

Chemical diversity and complexity of scaffolds are key for the design- or screening-based discovery of useful materials. Series of compounds based on a particular scaffold are often used to optimize properties. Although scaffolds are traditionally built up by sequential multistep syntheses, a convergent alternative pathway involves one-pot multicomponent reactions (MCRs).[1] A useful definition of the scaffold in the context of MCR has been introduced recently by us, and accordingly it is defined “as the smallest atomic denominator and its connectivity as resulting from a reaction or reaction sequence by using starting materials with common functional groups.”[2] A very large variety of MCR scaffolds have been described in the past based on different strategies.[3] For example, there are more than 40 piperazine scaffolds accessible by MCR.[2] The two-step Ugi MCRs/ring-forming Pictet–Spengler reaction is of particular interest, because it gives access to rather rigid, complex polycyclic products, which can lead to entropically preferred receptor binding (Scheme 1). This reaction sequence has been used for the synthesis of drugs and natural products, as well as for chemical-biology tools.
Scheme 1.

Some previously described molecular skeletons assembled by the Ugi–Pictet–Spengler sequence. The isocyanide component, the amine component, and the oxo/acid components are marked in red, blue, and black, respectively.
We described the shortest scalable synthesis of the schistosomiasis drug praziquantel 1 by an Ugi–Pictet–Spengler sequence.[4] The same route can be used for the synthesis of bioactive Praziquantel derivatives otherwise not amenable.[5] Almorexant 2, a selective and orally bioavailable orexin I antagonist recently began phase III clinical trials and was discovered by a sequence Ugi–Pictet–Spengler reactions.[6] By using a bifunctional oxocarboxylic acid, compounds, such as 3 with even more condensed cycles, can be conveniently synthesized.[7] Tyagi et al. performed the Ugi reaction in the Groebcke–Blackburn–Bienaymé variation followed by a Pictet–Spengler reaction to give several distinct skeletons of fused ring systems, for example, 4.[8] Phenylethylamine-derived isocyanides can be converted in just two steps to polycyclic products with unprecedented complexity (5).[9] Lesma et al. described the synthesis of 1,2,3,4-tetrahydro-β-carboline (THBC)-based tetracyclic peptidomimetics from an Ugi–Pictet–Spengler reaction sequence with β-turn-like conformations.[10] El Ka m et al. described the synthesis of 1,4-diketopiperazines (6) by using α-ketocarboxylic acids and homoveratryl isocyanide in the Ugi–Pictet–Spengler sequence.[11] Fukuyama et al. used an Ugi–Pictet–Spengler reaction in their total synthesis of the anticancer natural-product drug ecteinascidin 784 and the related naphthyridinomycin and lemonomycin.[12] Cano-Herrera and Miranda described an expedient entry to the piperazinohydroisoquinoline ring system (7) present in the tetrahydroisoquinoline antitumor alkaloids family by a sequential Ugi–Pictet–Spengler/reductive methylation reaction protocol.[13] Znabet et al. described an interesting biocatalytic desymmetrization of 3,4-cis-substituted meso-pyrrolidines (8) with an Ugi-type multi-component reaction followed by an in situ Pictet–Spengler-type cyclization-reaction sequence for the rapid asymmetric synthesis of alkaloid-like polycyclic compounds.[14] Other MCRs, such as the van Leusen reaction, have also been combined with the Pictet–Spengler reaction.[15] Most of the scaffolds are new and cannot be easily synthesized by methods other than by MCR chemistry methods. They thus provide an excellent source for the search for bioactive compounds.
Recently, We introduced an extension of the Ugi reaction of α-amino acids by introducing primary or secondary amines as additional reactants, thus rendering the U-5C-4CR into a truly four-component reaction with four highly variable starting materials.[16] Herein, chiral iminodicarboxamides can be formed by the variation of an α-amino acid, an oxocomponent, an isocyanide, and a primary or secondary amine as unprecedented components for this Ugi variation. The reaction is compatible with many functional groups and different fragments. For example, we have previously shown in one example that aminoacetaldehyde dimethylacetale can react together with the electron-rich aromatic amino acid tryptophan to give an unprecedented polycyclic ring system after a Pictet–Spengler ring closure in one pot.[16] We believe that such compounds are of interest to natural-product synthesis and for medicinal chemists, and therefore we would like to give full experimental detail, report the scope and stereochemistry of this new reaction by variation of different amino acids, isocyanides and oxocomponents, and several molecular structures (Scheme 2).
Scheme 2.
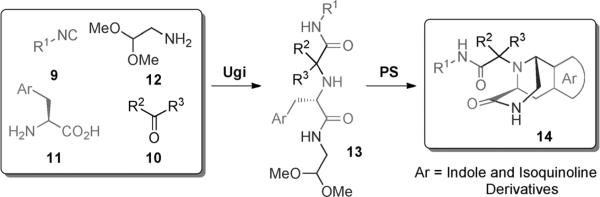
Synthesis of Ugi–Pictet–Spengler indole and isoquinoline derivatives.
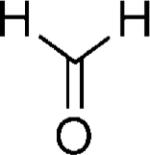
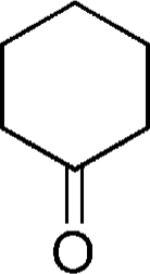
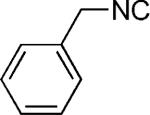
We first reacted tryptophan amino acid (11), a ketone (10), an isocyanide (9), and aminoacetaldehyde dimethyl acetal (12) in MeOH/H2O (4:1) at room temperature to form the U-5C-4CR product 13 as shown in Scheme 2. The Pictet–Spengler reaction product was not obtained by using various conditions, such as sulfonic acid, para-toluenesulfonic acid, and camphorsulfonic acid in toluene, however, by using concentrated formic acid at room temperature surprisingly gave the Pictet–Spengler cyclization product 14. The reaction sequence was also conveniently performed in one pot without loss of yield of final product by evaporation of the crude U-5C-4CR mixture followed by addition of formic acid. Furthermore, we found that the reaction also worked with 5-hydroxy tryptophan according to Scheme 3. We made variations by using different isocyanides and ketones or aldehydes as shown in Table 1, and all reactions worked giving satisfactory yields. Use of benzaldehyde (28) and cyclopentanecarbaldehyde (29) produced two diastereomers (4:1 by 1H NMR analysis, Table 1). In the case of valeraldehyde (36), two diastereomers in a ratio of 3.6:1 (by HPLC analysis, Table 1) were formed. All stereoisomers could be easily separated by using preparative supercritical fluid chromatography (SFC) HPLC analysis. By using TMS-oxy ethyl isocyanide, ethyl formate polycyclic compound 32 was obtained in good yield likely by an acid-catalyzed deprotection/esterification. All reactions are believed to work by the proposed mechanism shown in Scheme 3.[17] The reaction mechanism might occur by an initial deprotection step of the acetal groups in acidic condition to form the iminium ion (17) followed by electrophilic substitution (18). After subsequent deprotonation, the desired product (16) was obtained.
Scheme 3.

Proposed reaction mechanism of Pictet–Spengler reaction for indole derivatives.
Table 1.
Synthesis of Ugi–Pictet–Spengler reaction for indole derivative.
| ||||
|---|---|---|---|---|
| Compd. | R1-NC |
|
X | Yield[a] [%] |
| 19 |
|
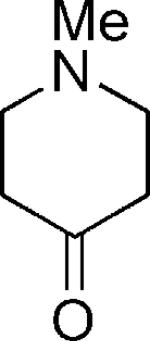
|
H | 42 |
| 20 |
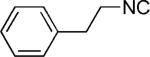
|
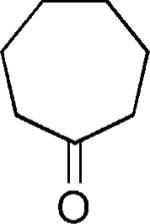
|
H | 52 |
| 21 |
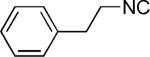
|
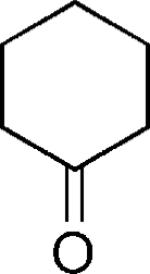
|
H | 48 |
| 22 |
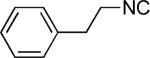
|
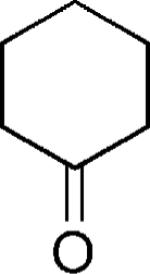
|
OH | 35 |
| 23 |
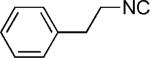
|
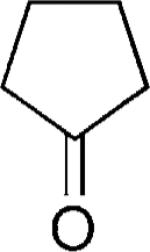
|
H | 27 |
| 24 |
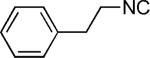
|
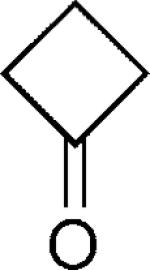
|
OH | 35 |
| 25 |
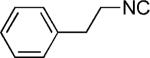
|
|
H | 28 |
| 26 |
|
|
OH | 44 |
| 27 |
|
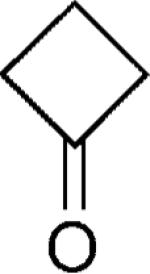
|
OH | 29 |
| 28 |
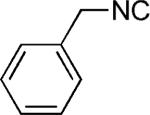
|
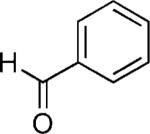
|
H | 61 (4:1)[b] |
| 29 |
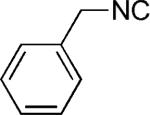
|
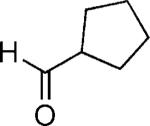
|
OH | 38 (4:1)[c] |
| 30 |
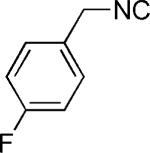
|
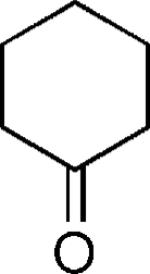
|
OH | 40 |
| 31 |
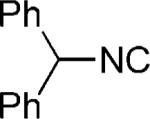
|
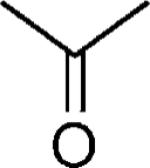
|
OH | 27 |
| 32 |
|
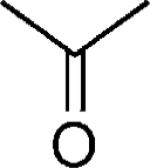
|
OH | 51 |
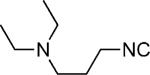
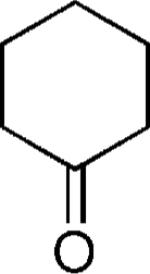
| 33 |
|
|
H | 25 |
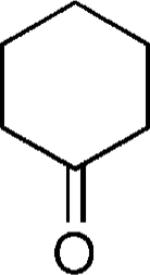
| 34 |
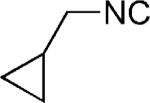
|
|
H | 19 |
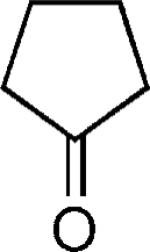
| 35 |
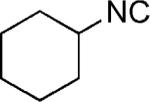
|
|
OH | 24 |
| 36 |
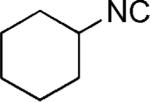
|
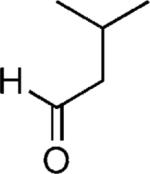
|
H | 52 (3.6:1)[c] |
Overall yield in two steps (crude Ugi reaction product was directly used for the Pictet–Spengler reaction).
Diastereomeric ratio (d.r.) was determined by 1H NMR spectroscopy.
d.r. was determined by UV spectroscopy.
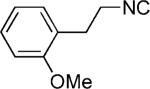
We also investigated the use of phenylalanine amino acid derivatives to form the Ugi–Pictet–Spengler isoquinoline (39). Although the U-5C-4CR product was formed in all cases, only when using 3-methoxy phenylalanine and 3,4-dimethoxy phenylalanine, the Pictet–Spengler isoquinoline product (39) was produced. However, if no electron-donating group is available on the meta position of the phenyl ring, such as phenylalanine or 4-methoxy phenylalanine, the Pictet–Spengler isoquinoline was not formed. The reaction might occur as shown in Scheme 4. The acetal group is removed in acidic condition to form the iminium ion (38) followed by electrophilic substitution and deprotonation to give the desired product (39).[17] Several analogous reactions were applied, and all analogs gave satisfactory yield even with diethyl (cyanomethyl)phosphonate (47) as shown in the Table 2.
Scheme 4.

Proposed reaction mechanism of the Pictet-Spengler isoquino-line derivative formation.
Table 2.
Synthesis of Ugi–Pictet–Spengler isoquinoline derivatives.
| ||||
|---|---|---|---|---|
| Compd. | R1-NC |
|
X | Yield[a] [%] |
| 40 |
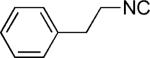
|
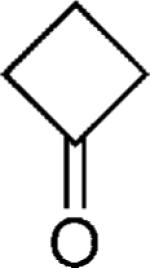
|
OMe | 64 |
| 41 |
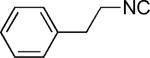
|
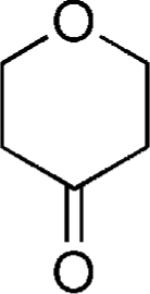
|
OMe | 59 |
| 42 |
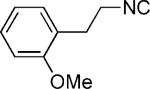
|
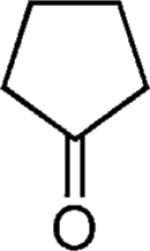
|
OMe | 62 |
| 43 |
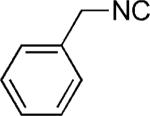
|
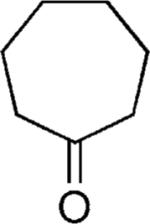
|
H | 35 |
| 44 |
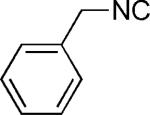
|
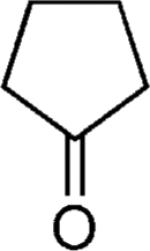
|
OMe | 72 |
| 45 |
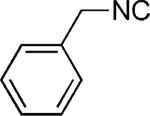
|
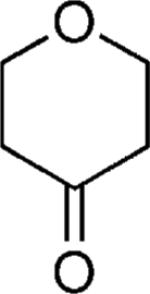
|
OMe | 49 |
| 46 |
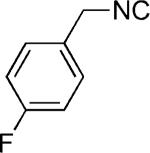
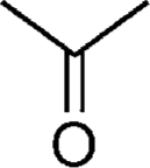
|
|
H | 44 |
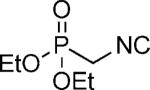
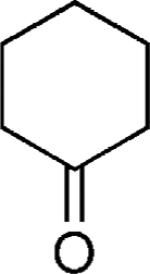
| 47 |
|
|
OMe | 76 |
| 48 |
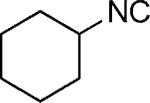
|
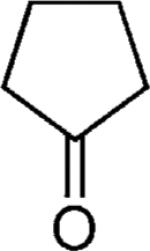
|
H | 47 |
| 49 |
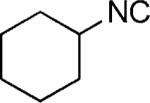
|
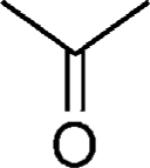
|
OMe | 52 |
Yield in two steps (crude Ugi reaction product was directly used for the Pictet–Spengler reaction).
To investigate the absolute structure, the relative stereochemistry and intermolecular contacts with respect to potential receptor–ligand interactions, several compounds were crystallized, and their single-crystal structures were solved (Figure 1).
Figure 1.

Structure of Ugi–Pictet–Spengler product 32 in solid state.
Although there have been several previous attempts to use the Ugi–Pictet–Spengler combination in natural-product chemistry, we believe its potential is far from being fully taped. A substructure search of the herein formed molecular skeletons in alkaloid- and natural-product databases revealed several potentially interesting targets, for example, the antitumor quinocarcin or the insecticidal notoamide B (Scheme 5).
Scheme 5.

Substructures in potential natural-product targets.
The advantages of our new combination of Ugi–Pictet–Spengler reactions is the expedited and convergent access to polycyclic natural-product-like skeletons. The yields over two steps are good to acceptable. The stereochemistry of the amino acid is conserved, and therefore stereochemically pure (symmetrical ketones, formaldehyde) or diastereomeric (aldehyde other than formaldehyde) compounds were obtained. Many variations in the oxo and isocyanide starting materials can lead to synthetic handles to further elaborate the primary synthetic structures. Work is ongoing to investigate the further synthetic applications and biological properties of the new compounds.
Experimental Section
General procedure of Ugi–Pictet–Spengler reaction
A mixture of l-amino acid (0.5 mmol), ketone (0.5 mmol), isocyanide (0.5 mmol), and aminoacetaldehyde dimethyl acetal (0.5 mmol) in 0.1m of MeOH/H2O (4:1) were stirred for 24–72 h at RT (in the case of 3,4-dimethoxy-l-phenylalanine, the reaction mixture was heated at 50°C for 72 h). Solvents were evaporated under reduced pressure. The crude Ugi product was dissolved in formic acid (2 mL) and stirred for another 16 h at RT. Reaction mixture was diluted with CH2Cl2, and solid K2CO3 was added in portion to neutralize the mixture. The excess K2CO3 was filtered off by using Celite, and solvents were evaporated to get crude product, which was purified by supercritical fluid chromatography, column-flash chromatography, or preparative TLC to give title compound in decent yield. Characterization of compound 20: 1H NMR (600 MHz, [D6]DMSO): δ = 1.25–1.55 (m, 9H), 1.73–178 (m, 1H), 1.95–2.04 (m, 2H), 2.68–2.75 (m, 3H), 2.93–2.98 (m, 2H), 3.28 (q, J = 13.8, 7.2 Hz, 2H), 3.49 (dd, J = 11.4, 4.2 Hz, 1H), 3.85 (d, J = 5.4 Hz, 1H), 4.04 (d, J = 3.6 Hz, 1H), 6.94 (t, J = 7.2 Hz, 1H), 7.03 (t, J = 7.2 Hz, 1H), 7.16–7.21 (m, 3H), 7.26–7.31 (m, 3H), 7.35 (d, J = 7.8 Hz, 1H), 7.39 (d, J = 3.6 Hz, 1H), 7.86 (t, J = 5.4 Hz, 1H), 10.73 ppm (s, 1H); 13C NMR (150 MHz, [D6]DMSO): δ = 23.0, 33.8, 24.8, 29.3, 29.5, 32.5, 35.1, 35.3, 40.5, 46.3, 47.2, 54.6, 68.6, 106.9, 111.2, 117.5, 118.5, 120.8, 126.0, 126.5, 128.3, 128.5, 134.3, 135.4, 139.5, 171.5, 175.4 ppm. SFCMS (atmospheric pressure chemical ionization (APCI)) m/z calcd for C29H34N4O2: 471.61 [M]+; found: 471.25.
Supplementary Material
Acknowledgements
This work was financially supported by the NIH (1R21 M087617, 1R01M097082, and 1P41M094055 for A.D.). K.K. acknowledges the ACS Medicinal Chemistry predoctoral fellowship (2011–2012).
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201300962. It contains experimental details, synthesis, and NMR spectroscopy characterization data for all compounds, as well as X-ray analysis data of compound 32.
References
- 1.a Ugi I, Dçmling A, Horl W. Endeavour. 1994;18:115–122. [Google Scholar]; b Zhu J, Bienaym H, editors. Multicomponent Reactions. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- 2.Dçmling A, Huang Y. Synthesis. 2010;42:2859–2883. [Google Scholar]
- 3.a Dçmling A. Curr. Opin. Chem. Biol. Curr. Opin. Chem. Bio. 2000;4:318–323. doi: 10.1016/s1367-5931(00)00095-8. [DOI] [PubMed] [Google Scholar]; b Hulme C, Gore V. Curr. Med. Chem. 2003;10:51–80. doi: 10.2174/0929867033368600. [DOI] [PubMed] [Google Scholar]; c Ugi I, Dçmling A, Gruber B, Almstetter M. Croat. Chem. Acta. 1997;69:631–647. [Google Scholar]; d Weber L. Curr. Opin. Chem. Biol. Curr. Opin. Chem. Bio. 2000;4:295–302. doi: 10.1016/s1367-5931(00)00092-2. [DOI] [PubMed] [Google Scholar]; e Weber L, Wallbaum S, Broger C, Gubernator K. Angew. Chem. 1995;107:2452–2454. [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1995;34:2280–2282. [Google Scholar]; f Ruijter E, Scheffelaar R, Orru RVA. Angew. Chem. 2011;123:6358–6371. doi: 10.1002/anie.201006515. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2011;50:6234–6246. doi: 10.1002/anie.201006515. [DOI] [PubMed] [Google Scholar]
- 4.Cao H, Liu H, Dçmling A. Chem. Eur. J. 2010;16:12296–12298. doi: 10.1002/chem.201002046. [DOI] [PubMed] [Google Scholar]
- 5.Liu H, William S, Herdtweck E, Botros S, Dçmling A. Chem. Biol. Drug Des. 2012;79:470–477. doi: 10.1111/j.1747-0285.2011.01288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a Brisbare-Roch C, Dingemanse J, Koberstein R, Hoever P, Aissaoui H, Flores S, Mueller C, Nayler O, van Gerven J, de Haas SL, Hess P, Qiu C, Buchmann S, Scherz M, Weller T, Fischli W, Clozel M, Jenck F. Nat. Med. 2007;13:150. doi: 10.1038/nm1544. [DOI] [PubMed] [Google Scholar]; b Aissaoui H, Cappi M, Clozel M, Fischli W, Koberstein R. 2001 in Patent W02001068609.
- 7.Wang W, Herdtweck E, Domling A. Chem. Commun. 2010;46:770–772. doi: 10.1039/b917660h. [DOI] [PubMed] [Google Scholar]
- 8.Tyagi V, Khan S, Bajpai V, Gauniyal HM, Kumar B, Chauhan PMS. J. Org. Chem. 2012;77:1414–1421. doi: 10.1021/jo202255v. [DOI] [PubMed] [Google Scholar]
- 9.Liu H, Dçmling A. J. Org. Chem. 2009;74:6895–6898. doi: 10.1021/jo900986z. [DOI] [PubMed] [Google Scholar]
- 10.Lesma G, Cecchi R, Crippa S, Giovanelli P, Meneghetti F, Musolino M, Sacchetti A, Silvani A. Org. Biomol. Chem. 2012;10:9004–9012. doi: 10.1039/c2ob26301g. [DOI] [PubMed] [Google Scholar]
- 11.El Kaim L, Gageat M, Gaultier L, Grimaud L. Synlett. 2007;2007:0500–0502. [Google Scholar]
- 12.a Mori K, Rikimaru K, Kan T, Fukuyama T. Org. Lett. 2004;6:3095–3097. doi: 10.1021/ol048857e. [DOI] [PubMed] [Google Scholar]; b Rikimaru K, Mori K, Kan T, Fukuyama T. Chem. Commun. 2005:394–396. doi: 10.1039/b415030a. [DOI] [PubMed] [Google Scholar]; c Endo A, Yanagisawa A, Abe M, Tohma S, Kan T, Fukuyama T. J. Am. Chem. Soc. 2002;124:6552–6554. doi: 10.1021/ja026216d. [DOI] [PubMed] [Google Scholar]
- 13.Cano-Herrera MA, Miranda LD. Chem. Commun. 2011;47:10770–10772. doi: 10.1039/c1cc10759c. [DOI] [PubMed] [Google Scholar]
- 14.Znabet A, Zonneveld J, Janssen E, De Kanter FJJ, Helliwell M, Turner NJ, Ruijter E, Orru RVA. Chem. Commun. 2010;46:7706–7708. doi: 10.1039/c0cc02938f. [DOI] [PubMed] [Google Scholar]
- 15.Kundu B, Sawant D, Partani P, Kesarwani AP. J. Org. Chem. 2005;70:4889–4892. doi: 10.1021/jo050384h. [DOI] [PubMed] [Google Scholar]
- 16.Khoury K, Sinha MK, Nagashima T, Herdtweck E, Dçmling A. Angew. Chem. 2012;124:10426–10429. doi: 10.1002/anie.201205366. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2012;51:10280–10283. doi: 10.1002/anie.201205366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a Pictet A, Spengler T. Ber. Dtsch. Chem. Ges. 1911;44:2030–2036. [Google Scholar]; b Larghi EL, Kaufman TS. Synthesis. 2006;37:187. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.