

Abstract
Voltage-gated ion channels generate dynamic ionic currents that are vital to the physiological functions of many tissues. These proteins contain separate voltage-sensing domains, which detect changes in transmembrane voltage, and pore domains, which conduct ions. Coupling of voltage sensing and pore opening is critical to the channel function and has been modeled as a protein–protein interaction between the two domains. Here, we show that coupling in Kv7.1 channels requires the lipid phosphatidylinositol 4,5-bisphosphate (PIP2). We found that voltage-sensing domain activation failed to open the pore in the absence of PIP2. This result is due to loss of coupling because PIP2 was also required for pore opening to affect voltage-sensing domain activation. We identified a critical site for PIP2-dependent coupling at the interface between the voltage-sensing domain and the pore domain. This site is actually a conserved lipid-binding site among different K+ channels, suggesting that lipids play an important role in coupling in many ion channels.
Keywords: channelopathy, electromechanical coupling, KCNQ, voltage channel, phosphoinositide
Voltage-gated ion channels are integral membrane proteins that sense membrane voltage and respond by opening or closing a transmembrane pore. Ionic currents carried by voltage-gated ion channels control contraction in muscle, encode information in the nervous system, and trigger secretion in neurohormonal tissues. Voltage-gated ion channels contain four voltage-sensing domains (VSDs) and a central pore domain (PD) that are structurally distinct (1, 2). In voltage-gated potassium (Kv) channels, the first four transmembrane segments (S1–S4) of each α-subunit forms a VSD. In response to changes in transmembrane voltage, the VSD undergoes a conformational change, called activation, during which membrane depolarization moves the S4 segment outward (3). The PD is formed by the last two transmembrane segments (S5, S6) from four α-subunits and undergoes a mainly voltage-independent conformational change during which the intracellular ends of the S6 segments bend, opening the ionic pore (4, 5). Interestingly, the PD and VSD can exist in pore-only (6) and voltage sensor-only proteins, respectively, where they function independently (7, 8). Confining sensitivity to voltage, or to other stimuli, within a domain diversifies the ion channel properties that can be achieved by partnering different pore and sensor domains. However, this modular architecture also raises a fundamental question as to how VSD activation is transmitted to the PD. Previous studies of this coupling process have revealed the importance of direct protein–protein interactions at the VSD–PD interface (9–13); however, the possible role of membrane lipids in VSD–PD coupling remains undetermined.
The membrane lipid phosphatidylinositol 4,5-bisphosphate (PIP2) modulates the activity of many ion channels, including some voltage-gated channels (14). Notably, all members of the Kv7 family (Kv7.1–Kv7.5), which play important physiological roles in the cardiac (15) or the nervous (16) systems, require PIP2 to be opened by membrane depolarization (17). In fact, silencing of Kv7 channels by PIP2 depletion is known to underlie the modulation of neuronal excitability by hormone receptor signaling (16). Despite this relevance to human health, it remains unclear where PIP2 binds and why PIP2 binding is required for voltage-dependent gating. We sought to determine, in Kv7.1 channels, whether PIP2 is required for any of three fundamental gating processes: activation of the VSD by membrane depolarization, coupling of VSD activation and PD opening, or opening of the PD to allow passage of ions.
Results
PIP2 Is Not Required for VSD Activation.
Using voltage-clamp fluorometry on channels expressed in Xenopus oocytes, we determined the effect of PIP2 depletion on VSD activation and PD opening simultaneously. In voltage-clamp fluorometry, fluorescent labeling of the S3–S4 linker generates measurable changes in fluorescence intensity that are correlated with S4 movement during VSD activation; meanwhile, measurement of the whole-cell ionic current detects PD opening (Fig. 1A) (18). As reported previously, we used pseudo–wild-type (psWT)—C214A/G219C/C331A—Kv7.1 channels to avoid nonspecific labeling of native Cys214 and Cys331 and labeled position G219C with Alexa 488 C5 maleimide (19). To deplete PIP2, we expressed the voltage-sensing phosphatase from Ciona intestinalis (CiVSP), a lipid phosphatase that rapidly dephosphorylates PIP2 upon membrane depolarization (20, 21). When we activated CiVSP with a train of six depolarizing (+60-mV) pulses (Fig. 1B), the psWT channel current amplitude was dynamically and severely decreased within the first pulse to only 18 ± 2% that of oocytes expressing psWT channels alone and recorded on the same day. Application of five subsequent pulses produced yet further inhibition (−CiVSP: I6/I1 = 88 ± 1%; +CiVSP: I6/I1 = 31 ± 2%; P < 0.0001) leaving only 4 ± 3% of the current amplitude in the same day controls (Fig. 1C). In clear contrast, CiVSP activation did not decrease the magnitude of the fluorescence signal change (ΔF/F) during the first pulse or upon subsequent pulses (−CiVSP: ΔF6/ΔF1 = 88 ± 1%; +CiVSP: ΔF6/ΔF1 = 86 ± 1%; P = 0.246; Fig. 1C), indicating that VSD activation still occurs after PIP2 depletion. As an alternative method to voltage-clamp fluorometry we detected VSD activation using 2-sulfo-natoethyl methanethiosulfonate (MTSES) modification of I230C in S4, which is only accessible to MTSES when the VSD is activated (3, 22) (Figs. S1–S3). This experiment confirmed that VSD activation occurs in the absence of PIP2. After PIP2 depletion, we found that the steady-state voltage dependence of VSD activation, reflected in the fluorescence–voltage (F–V) relationship, was unchanged (Fig. S4 A–H), whereas the ionic currents were inhibited. Of note, the kinetics of the fluorescence signal changes did show a mild dependence on PIP2 (Fig. S4 E, F, and I). Altogether, these results demonstrate that PIP2 is not required for detection of membrane voltage within the VSD.
Fig. 1.

PIP2 depletion suppresses pore opening but not VSD activation. (A) Cartoon of channel motions measured by voltage-clamp fluorometry. (B) Voltage protocol. (C) Ionic currents (Upper) and relative fluorescent intensity changes (ΔF/F; Lower) from cells expressing psWT Kv7.1 alone (black) or with CiVSP (blue). Example raw data traces (Left, Center). Average value at the end of each 60-mV pulse normalized to that at the first pulse (Right). Error bars represent SEM and n = 3–20 for all figures.
PIP2 Is Required for Coupling.
We tested whether PIP2 is required for coupling. Coupling can be quantified by measuring the effect of VSD activation on PD opening or by measuring how PD opening affects VSD activation (23, 24). We chose the latter approach because we could still measure VSD activation after PIP2 depletion when PD opening became undetectable (Fig. 1). To promote pore opening, we introduced the mutation L353K into the S6 gate (25). Leu353 is located near the putative bundle crossing, and previous studies have shown that introduction of a positive or negative amino acid at this position results in a channel that remains open (25) as if the mutual electrostatic repulsion among the introduced charges destabilizes the closed state of the PD. psWT/L353K channels conducted instantaneous current at every voltage we applied (Fig. 2 A and C), and these currents were not inhibited when we expressed and activated CiVSP with a train of six depolarizing pulses (+CiVSP: I6/I1 = 102 ± 2.4%; −CiVSP: I6/I1 = 99 ± 0.7%; P = 0.185; Fig. 2A). Furthermore, psWT/L353K currents reversed near the K+ equilibrium (Fig. 2C) and were reduced when we applied the Kv7.1 pore blocker chromanol 293B (26) (Fig. 2B), providing evidence that the observed constitutive currents were indeed conducted by expressed psWT/L353K channels. By comparing the VSD activation of psWT and locked open psWT/L353K channels, we were able to detect VSD–PD coupling directly as a mild, but statistically significant (P < 0.0001), leftward shift in the F–V relationship (psWT/L353K: V1/2 = −72.1 ± 1.5 mV; psWT: V1/2 = −57.1 ± 0.8 mV; Fig. 2C, Left). This shift is consistent with the positive coupling between VSD activation and PD opening, i.e., less energy is required to activate the VSD if the PD is open. This shift is similar in magnitude to that observed in other channels locked open using metal bridging (24). When we depleted PIP2, using CiVSP, the psWT/L353K channels remained open, but their F–V relationship no longer differed (P = 0.117) from psWT channels (psWT + CiVSP: V1/2 = −60.4 ± 0.6 mV; psWT/L353K + CiVSP: V1/2 = −58.5 ± 1.1 mV; Fig. 2C, Right). This result indicates that PIP2 is required for PD opening (by L353K) to promote VSD activation. We tested whether other mutations also require PIP2 to affect VSD activation and identified two such mutations (S349A, G350A) (Fig. S5). Ser349, Gly350, and Leu353 are located in the S6 gate, and homology modeling predicted that they do not interact with the VSD or the S4–S5 linker (Fig. S5E). This is consistent with the idea that these mutations directly alter PD opening and indirectly affect VSD activation through PIP2-dependent coupling. Thus, our experiments show that PIP2 is required both for VSD activation to cause PD opening (Fig. 1) and for PD opening to affect the activation of the VSD (Fig. 2). These findings show that a membrane lipid can be required for the functional coupling between the VSD and the PD.
Fig. 2.

VSD–PD coupling requires PIP2. Voltage-clamp fluorometry of psWT (black), psWT + CiVSP (blue), psWT/L353K (magenta), and psWT/L353K + CiVSP (orange). (A) Raw currents and normalized current amplitude as in Fig. 1. (B) Inhibition of current by 200 μM Chromanol 293B. (C) Steady-state current–voltage (Upper) and fluorescence–voltage (F–V) (Lower) relationships. For details of voltage protocol, see Fig. S4.
We devised an equilibrium model of Kv7.1 gating by PIP2 and voltage in which bound PIP2 couples VSD and PD so that VSD activation enhances PD opening by a factor θ, and vice versa (Fig. 3A, Fig. S6, and SI Materials and Methods for full model derivation and fitting parameters). Our data regarding the voltage and PIP2 dependence of the ionic current and VSD activation were fit well with this model (Fig. 3 B and C). The model does not include any direct effects of PIP2 on the opening of the PD or the activation of the VSD, thus demonstrating that the requirement of PIP2 for VSD–PD coupling alone is sufficient to quantitatively recapitulate the channel behavior. However, the possibility that PIP2 may also have a minor direct effect on the opening of the PD cannot be excluded. Importantly, as observed in our experiments, the model simulation predicted that PIP2 depletion would not generate a measurable shift in the psWT F–V. This occurs because the intrinsic open probability of the PD is low and the coupling is weak, as indicated by several recent papers (27, 28).
Fig. 3.

Model of PIP2-mediated coupling predicts observed gating behavior. (A) An equilibrium model of PIP2- and voltage-dependent gating (Fig. S6 and SI Materials and Methods) used to fit (dotted lines) (B) the PIP2-response curve of WT channels (data from ref. 32) and (C) the conductance–voltage (G–V; Upper) and F–V relationships (Lower) of psWT (black), psWT + CiVSP (blue), psWT/L353K (magenta), and psWT/L353K + CiVSP (orange). Symbols represent experimental data. In the model, θ represents the coupling, whereas Kv and Kg describe the voltage sensor activation and pore opening, respectively, in the absence of PIP2.
PIP2 Binds at the VSD–PD Interface.
A critical step to understanding how PIP2 binding mediates VSD–PD coupling is to identify its binding site. The most intuitive location is at the VSD–PD interface, a region that has already been shown to contain protein–protein interactions important for Kv channel coupling (9–13). PIP2 binding sites in proteins generally contain multiple basic residues located near the intracellular leaflet of the membrane to interact electrostatically with the negatively charged PIP2 head group (29). Accordingly, using a Kv7.1 homology model, we identified 16 basic residues that are located near VSD–PD interface (30). These residues are highly conserved among the Kv7 channels, which require PIP2 for voltage-dependent gating (17), but are poorly conserved among other Kv channels that do not require PIP2 (31) (Fig. 4A). Using site-directed mutagenesis, we individually neutralized each of these basic residues in WT Kv7.1 and measured the effect on the expressed current amplitude, using two-electrode voltage clamp. Because the endogenous level of PIP2 in the oocyte membrane is within the sensitive range of Kv7.1 (32), we expected that a mutation that disrupts PIP2 binding would decrease the current. Indeed, we found that eight mutations (R190Q, R195Q, H258N, R259Q, K354N, R360Q, H363N, R366Q) severely decreased (>50% reduction; Fig. 4 B and C, blue) the whole-cell current amplitude, and four mutations (R192Q, R243Q, K358N, K362N) had a milder reduction (<50% reduction; Fig. 4 B and C, green). However, four neutralizing mutations (R181Q, K183N, K196N, R249Q; Fig. 4 B and C, red) and three additional charge-reversing mutations (R249E, K358E, R360E; Fig. S7) actually increased the current amplitude. Importantly, we detected robust surface expression of the loss-of-current mutants by Western blot analysis of proteins labeled by extracellular biotin (Fig. 4D). This result shows that loss of current expression is not due to reduced protein synthesis or membrane trafficking. Furthermore, we detected fluorescent signal changes using voltage-clamp fluorometry for all of the loss-of-current mutations (Fig. 4E), indicating that these mutant channels not only expressed to the membrane but also retained VSD activation. Therefore, the loss-of-current mutations affected channel gating by decreasing coupling, consistent with decreased PIP2 binding, or by decreasing PD opening.
Fig. 4.

Mutations at the VSD–PD interface alter channel function. (A) Sequence alignment of Kv channels S2–S3, S4–S5 linkers, and proximal C terminus. Highlighted residues were individually neutralized in WT Kv7.1. (B) Raw current at −100 to +40 mV. (C) Current amplitude (at +20 mV) normalized to WT (I/Iwt). Color code: blue, I/Iwt < 0.5; green, 0.5 < I/Iwt < 1; red, I/Iwt > 1. (D) Western blot of biotinylated membrane proteins. CTL Mem, CTL Lys: membrane fraction and whole-cell lysate from uninjected cells. (E) ΔF/F (−80 to +60 mV).
We tested whether our mutations affected current amplitude by changing the apparent PIP2 affinity. We measured the time course of the current rundown that occurs spontaneously as phosphoinositides are lost from excised membrane patches. Previous studies have shown that currents from channels with a higher apparent affinity for PIP2 run down slower than those with a lower apparent affinity (32). Although we could not record the current rundown for the severe loss-of-current mutations, we found that the effects of other mutations (K358N, K362N, R181Q, K183N, R249Q, R249E) on the time course of rundown after patch excision were correlated with their effects on expressed current amplitude. That is, relative to WT channels, the rundown was faster for the mild loss-of-current mutations and slower for the gain-of-current mutations (Fig. 5). These results further support the hypothesis that the mutations of conserved basic residues at the VSD–PD interface affect a PIP2-mediated process that is required for channel function.
Fig. 5.
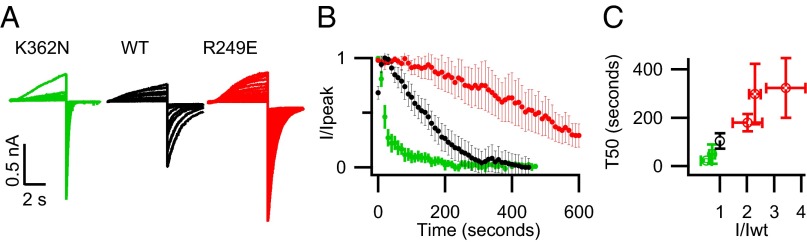
Mutations at the VSD–PD interface alter apparent PIP2 affinity. (A) Raw currents in response to repeated +80-mV pulses following excision of inside-out membrane patches. (B) Mean normalized tail current amplitude versus time following patch excision for K362N (green), WT (black), and R249E (red). (C) Time to 50% rundown versus I/Iwt. From Left to Right: K362N, K358N, WT, R249Q, K183N, R249E. All experiments were done in the presence of the KCNE1 subunit to slow current rundown (32) for better resolution of rundown time course.
We directly quantified the strength of VSD–PD coupling for several of the mutations (R192Q, R195Q, K196E, K358N, K358E, R360Q, R360E, K362N, H363N, R366Q) by measuring the F–V shift caused by locking the PD open with L353K. We found that the mutations influenced VSD–PD coupling in a manner that was correlated with their effect on the expressed current amplitude: i.e., the loss-of-current mutations decreased the magnitude of the F–V shift, whereas the gain-of-current mutations increased the F–V shift (Fig. 6). Strikingly, our equilibrium model predicted this relationship when we simply varied the binding constant for PIP2 (KPIP2) (Fig. 6D, black line). Taken together, our results suggest that these mutations disrupt VSD–PD coupling and decrease channel current by directly affecting PIP2 binding.
Fig. 6.

Mutations at the VSD–PD interface alter coupling. Colors as in Fig. 4C. (A–C) F–V relationships of psWT/mutation (solid, filled) and psWT/L353K/mutation (dotted, open). (D) The change in z*V1/2 between psWT/mutation and psWT/L353K/mutation versus I/Iwt. From Left to Right: H363N, R195Q, R360Q, R366Q, K362N, R192Q, K358N, WT, K3358E, R360E, K196N. z*V1/2 is a measure of the energy required to activate the VSD, where z and V1/2 were obtained by fitting the Boltzmann equation (see Materials and Methods). The black line represents simulated data generated by varying the PIP2 equilibrium constant (KPIP2) in the model shown in Fig. 3A.
We mapped our data onto a Kv7.1 homology model (30) and found a cluster of severe loss-of-current residues at the VSD–PD interface that includes Arg190 and Arg195 in the S2–S3 linker, His258 and Arg259 in the S4–S5 linker, and Lys354 in the proximal C terminus (Fig. 7, blue). In our experiments, these mutations mimicked the effects of PIP2 depletion on psWT channels: they severely inhibited the ionic current, did not prevent VSD activation, and greatly diminished the VSD–PD coupling. Therefore, this cluster of basic residues constitutes a critical interaction site for PIP2-mediated coupling. Remarkably, when we aligned the crystal structure of the voltage-independent Kir2.2 channel bound to PIP2 (33) and the homology model of the activated–open state of Kv7.1, the PIP2 head group from the Kir2.2 structure was centered right in the cluster (Fig. 7 A and B, and Fig. S8). Furthermore, the high-impact residues His258, Arg259, and Lys354 corresponded spatially to the PIP2 binding residues on the slide helix and the C linker of the Kir2.2 structure (Fig. S8C). These findings provide compelling evidence that our identified site is actually a PIP2 binding site that is conserved among voltage-dependent and voltage-independent K+ channels. The gain-of-current residues surround the PIP2 head group when we align the Kir2.2 crystal structure with the resting–closed state model of Kv7.1 (Fig. 7C). This result suggests that neutralizing mutations at these positions increase the channel current by disrupting PIP2 interactions that occur in the closed state. This may increase the channel current by destabilizing the closed state, or by making PIP2 more available to bind in the pocket present in the activated–open state. The mild loss-of-current residues are located outside the conserved binding site (Fig. 7A) and likely indirectly affect PIP2 binding at the high-impact cluster.
Fig. 7.

Structural alignment identifies a conserved PIP2 binding site. (A–C) Homology models of the Kv7.1 activated–open (A and B) and resting–closed (C) states (30) were aligned with the Kir2.2-PIP2 crystal structure (33) (Fig. S8) to position the PIP2 head group (magenta) within the models. (A) Bottom view of the activated–open model with data from Fig. 4C mapped. (B and C) Side view of one subunit from the activated–open model (B) or resting–closed model (C) with putative PIP2 head group binding residues (blue: Arg190, Arg195, His258, Arg259, Lys354) and gain-of-current residues (red: Arg181, Lys183, Lys196, Arg249) shown.
Discussion
Coupling is a critical process for ion channel function by which detection of a physiological stimulus within a sensor domain is transmitted to the PD to modulate channel opening. Our study identifies PIP2 as a required factor for coupling between the VSD and PD in Kv7.1, thus expanding the current model of VSD–PD coupling to include lipid molecules. We find that PIP2 is required for the communication of conformational energy from the VSD to the PD and from the PD to the VSD. Consistent with this finding, we identify a PIP2 binding site at the interface between the VSD and the PD. We expect that PIP2 binding resolves the electrostatic repulsion between the two domains, caused by the basic residues within the site, and holds the VSD in close proximity to the PD to potentiate the coupling between the two domains. The details of how PIP2 binding potentiates coupling and precisely how VSD activation is transmitted to the pore will require further studies.
Various ion channels, equipped with different sensor domains, require PIP2 to open in response to stimulation. Previous studies have posited that PIP2 stabilizes the open state of the PD, a conclusion that makes sense considering that the PD is the common element among voltage-dependent and voltage-independent K+ channels that required PIP2. However, another common feature is that coupling between the conserved pore and modular sensor domains is necessary for channel function. In this study, we found that PIP2 is required for VSD–PD coupling in the voltage-dependent Kv7.1 channel. Structural studies have suggested that PIP2 couples the conformation of the cytosolic domain to that of the PD in voltage-independent Kir channels (33, 34). Furthermore, PIP2 has been shown to potentiate the functional coupling in CiVSP between the VSD activation and the enzymatic activity of the cystosolic phosphatase domain (35). Therefore, we propose a general model for PIP2 regulation of ion channels, and of some nonchannel proteins, whereby PIP2 binding at the interface between the modular sensor and effector domains mediates functional coupling. It should be noted that not all Kv channels are PIP2 dependent (31). Interestingly, the crystal structure of the voltage-dependent Kv1.2–2.1 chimera channel, solved in the presence of phospholipids, reveals an anionic phospholipid bound at the VSD–PD interface (36) in a position very close to our identified Kv7.1 PIP2 interaction site (Fig. S8D). This suggests that other anionic lipids mediate VSD–PD coupling in Kv channels that do not require PIP2.
We were able to fit our experimental data with a simple model consisting of one VSD, one PD, and one PIP2 binding site. Initially, this was a surprising result considering that the channel is tetrameric and contains four VSDs, one PD, and, presumably, four PIP2 sites as seen in the Kir-PIP2 structures (33, 34). However, our data demonstrate that PIP2 is required to link the activation of the VSD to the PD; therefore, if fewer than four PIP2 binding sites are occupied, the PD will not be functionally coupled to all four VSDs. Indeed, the activation of Kv7.1 is far from saturated by the endogenous level of PIP2 available in the oocyte membrane (32) such that most of the channel population may have only one PIP2 bound and one VSD coupled. In addition, a recent study suggested that the voltage-dependent activation of Kv7.1 can be explained by an allosteric model where activation of only one VSD can open the channel (27). Thus, although our simple model is structurally inaccurate, it is a reasonable model of the channel gating at our experimental conditions and recapitulates the important features of the data.
PIP2-mediated coupling in Kv7.1 has direct implications for human pathophysiology. In the heart, Kv7.1 subunits coassemble with KCNE1 accessory subunits to generate the slow delayed rectifier current (IKs) (37, 38) that regulates the duration of the cardiac action potential. Inherited loss-of-function mutations of Kv7.1 and KCNE1 are associated with long QT syndrome (LQT), in which the ventricular action potential duration is prolonged, resulting in a high risk of ventricular arrhythmias and sudden death. Eleven previously reported LQT-associated mutations (39–46) affect basic residues in the PIP2 pocket (Table S1). For these mutations, loss of VSD–PD coupling may compromise the IKs channel function and create a substrate for cardiac arrhythmias. Our results demonstrate that modifiers of VSD activation will not rescue channel function for these mutations. Furthermore, drugs that force the PD open will abolish the voltage and time dependence of the IKs current that is critical for the timing of the action potential (47). However, if a drug could target the PIP2-dependent coupling, it would amplify the current without losing these physiologically important characteristics. This is an exciting rationale for drug design that may be applicable to other PIP2-related ion channel diseases (48).
Materials and Methods
Mutagenesis.
Site-directed mutations were introduced using overlap extension and high-fidelity PCR. DNA sequencing confirmed each mutation. RNA was made by in vitro transcription using the mMessage mMachine T7 polymerase kit (Applied Biosystems).
Channel Expression.
A total of 9.2 ng of channel cRNA was injected, using Nanoject (Drummond), into each of stage V–VI, defolliculated oocytes from Xenopus laevis. For expression of CiVSP, 2.3 ng of CiVSP RNA was injected simultaneously. The cells were incubated at 18 °C for 4–7 d for robust expression in ND96 solution (in mM: 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 5 Hepes, 0.3 K2EDTA).
Electrophysiology
Two-electode voltage clamp.
Whole-cell currents were recorded from oocytes bathed in ND96 solution using a CA-B amplifier (Dagan) in two-electrode voltage-clamp mode. Microelectrodes were pulled to a resistance of 0.3–3 ΜΩ and filled with 3 mM KCl. Signals were sampled at 1 kHz using the Patchmaster acquisition software (HEKA). The holding potential was set to −80 mV throughout, unless otherwise specified.
Voltage-clamp fluorometry.
For voltage-clamp fluorometry, oocytes were labeled on ice for 45 min in 10 μM Alexa 488 C5 maleimide (Life Technologies) in high-potassium depolarizing solution (in mM: 98 KCl, 1.8 CaCl2, 1 MgCl2, 5 Hepes, pH 7.6). The cells were washed with ND96 and kept on ice until recording. Fluorescent signals were recorded simultaneously with the whole-cell (TEVC) currents in ND96 solution, using a DLMFS (Leica) upright microscope through a FITC filter cube (Leica). Light from a standard 100-W halogen bulb was focused onto the animal pole of the oocyte and emission from the cube was focused on a P20A photodiode. The current from the photodiode was amplified using an EPC10 patch amplified (HEKA), low-pass filtered at 200 Hz, and sampled at 1 kHz using Patchmaster (HEKA).
Patch clamp.
Inside-out membrane patches were formed using patch electrodes pulled to 0.5–1 ΜΩ and filled with pipette solution (in mM: 140 KMeSO3, 20 Hepes, 2 KCl, 2 MgCl2, pH 7.2) and excised into the bathing solution (in mM: 140 KMeSO3, 20 Hepes, 2 KCl, 5 EGTA, 1.5 mM MgATP, pH 7.2). Macroscopic currents were recorded, at room temperature, using an Axopatch 200-B amplifier (Axon Instruments) driven by the Pulse (HEKA) acquisition software. Current were digitized at 1 kHz.
All recordings were made in room temperature (20–22 °C).
Data Analysis.
The baseline fluorescence was fit with a line during the 2 s at the −80-mV holding potential that preceded each test pulse. This linear baseline approximation was extrapolated to the duration of the pulse and ΔF/F was calculated as (F(t) − Fbaseline(t))/Fbaseline(t), where F(t) is the fluorescent intensity at time t (in arbitrary units) and Fbaseline(t) is the extrapolated baseline value at time t.
The Boltzmann equation was used to fit the fluorescence–voltage relationships: Normalized ΔF(V) = PVa(V) = 1/(1 + exp(−z*F*(V − V1/2)/RT), where PVa is the voltage-dependent probability of the voltage sensor assuming the activated state, V is the test voltage (in volts), V1/2 is the voltage of half-maximal voltage sensor activation, z is the number of elementary charges translocated across the membrane upon VSD activation, R is the gas constant (in joules per kelvin per mole), and F is the faraday constant (in coulombs per mole).
Statistical comparison was done using a Student t test. Errors represent SEM throughout.
Chemical Modification.
MTSES (Toronto Research Chemicals) was dissolved in DMSO at 100 mM, aliquoted, and frozen immediately. Aliquots were thawed just before use and added directly to bath solution in a bolus to achieve the desired final concentration.
Biotinylation and Western Blot.
Membrane expression was detected through biotinylation and Western blot (49). Cell surface proteins of intact oocytes were labeled with 1 mg/mL sulfo-NHS-SS-biotin (Thermo Scientific). The cells were washed, homogenized, and incubated with NeutrAvidin beads (Thermo Scientific) to pull down biotin-labeled proteins. The pulled-down proteins were probed via Western blot using a Kv7.1 antibody (Santa Cruz Biotechnology) or a Gβ antibody to test for labeling of cytosolic proteins.
Supplementary Material
Acknowledgments
We thank Dr. Y. Okamura (Osaka University) for the CiVSP clone and Dr. S. Goldstein (Brandeis University) for the KCNQ1 clone. We also thank Drs. J. Nerbonne and A. Federman for review of the manuscript, and A. Krumholz for many excellent discussions throughout this project. This study was funded by an Established Investigator Award 0440066N from the American Heart Association, Grants R01-HL70393 and R01-NS060706 from National Institutes of Health (to J.C.), Burroughs Welcome Fund 1010299 (to J.R.S.), and American Heart Association Predoctoral Fellowship 11PRE5720009 (to M.A.Z.). J.C. is the Professor of Biomedical Engineering on the Spencer T. Olin Endowment.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1305167110/-/DCSupplemental.
References
- 1.Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309(5736):897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 2.Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475(7356):353–358. doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Larsson HP, Baker OS, Dhillon DS, Isacoff EY. Transmembrane movement of the shaker K+ channel S4. Neuron. 1996;16(2):387–397. doi: 10.1016/s0896-6273(00)80056-2. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y, Holmgren M, Jurman ME, Yellen G. Gated access to the pore of a voltage-dependent K+ channel. Neuron. 1997;19(1):175–184. doi: 10.1016/s0896-6273(00)80357-8. [DOI] [PubMed] [Google Scholar]
- 5.Jiang Y, et al. The open pore conformation of potassium channels. Nature. 2002;417(6888):523–526. doi: 10.1038/417523a. [DOI] [PubMed] [Google Scholar]
- 6. Shaya D, et al. (2011) Voltage-gated sodium channel (NaV) protein dissection creates a set of functional pore-only proteins. Proc Natl Acad Sci USA 108(30):12313–12318. [DOI] [PMC free article] [PubMed]
- 7.Sasaki M, Takagi M, Okamura Y. A voltage sensor-domain protein is a voltage-gated proton channel. Science. 2006;312(5773):589–592. doi: 10.1126/science.1122352. [DOI] [PubMed] [Google Scholar]
- 8.Ramsey IS, Moran MM, Chong JA, Clapham DE. A voltage-gated proton-selective channel lacking the pore domain. Nature. 2006;440(7088):1213–1216. doi: 10.1038/nature04700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ledwell JL, Aldrich RW (1999) Mutations in the S4 region isolate the final voltage-dependent cooperative step in potassium channel activation. J Gen Physiol 113(3):389–414. [DOI] [PMC free article] [PubMed]
- 10.Lu Z, Klem AM, Ramu Y. Ion conduction pore is conserved among potassium channels. Nature. 2001;413(6858):809–813. doi: 10.1038/35101535. [DOI] [PubMed] [Google Scholar]
- 11.Long SB, Campbell EB, Mackinnon R. Voltage sensor of Kv1.2: Structural basis of electromechanical coupling. Science. 2005;309(5736):903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- 12.Soler-Llavina GJ, Chang T-H, Swartz KJ. Functional interactions at the interface between voltage-sensing and pore domains in the Shaker Kv channel. Neuron. 2006;52(4):623–634. doi: 10.1016/j.neuron.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 13. Grabe M, Lai HC, Jain M, Nung Jan Y, Yeh Jan L (2007) Structure prediction for the down state of a potassium channel voltage sensor. Nature 445(7127):550–553. [DOI] [PubMed]
- 14.Suh B-C, Hille B. PIP2 is a necessary cofactor for ion channel function: How and why? Annu Rev Biophys. 2008;37:175–195. doi: 10.1146/annurev.biophys.37.032807.125859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Q, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12(1):17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- 16.Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6(11):850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- 17. Zhang H, et al. (2003) PIP2 activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 37(6):963–975. [DOI] [PubMed]
- 18.Mannuzzu LM, Moronne MM, Isacoff EY. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 1996;271(5246):213–216. doi: 10.1126/science.271.5246.213. [DOI] [PubMed] [Google Scholar]
- 19. Osteen JD, et al. (2010) KCNE1 alters the voltage sensor movements necessary to open the KCNQ1 channel gate. Proc Natl Acad Sci USA 107(52):22710–22715. [DOI] [PMC free article] [PubMed]
- 20.Murata Y, Iwasaki H, Sasaki M, Inaba K, Okamura Y. Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature. 2005;435(7046):1239–1243. doi: 10.1038/nature03650. [DOI] [PubMed] [Google Scholar]
- 21. Iwasaki H, et al. (2008) A voltage-sensing phosphatase, Ci-VSP, which shares sequence identity with PTEN, dephosphorylates phosphatidylinositol 4,5-bisphosphate. Proc Natl Acad Sci USA 105(23):7970–7975. [DOI] [PMC free article] [PubMed]
- 22. Rocheleau JM, Kobertz WR (2008) KCNE peptides differently affect voltage sensor equilibrium and equilibration rates in KCNQ1 K+ channels. J Gen Physiol 131(1):59–68. [DOI] [PMC free article] [PubMed]
- 23. Arcisio-Miranda M, Muroi Y, Chowdhury S, Chanda B (2010) Molecular mechanism of allosteric modification of voltage-dependent sodium channels by local anesthetics. J Gen Physiol 136(5):541–554. [DOI] [PMC free article] [PubMed]
- 24. Ryu S, Yellen G (2012) Charge movement in gating-locked HCN channels reveals weak coupling of voltage sensors and gate. J Gen Physiol 140(5):469–479. [DOI] [PMC free article] [PubMed]
- 25. Boulet IR, Labro AJ, Raes AL, Snyders DJ (2007) Role of the S6 C-terminus in KCNQ1 channel gating. J Physiol 585(Pt 2):325–337. [DOI] [PMC free article] [PubMed]
- 26.Lerche C, et al. Chromanol 293B binding in KCNQ1 (Kv7.1) channels involves electrostatic interactions with a potassium ion in the selectivity filter. Mol Pharmacol. 2007;71(6):1503–1511. doi: 10.1124/mol.106.031682. [DOI] [PubMed] [Google Scholar]
- 27.Osteen JD, et al. Allosteric gating mechanism underlies the flexible gating of KCNQ1 potassium channels. Proc Natl Acad Sci USA. 2012;109(18):7103–7108. doi: 10.1073/pnas.1201582109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruscic KJ, et al. IKs channels open slowly because KCNE1 accessory subunits slow the movement of S4 voltage sensors in KCNQ1 pore-forming subunits. Proc Natl Acad Sci USA. 2013;110(7):E559–E566. doi: 10.1073/pnas.1222616110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rosenhouse-Dantsker A, Logothetis DE (2007) Molecular characteristics of phosphoinositide binding. Pflugers Arch 455(1):45–53. [DOI] [PubMed]
- 30.Smith J, Vanoye CG, George AL, Jr, Meiler AL, Sanders CR. Structural models for the KCNQ1 voltage-gated potassium channel. Biochemistry. 2007;46(49):14141–14152. doi: 10.1021/bi701597s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kruse M, Hammond GRV, Hille B (2012) Regulation of voltage-gated potassium channels by PI(4,5)P2. J Gen Physiol 140(2):189–205. [DOI] [PMC free article] [PubMed]
- 32.Li Y, et al. KCNE1 enhances phosphatidylinositol 4,5-bisphosphate (PIP2) sensitivity of IKs to modulate channel activity. Proc Natl Acad Sci USA. 2011;108(22):9095–9100. doi: 10.1073/pnas.1100872108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hansen SB, Tao X, MacKinnon R. Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature. 2011;477(7365):495–498. doi: 10.1038/nature10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whorton MR, MacKinnon R. Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell. 2011;147(1):199–208. doi: 10.1016/j.cell.2011.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kohout SC, et al. (2010) Electrochemical coupling in the voltage-dependent phosphatase Ci-VSP. Nat Chem Biol 6(5):369–375. [DOI] [PMC free article] [PubMed]
- 36.Long SB, Tao X, Campbell EB, MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450(7168):376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 37.Sanguinetti MC, et al. Coassembly of KVLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature. 1996;384(6604):80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 38.Barhanin J, et al. KVLQT1 and lsK (minK) proteins associate to form the IKs cardiac potassium current. Nature. 1996;384(6604):78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 39.Chouabe C, et al. Novel mutations in KvLQT1 that affect Iks activation through interactions with Isk. Cardiovasc Res. 2000;45(4):971–980. doi: 10.1016/s0008-6363(99)00411-3. [DOI] [PubMed] [Google Scholar]
- 40.Napolitano C, et al. Genetic testing in the long QT syndrome: Development and validation of an efficient approach to genotyping in clinical practice. JAMA. 2005;294(23):2975–2980. doi: 10.1001/jama.294.23.2975. [DOI] [PubMed] [Google Scholar]
- 41.Millat G, et al. Spectrum of pathogenic mutations and associated polymorphisms in a cohort of 44 unrelated patients with long QT syndrome. Clin Genet. 2006;70(3):214–227. doi: 10.1111/j.1399-0004.2006.00671.x. [DOI] [PubMed] [Google Scholar]
- 42.Kubota T, Shimizu W, Kamakura S, Horie M. Hypokalemia-induced long QT syndrome with an underlying novel missense mutation in S4–S5 linker of KCNQ1. J Cardiovasc Electrophysiol. 2000;11(9):1048–1054. doi: 10.1111/j.1540-8167.2000.tb00178.x. [DOI] [PubMed] [Google Scholar]
- 43.Choi G, et al. Spectrum and frequency of cardiac channel defects in swimming-triggered arrhythmia syndromes. Circulation. 2004;110(15):2119–2124. doi: 10.1161/01.CIR.0000144471.98080.CA. [DOI] [PubMed] [Google Scholar]
- 44.Splawski I, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102(10):1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 45.Splawski I, et al. Genomic structure of three long QT syndrome genes: KVLQT1, HERG, and KCNE1. Genomics. 1998;51(1):86–97. doi: 10.1006/geno.1998.5361. [DOI] [PubMed] [Google Scholar]
- 46.Tanaka T, et al. Four novel KVLQT1 and four novel HERG mutations in familial long-QT syndrome. Circulation. 1997;95(3):565–567. doi: 10.1161/01.cir.95.3.565. [DOI] [PubMed] [Google Scholar]
- 47.Silva J, Rudy Y. Subunit interaction determines IKs participation in cardiac repolarization and repolarization reserve. Circulation. 2005;112(10):1384–1391. doi: 10.1161/CIRCULATIONAHA.105.543306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Logothetis DE, Petrou VI, Adney SK, Mahajan R. Channelopathies linked to plasma membrane phosphoinositides. Pflugers Arch. 2010;460(2):321–341. doi: 10.1007/s00424-010-0828-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu D, et al. State-dependent electrostatic interactions of S4 arginines with E1 in S2 during Kv7.1 activation. J Gen Physiol. 2010;135(6):595–606. doi: 10.1085/jgp.201010408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.