects (SI Appendix, Fig. S7B). This finding raises the possibility that Pds5 might also affect Scc1 protein stability in the long run. However, this phenomenon cannot explain the effect of V137K on Smc3 acetylation as cells enter S phase.
Smc3 De-Acetylation Is Not Triggered by Cohesin’s Dissociation from Chromosomes.
Given the potential importance of Smc3 acetylation in preventing cohesin’s release from chromatin, a key question concerns how the deacetylase Hos1/HDAC8 is prevented from de-acetylating Smc3 from the end of S phase until cleavage of Scc1 at the beginning of anaphase (27). The finding that de-acetylation can be triggered by cleavage of Scc1 by TEV protease in G2/M-arrested cells (27), when Hos1 is normally inactive, raised the possibility that it is cohesin’s association with chromatin that keeps Hos1 in check. If true, then ring-opening by cleavage of Smc3’s coiled coil (38) should also trigger de-acetylation. Using an improved method of expressing TEV protease from the GAL promoter, complete severance of Smc3’s coiled coil occurred within 40 min of galactose induction (Fig. 3A, Top and Middle) and this was accompanied by cohesin’s dissociation from chromatin (SI Appendix, Fig. S12). Surprisingly, cleavage of Smc3 in this manner had little or no effect on Smc3 acetylation (Fig. 3A), which contrasts with the rapid de-acetylation upon Scc1 cleavage (Fig. 3B).
Fig. 3.
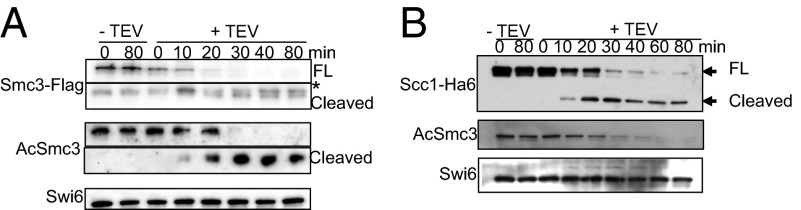
Cohesin dissociation from DNA does not trigger Smc3 deactylation. Release of cohesin from chromatin via Scc1 (B) but not Smc3 cleavage trigger de-acetylation (A). Strains K20018, K20019, K20021, and K20022 were grown in medium lacking tryptophan [2% (wt/vol) glucose] and then transferred in YEP medium containing 2% raffinose for 5 h. Subsequently, cells were arrested in metaphase by nocodazole for 2 h and then 2% galactose was added to induce the expression of TEV. Western blots against the indicated proteins were performed on whole-cell extracts. Full pictures of A are shown in SI Appendix, Fig S12C. An asterisk indicates a-specific bands.
Pds5 Protects Smc3 from De-Acetylation by Hos1.
Our finding that cleavage of Scc1 but not Smc3 triggers the latter’s de-acetylation raises the possibility that Hos1 is inhibited during G2/M by factors associated with cohesin’s kleisin subunit. Pds5 is a good candidate because it binds close to the domain of α-kleisin associated with Smc3’s NBD (Fig. 1). Indeed, the observation that Smc3 acetylation greatly reduces Pds5’s turnover within chromosomal cohesin (21) suggests that Pds5 might interact directly with acetylated Smc3 NBDs. To address Pds5’s role in protecting acetylated Smc3 from Hos1, we investigated the behavior of two temperature-sensitive alleles, namely pds5-101 and pds5-99. Both alleles are defective in establishing sister-chromatid cohesion (3) and in promoting Smc3 acetylation (SI Appendix, Figs. S11A and S13) during S phase at the restrictive temperature.
To test the effect of inactivating these alleles after S phase, wild-type and mutant cultures were shifted to the restrictive temperature only after they had first been arrested in the M phase following incubation in the presence of nocodazole at the permissive temperature. Strikingly, acetylated Smc3 declined rapidly in a Hos1-dependent manner in pds5-101 but not in wild-type or pds5-99 cells (Fig. 4A). This de-acetylation is not caused by the loss of Scc1 integrity because there is no measurable effect on Scc1 level during this incubation time (SI Appendix, Fig. S14).
Fig. 4.

Pds5 protects acetylated Smc3. Inactivation of pds5-101 but not pds5-99 ts mutants causes Hos1-dependent de-acetylation in G2/M cells. (A)Yeast strains K12757, K17780, K17778, and K17774 were arrested with nocodazole at 25 °C (90 min) and then incubated at 35 °C. Proteins were extracted at indicated time points, and the levels of the indicated proteins were monitored. (B) Inactivation of pds5-101 but not pds5-99 ts mutants in G2/M causes loss of cohesion maintenance. Cells growing in synthetic medium lacking methionine were arrested at 25 °C in G1. Cells were then released in YEPD medium supplemented with 2 mM methionine to induce metaphase. Subsequently, the cultures were incubated at 35 °C. Graphs showing percentage of cells with single or double (split) GFP dots and the average split dots distance were shown in K15024, K15073, and K20119 strains.
To test whether this decline is accompanied by a loss of sister chromatid cohesion, we synchronized cells growing at the permissive temperature in metaphase by releasing them from a pheromone-induced G1 arrest in the presence of methionine and then shifted to the restrictive temperature. Sister chromatid cohesion was quantified by measuring the distance between sister URA3 GFP dots. Both the fraction of cells with two distinct URA3 GFP dots and the distance between these increased in pds5-101 but not in wild-type or pds5-99 cells (Fig. 4C), a result that confirms the previous conclusion that pds5-101 but not pds5-99 is defective in maintaining sister chromatid cohesion (3). These data suggest that Pds5 not only promotes Smc3 acetylation during S phase but also protects it from Hos1 thereafter. To confirm this finding, we analyzed the effect of inducing depletion of Pds5-AID during G2/M by the addition of auxin (SI Appendix, Fig. S15). Because this process also induced a rapid decline in Smc3 acetylation without having any measureable effect on Scc1 abundance (SI Appendix, Fig. S15), we conclude that one of Pds5’s several functions is to protect acetylated Smc3 from Hos1.
Protecting Acetylated Smc3 from Hos1 Contributes to Maintenance of Cohesion.
Our observation that pds5-101 cells fail to maintain both acetylation and cohesion while pds5-99 cells maintain both properties upon shift to the restrictive temperature in G2/M is consistent with the idea that cohesion maintenance depends on persistent acetylation (21). According to this notion, de-acetylation could be a cause of cohesion loss in G2/M pds5-101 cells and deletion of HOS1 should rescue this. To test this theory, we measured both the fraction of cells with split GFP dots as well as their average separation in wild-type, pds5-101, and pds5-101 hos1Δ cells upon shift of metaphase-arrested cells to the restrictive temperature. Striking upon shift to the restrictive temperature, hos1Δ both delays dot splitting and reduces their degree of separation (Fig. 5). This finding implies that de-acetylation by Hos1 does indeed contribute to loss of cohesion in G2/M pds5-101 cells and is further evidence that maintenance of cohesion depends on continued acetylation. It is nevertheless important to note that the loss of cohesion in pds5-101 hos1Δ cells remains greater than that of hos1Δ single mutants (SI Appendix, Fig. S16), showing that de-acetylation is not solely responsible for cohesion loss in pds5-101 cells. This finding implies that Pds5 maintains cohesion in G2/M cells not only by protecting acetylated but also by an additional hitherto mysterious mechanism. Because Pds5 does more than just maintain acetylation during G2/M, it will be difficult to assess more precisely the importance of acetylation protection without new alleles defective solely in this process. One explanation for the different behavior of pds5-99 and pds5-101 mutants is that the former is defective in de novo acetylation but not in protecting acetylation, but the latter is defective in both functions.
Fig. 5.

The absence of Hos1 partially suppresses the loss of G2/M cohesion in the pds5 mutant. Yeast strains K15024, K15073, and K20054 were arrested in metaphase at 25 °C (as shown in Fig. 4). Then, the cultures were incubated at 35 °C. Sister chromatid cohesion was monitored by fluorescence microscopy. (A) Distance between split dots. (B) Increased population with split dots after shifting to 35 °C. (C) Mean distance of split dots.
It is important to point out that because Pds5 has multiple functions, both promoting cohesin’s release from chromatin and its maintenance, the consequences of its inactivation are difficult to predict. For example, loss of acetylation because of Pds5’s inactivation should be accompanied by a loss of releasing activity, which should mitigate the consequences of acetylation loss, assuming of course that the function of acetylation is merely to block cohesin’s release from chromatin. To explain our finding that a decline in acetylation contributes to the loss cohesion in pds5-101 cells, we suggest that these cells might retain some releasing activity, at least for a limited period after a shift to the restrictive temperature, and that this acts on those cohesin complexes, the acetylation of which has been removed by Hos1.
Curiously, we find little or no evidence for any rescue of the cohesion defects of pds5-101 by wpl1Δ (SI Appendix, Fig. S17), which might be expected to inactivate releasing activity completely. We suggest that there are two explanations for this. First, wpl1Δ on its own causes quite severe cohesion defects (22) for reasons that are currently unclear, a phenomenon that would obscure any rescue. Second, because wpl1Δ only partially compensates for the lack of acetylation mediated by Eco1 (22, 24, 25, 35), any rescue of cohesion loss in pds5-101 cells would be predicted to be less effective than that by hos1Δ, which should fully reverse any adverse effects caused by precocious de-acetylation. Additionally, detecting rescue—however weak or strong—is clearly hampered by the fact that Pds5 has a role in maintaining cohesion that does not involve protecting acetylation. If, as envisaged by the ring model, cohesion is mediated solely by entrapment of sister DNAs inside cohesin rings, then this fourth Pds5 activity presumably also prevents ring opening. It might do so either by controlling dissociation of the Smc3/kleisin interface or by controlling dissociation of one of the ring’s other two interfaces. It may be relevant in this regard that there is a suggestion from FRET studies that Pds5 might interact with the Smc1/Smc3 hinge interface (39), although whether this has any functional significance is not known.
Discussion
Pds5 is a highly conserved cohesin subunit, the functions of which are poorly understood. We show here that Pds5 is recruited to cohesin rings in yeast by binding sequences centered around a valine at position 137 (V137) within their kleisin subunit, a domain that is adjacent to the N-terminal α-helices involved in binding to Smc3 NBDs. Although it was concluded that Pds5 and Wapl exert their effects on Xenopus cohesin by binding to the central domain of its kleisin subunit, we note that the interaction between Pds5 and kleisin was markedly reduced when sequences equivalent to V137 in yeast α-kleisin were deleted (40). Thus, an association between Pds5 and sequences close to α-kleisin’s N-terminal Smc3-binding domain appears to be a conserved feature. Interestingly, these sequences are highly conserved in animal cells.
Our analysis of cohesin complexes that cannot recruit Pds5 because of mutation of V137, as well as the effects of Pds5 depletion or mutation, has revealed several important insights into the role of this enigmatic subunit. Although Pds5’s association with cohesin is unnecessary for loading cohesin onto chromatin, it is crucial for subsequent release, a function mediated partly by Pds5’s recruitment of Wapl and partly through more a more direct participation of Pds5 in the process of release (21). If, as currently thought, release involves escape of DNAs from entrapment inside the cohesin ring via an “exit gate” created by transient dissociation of kleisin’s N-terminal domain from the Smc3 NBD, then by binding very close to the former, Pds5 would be well placed to facilitate dissociation and thereby DNA escape. Our finding that Pds5’s recruitment to cohesin is required for the latter’s release from chromatin is consistent with the observation that depletion of Pds5 from Xenopus extracts leads to a greater retention of cohesin on chromosome arms during prophase (40). This finding is also consistent with the existence of Pds5 alleles in yeast that are defective in chromatin release (21) and with the finding that alleles of Wapl that are unable to bind Pds5 in vitro are also defective as a result of cohesin’s dissociation during prophase (40).
Remarkably, Pds5 has two other functions whose role is to mitigate the release mediated by Pds5 together with Wapl and Scc3. If stable cohesion depends on the permanent entrapment of sister DNAs inside cohesin rings, then cells must possess a mechanism that prevents opening of cohesin’s DNA exit gate. This mechanism involves acetylation of K112 and K113 within Smc3’s NBD by the Eco1, which is thought to prevent dissociation of kleisin’s N-terminal domain from Smc3 (21). We show here that Pds5 has roles not only in promoting de novo Smc3 acetylation during S phase, but also in preventing de-acetylation mediated by Hos1 during G2/M. It is well known that de novo acetylation is an essential process, but whether its subsequent protection from Hos1 is equally vital in maintaining cohesion is less well established. Our finding that deletion of HOS1 partially rescues the loss of cohesion caused by inactivating Pds5 during G2/M is consistent with the notion that persistent acetylation (because of Pds5’s protection) is crucial for keeping cohesin rings locked shut. Further proof of this concept will require identification of the HEAT repeats within Pds5 that are specifically concerned with acetylation protection and the creation of mutant specifically defective in this process.
Our finding that Pds5 promotes Smc3 acetylation may explain why Pds5 overexpression partially suppresses the temperature sensitivity of certain thermosensitive eco1 mutants in budding yeast (41). How does Pds5 perform this crucial function? One possibility is that Pds5 actually binds Eco1 and thereby brings the latter into close proximity with its targets within the Smc3 NBD. Consistent with this notion is the finding that S. pombe’s Eco1 ortholog Eso1 interacts with Pds5 in a yeast two-hybrid assay (23). However, neither the existence nor nature of this interaction has been elucidated by biochemical studies. An alternative is that Pds5 associates with cohesin in a manner that alters the conformation of Smc3’s NBD, exposing K112/K113 to Eco1 bound to proliferating cell nuclear antigen (42) at the replication fork. Both alternatives are consistent with our finding that Pds5 binds to a region of α-kleisin that is in close proximity to its N-terminal domain, which in turn is tightly associated with Smc3’s NBD.
We note that the proximity of the Smc3 NBD to the site of Pds5 binding on α-kleisin might also enable Pds5 to protect acetylation from Hos1. It is striking in this regard that the cohesin acetylation established and maintained by Pds5 subsequently affects the nature of Pds5’s association with chromosomal cohesin rings. Acetylation by Eco1 increases the residence time of Pds5’s association with chromosomal cohesin from approximate 90 s to over 10 min (21). The fact that Pds5 protects acetylated K112 and K113 from Hos1, whereas acetylation of K112 and K113 greatly stabilizes Pds5’s association with cohesin, suggests that Pds5 might directly bind to acetylated K112 and K113. Further investigation is needed to determine whether other proteins are implicated in this mechanism. Scc3 may be part of this process, as yeast two-hybrid data suggest that Hos1 might interact with Scc3 (43).
Our finding that Pds5 is essential for de novo acetylation of Smc3 in Saccharomyces cerevisiae is consistent with a recent report that it is similarly required in S. pombe (37), and this feature may therefore be a highly conserved aspect of Pds5. Importantly, the demonstration that Pds5 has a crucial role promoting acetylation mediated by Eco1 demonstrates once and for all that Pds5 is intimately involved in establishing cohesion as well as maintaining it, contrary to previous reports (41, 44).
Because acetylation is clearly an essential function, it is curious that Pds5 is not an essential gene in S. pombe. We suggest that this enigma arises because Pds5 in S. pombe, like its S. cerevisiae counterpart, also has a crucial role in releasing cohesin from chromatin. Because one of the main functions of acetylation is to block release, S. pombe cells lacking Pds5 no longer require acetylation, explaining why pds5Δ rescues the lethality caused by eso1Δ (23). The viability of pds5Δ mutants in S. pombe is therefore misleading in so far that it suggested Pds5 does not have any essential function. We predict that mutations that specifically affect Pds5’s ability to promote cohesin acetylation in S. pombe without affecting its releasing activity will prove to be lethal.
Our finding that Pds5 is required for maintaining Smc3 acetylation provides only a partial explanation for how this protein helps maintain sister chromatid cohesion during the G2/M phase in S. cerevisiae. The reason for this is that despite fully preventing Smc3 de-acetylation, hos1Δ only partially suppresses loss of cohesion because of inactivation of pds5-101 in the G2/M phase. Indeed, were protecting acetylated Smc3 from Hos1 the sole function during G2/M, then hos1Δ should suppress the lethality caused by pds5-101 but it does not do so. We conclude that Pds5 has yet another activity, namely to maintain cohesion via a mechanism not involving protection of acetylated Smc3. If, as envisaged by the ring model, cohesion is mediated solely by entrapment of sister DNAs inside cohesin rings, then this fourth Pds5 activity presumably also prevents ring opening. Pds5 might do so either by controlling dissociation of the Smc3/kleisin interface, a process known to be regulated by Pds5 (21), or by controlling dissociation of one of the ring’s other two interfaces. It may be relevant in this regard that there is a suggestion from FRET studies that Pds5 might interact with the hinge interface (39), although whether this has any functional impact is not known.
Conclusion
We have investigated the functions of Pds5 (3, 4) and showed that by binding to a small domain immediately adjacent and C-terminal to the N-terminal helices that bind Smc3 NBDs, it promotes cohesin’s release from chromatin in cooperation with Wapl and Scc3. Remarkably, Pds5 has two other functions that together mitigate the potentially lethal consequences of its participation in releasing activity. Pds5 promotes acetylation of Smc3 by Eco1 during S phase and prevents de-acetylation by Hos1 during G2 and M phases (SI Appendix, Fig. S13). Interestingly, Pds5 has yet another, as yet mysterious, function in stabilizing cohesion besides maintaining Smc3 acetylation.
Materials and Methods
All yeast strains are derivative of W303 (K699). The genotypes of yeast stains are listed in the SI Appendix. Detailed experimental procedures for coimmunoprecipitation, live-cell imaging, iFRAP analysis, and in vivo site-specific cross-linking are described in the SI Appendix. Chip-sequencing and chip-qPCR are performed using exponential growing cells follow published protocols (12, 21).
Supplementary Material
Acknowledgments
We thank Sylvia Panizza for the initial screen of Pds5 mutants and all members in Nasmyth's group for valuable discussions. We specially thank Linda Warfield and Stephen Hahn for a kind gift of plasmids encoding an amber codon tRNA and a modified tRNA synthetase (pLH157), and Masato Kanemaki for plasmids and strains of the auxin-inducible degron system. K.-L.C. is supported by the Croucher Foundation fellowship; T.G. is supported by the European Molecular Biology Organization fellowship. The work was funded by the Medical Research Council, the Wellcome Trust, and Cancer Research UK.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1306900110/-/DCSupplemental.
References
- 1.Haering CH, Löwe J, Hochwagen A, Nasmyth K. Molecular architecture of SMC proteins and the yeast cohesin complex. Mol Cell. 2002;9(4):773–788. doi: 10.1016/s1097-2765(02)00515-4. [DOI] [PubMed] [Google Scholar]
- 2.Haering CH, Farcas AM, Arumugam P, Metson J, Nasmyth K. The cohesin ring concatenates sister DNA molecules. Nature. 2008;454(7202):297–301. doi: 10.1038/nature07098. [DOI] [PubMed] [Google Scholar]
- 3.Panizza S, Tanaka T, Hochwagen A, Eisenhaber F, Nasmyth K. Pds5 cooperates with cohesin in maintaining sister chromatid cohesion. Curr Biol. 2000;10(24):1557–1564. doi: 10.1016/s0960-9822(00)00854-x. [DOI] [PubMed] [Google Scholar]
- 4.Hartman T, Stead K, Koshland D, Guacci V. Pds5p is an essential chromosomal protein required for both sister chromatid cohesion and condensation in Saccharomyces cerevisiae. J Cell Biol. 2000;151(3):613–626. doi: 10.1083/jcb.151.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rankin S, Ayad NG, Kirschner MW. Sororin, a substrate of the anaphase-promoting complex, is required for sister chromatid cohesion in vertebrates. Mol Cell. 2005;18(2):185–200. doi: 10.1016/j.molcel.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 6.Schmitz J, Watrin E, Lénárt P, Mechtler K, Peters JM. Sororin is required for stable binding of cohesin to chromatin and for sister chromatid cohesion in interphase. Curr Biol. 2007;17(7):630–636. doi: 10.1016/j.cub.2007.02.029. [DOI] [PubMed] [Google Scholar]
- 7.Kueng S, et al. Wapl controls the dynamic association of cohesin with chromatin. Cell. 2006;127(5):955–967. doi: 10.1016/j.cell.2006.09.040. [DOI] [PubMed] [Google Scholar]
- 8.Tóth A, et al. Yeast cohesin complex requires a conserved protein, Eco1p(Ctf7), to establish cohesion between sister chromatids during DNA replication. Genes Dev. 1999;13(3):320–333. doi: 10.1101/gad.13.3.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gruber S, et al. Evidence that loading of cohesin onto chromosomes involves opening of its SMC hinge. Cell. 2006;127(3):523–537. doi: 10.1016/j.cell.2006.08.048. [DOI] [PubMed] [Google Scholar]
- 10.Nasmyth K. Cohesin: A catenase with separate entry and exit gates? Nat Cell Biol. 2011;13(10):1170–1177. doi: 10.1038/ncb2349. [DOI] [PubMed] [Google Scholar]
- 11.Ciosk R, et al. Cohesin’s binding to chromosomes depends on a separate complex consisting of Scc2 and Scc4 proteins. Mol Cell. 2000;5(2):243–254. doi: 10.1016/s1097-2765(00)80420-7. [DOI] [PubMed] [Google Scholar]
- 12.Hu B, et al. ATP hydrolysis is required for relocating cohesin from sites occupied by its Scc2/4 loading complex. Curr Biol. 2011;21(1):12–24. doi: 10.1016/j.cub.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arumugam P, et al. ATP hydrolysis is required for cohesin’s association with chromosomes. Curr Biol. 2003;13(22):1941–1953. doi: 10.1016/j.cub.2003.10.036. [DOI] [PubMed] [Google Scholar]
- 14.Arumugam P, Nishino T, Haering CH, Gruber S, Nasmyth K. Cohesin’s ATPase activity is stimulated by the C-terminal Winged-Helix domain of its kleisin subunit. Curr Biol. 2006;16(20):1998–2008. doi: 10.1016/j.cub.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 15.Weitzer S, Lehane C, Uhlmann F. A model for ATP hydrolysis-dependent binding of cohesin to DNA. Curr Biol. 2003;13(22):1930–1940. doi: 10.1016/j.cub.2003.10.030. [DOI] [PubMed] [Google Scholar]
- 16.Uhlmann F, Wernic D, Poupart MA, Koonin EV, Nasmyth K. Cleavage of cohesin by the CD clan protease separin triggers anaphase in yeast. Cell. 2000;103(3):375–386. doi: 10.1016/s0092-8674(00)00130-6. [DOI] [PubMed] [Google Scholar]
- 17.Gandhi R, Gillespie PJ, Hirano T. Human Wapl is a cohesin-binding protein that promotes sister-chromatid resolution in mitotic prophase. Curr Biol. 2006;16(24):2406–2417. doi: 10.1016/j.cub.2006.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishiyama T, et al. Sororin mediates sister chromatid cohesion by antagonizing Wapl. Cell. 2010;143(5):737–749. doi: 10.1016/j.cell.2010.10.031. [DOI] [PubMed] [Google Scholar]
- 19.Losada A, Hirano M, Hirano T. Identification of Xenopus SMC protein complexes required for sister chromatid cohesion. Genes Dev. 1998;12(13):1986–1997. doi: 10.1101/gad.12.13.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waizenegger IC, Hauf S, Meinke A, Peters JM. Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell. 2000;103(3):399–410. doi: 10.1016/s0092-8674(00)00132-x. [DOI] [PubMed] [Google Scholar]
- 21.Chan KL, et al. Cohesin’s DNA exit gate is distinct from its entrance gate and is regulated by acetylation. Cell. 2012;150(5):961–974. doi: 10.1016/j.cell.2012.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rowland BD, et al. Building sister chromatid cohesion: smc3 acetylation counteracts an antiestablishment activity. Mol Cell. 2009;33(6):763–774. doi: 10.1016/j.molcel.2009.02.028. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka K, Hao Z, Kai M, Okayama H. Establishment and maintenance of sister chromatid cohesion in fission yeast by a unique mechanism. EMBO J. 2001;20(20):5779–5790. doi: 10.1093/emboj/20.20.5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Unal E, et al. A molecular determinant for the establishment of sister chromatid cohesion. Science. 2008;321(5888):566–569. doi: 10.1126/science.1157880. [DOI] [PubMed] [Google Scholar]
- 25.Rolef Ben-Shahar T, et al. Eco1-dependent cohesin acetylation during establishment of sister chromatid cohesion. Science. 2008;321(5888):563–566. doi: 10.1126/science.1157774. [DOI] [PubMed] [Google Scholar]
- 26.Ivanov D, et al. Eco1 is a novel acetyltransferase that can acetylate proteins involved in cohesion. Curr Biol. 2002;12(4):323–328. doi: 10.1016/s0960-9822(02)00681-4. [DOI] [PubMed] [Google Scholar]
- 27.Beckouët F, et al. An Smc3 acetylation cycle is essential for establishment of sister chromatid cohesion. Mol Cell. 2010;39(5):689–699. doi: 10.1016/j.molcel.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borges V, et al. Hos1 deacetylates Smc3 to close the cohesin acetylation cycle. Mol Cell. 2010;39(5):677–688. doi: 10.1016/j.molcel.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 29.Xiong B, Lu S, Gerton JL. Hos1 is a lysine deacetylase for the Smc3 subunit of cohesin. Curr Biol. 2010;20(18):1660–1665. doi: 10.1016/j.cub.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 30.Deardorff MA, et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature. 2012;489(7415):313–317. doi: 10.1038/nature11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen HT, Warfield L, Hahn S. The positions of TFIIF and TFIIE in the RNA polymerase II transcription preinitiation complex. Nat Struct Mol Biol. 2007;14(8):696–703. doi: 10.1038/nsmb1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yeh E, et al. Pericentric chromatin is organized into an intramolecular loop in mitosis. Curr Biol. 2008;18(2):81–90. doi: 10.1016/j.cub.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mishra A, et al. Both interaction surfaces within cohesin’s hinge domain are essential for its stable chromosomal association. Curr Biol. 2010;20(4):279–289. doi: 10.1016/j.cub.2009.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kulemzina I, et al. Cohesin rings devoid of Scc3 and Pds5 maintain their stable association with the DNA. PLoS Genet. 2012;8(8):e1002856. doi: 10.1371/journal.pgen.1002856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J, et al. Acetylation of Smc3 by Eco1 is required for S phase sister chromatid cohesion in both human and yeast. Mol Cell. 2008;31(1):143–151. doi: 10.1016/j.molcel.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 36.Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods. 2009;6(12):917–922. doi: 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- 37.Vaur S, Feytout A, Vazquez S, Javerzat JP. Pds5 promotes cohesin acetylation and stable cohesin-chromosome interaction. EMBO Rep. 2012;13(7):645–652. doi: 10.1038/embor.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gruber S, Haering CH, Nasmyth K. Chromosomal cohesin forms a ring. Cell. 2003;112(6):765–777. doi: 10.1016/s0092-8674(03)00162-4. [DOI] [PubMed] [Google Scholar]
- 39.Mc Intyre J, et al. In vivo analysis of cohesin architecture using FRET in the budding yeast Saccharomyces cerevisiae. EMBO J. 2007;26(16):3783–3793. doi: 10.1038/sj.emboj.7601793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shintomi K, Hirano T. Releasing cohesin from chromosome arms in early mitosis: Opposing actions of Wapl-Pds5 and Sgo1. Genes Dev. 2009;23(18):2224–2236. doi: 10.1101/gad.1844309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Noble D, et al. Intersection between the regulators of sister chromatid cohesion establishment and maintenance in budding yeast indicates a multi-step mechanism. Cell Cycle. 2006;5(21):2528–2536. doi: 10.4161/cc.5.21.3405. [DOI] [PubMed] [Google Scholar]
- 42.Moldovan GL, Pfander B, Jentsch S. PCNA controls establishment of sister chromatid cohesion during S phase. Mol Cell. 2006;23(5):723–732. doi: 10.1016/j.molcel.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 43.Yu H, et al. High-quality binary protein interaction map of the yeast interactome network. Science. 2008;322(5898):104–110. doi: 10.1126/science.1158684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stead K, et al. Pds5p regulates the maintenance of sister chromatid cohesion and is sumoylated to promote the dissolution of cohesion. J Cell Biol. 2003;163(4):729–741. doi: 10.1083/jcb.200305080. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.