

Abstract
We designed, synthesized and screened a library of analogs of the organophosphate pesticide metabolite paraoxon against a recombinant variant of human serum paraoxonase-1. Alterations of both the aryloxy leaving group and the retained alkyl chains of paraoxon analogs resulted in substantial changes to binding and hydrolysis, as measured directly by spectrophotometric methods or in competition experiments with paraoxon. Increases or decreases in the steric bulk of the retained groups generally reduced the rate of hydrolysis, while modifications of the leaving group modulated both binding and turnover. Studies on the hydrolysis of phosphoryl azide analogs as well as amino-modified paraoxon analogs, the former being developed as photo-affinity labels, found enhanced tolerance of structural modifications, when compared with O-alkyl substituted molecules. Results from computational modeling predict a predominant active site binding mode for these molecules which is consistent with several proposed catalytic mechanisms in the literature, and from which a molecular-level explanation of the experimental trends is attempted. Overall, the results of this study suggest that while paraoxonase-1 is a promiscuous enzyme, there are substantial constraints in the active site pocket, which may relate to both the leaving group and the retained portion of paraoxon analogs.
I. Introduction
Paraoxonase-1 (PON1) is a serum protein associated with high-density lipoprotein (HDL) cholesterol particles. It is capable of catalyzing the hydrolysis of a wide array of organic esters and organophosphorus (OP) compounds; however, its physiological role remains enigmatic. Nevertheless, PON1 is able to hydrolyze pesticide metabolites such as paraoxon (1), chlorpyrifos oxon (2), G-series nerve agents like sarin (3), and V-series nerve agents like VR (4) and VX (5).1–4 Due to this property, and because it is a native human protein, PON1 is a suitable candidate for a bio-scavenger against OP agent exposure.3–5 However, the catalytic efficiency of the native protein is at least two orders of magnitude lower than what is desired for effective protection against these nerve agents and pesticides.6 While our group and others3,4,6 are attempting to engineer mutants of PON1 that will have improved catalytic efficiency, the uncertainty regarding the potential active site and mechanism of hydrolysis has slowed these efforts. Furthermore, the lack of a crystal structure of a ligand-bound PON1 variant has left many questions unanswered, and indeed, it remains unclear whether the protein uses multiple catalytic residues and even whether all substrates bind the same way to the protein.4,5
In order to improve our understanding of the PON1 active site and what constraints on substrates exist, we undertook a quantitative structure-activity relationship (SAR) study to obtain information on binding and catalytic turnover. The crystal structure of a bacterially expressible recombinant PON1 variant, known as G2E6, was solved by Tawfik, Sussman and colleagues (PDB ID 1V04).7 The sequences of G2E6 and human PON1 are identical in the putative active site, which is in the interior of the β-propeller fold of the enzyme.7 Most of the sequence differences are in the solvent-exposed surface of the protein and are predominantly non-polar to polar mutations, which are likely related to solubility and stability. Kinetic studies show generally similar behavior between the two proteins; however, the G2E6 variant hydrolyzes paraoxon more efficiently, while the human variant hydrolyzes the nerve agents VX and VR (Figure 1) more efficiently.7,8 Overall, the active site of G2E6 is expected to be similar to that of human PON1. PON1 has similar activity to bacterial phosphotriesterase, which has higher catalytic efficiency against paraoxon.9 However, we focused our efforts in engineering PON1 against OP compounds since PON1 is a human enzyme.
Figure 1.

Organophosphorus insecticides and nerve agents paraoxon (1), chlorpyrifos oxon (2), sarin (3), VR (4), and VX (5).
To date, the natural substrates of PON1 are unknown, but it has been proposed that its native role is a lactonase.10 Many researchers also believe that PON1 plays a role in regulation of LDL oxidation, and in prevention of atherosclerosis.11–13 Tawfik et al. studied a range of substrates of considerable diversity, to probe the hypothesis that PON1 is a lactonase (Figure 2).10 They also found that the basicity of the leaving group had little effect on the kcat or kcat/KM for good leaving groups, but worse leaving groups (pKa of the conjugate acid > 7) reacted more slowly with increasing basicity. Although they proposed a natural function for the enzyme, the experiments did not address the issue of the actual mechanism of hydrolysis of OP compounds. In an attempt to use structure-activity relationship (SAR) studies to probe substrate constraints on turnover and the active site tolerances of PON1, we studied a series of paraoxon analogs (Figure 3) grouped into five classes.
Figure 2.

Examples of PON1 substrates studied by Tawfik’s group.10
Figure 3.

Five classes of paraoxon analogs synthesized and tested in this study.
Group I: Paraoxon analogs
The molecules in this group are phosphoryl triester analogs of paraoxon with variations only on the O-alkyl side chains. The kinetic parameters for this library of molecules can help us understand the role of size of the substrate retained groups on binding and hydrolysis by PON1.
Group II: Phenoxy-substituted paraoxon analogs
Ligands of this group are phosphoryl triesters that contain an unsubstituted phenolate leaving group, and include considerable variety in the side chains as for the group I molecules. The group II compounds also sample the active site tolerances of PON1 for molecules that lack the electron-withdrawing para-nitro moiety on the leaving group.
Group III: Phosphoryl azides as photo-affinity labels (PALs)
This group includes phosphoryl azides, which upon irradiation can produce a highly reactive singlet nitrene that can covalently insert to the active site residues of the protein and thus act as photoaffinity labels.14 The kinetic parameters studied within this group will aid in the design of photoaffinity labels for PON1 and also explore the role of polar atoms on the retained group, which can potentially affect the interaction of ligand with polar residues in the active site.
Group IV: Paraoxon analogs containing other leaving groups
The molecules in this group are paraoxon analogs that contain substituted aromatic rings as the leaving group. Accordingly, the primary purpose of these molecules is to improve our understanding of the effects of the leaving group on the catalysis: whether it is purely an electronic effect of the leaving group substituents, or if there are effects related to the orientation of substrate in the active site, as well. Most prior studies (e.g., those of Tawfik et al.10) have primarily used paraoxon analogs that contain electron-withdrawing substituents on the aromatic ring, but very few were electron-donating groups.10,15 Analogs 30–32 have electron-donating substituents on the aromatic ring, and 33 has a weakly electron-withdrawing substituent.
Group V: Phosphoramidates
Molecules 34 and 35 are both inhibitors of DFPase, and they do not contain an aromatic leaving group.16 Molecule 36, on the other hand, is a phosphoramidate with phenolate leaving groups. Thus, this moiety primarily aids in examining leaving group effects, both in terms of the direct effect on the energetics of hydrolysis as well as in terms of potential interactions in the active site of the protein.
In this report, we describe experimental and computational structure-activity relationship studies aimed at identifying the constraints on hydrolysis of paraoxon analogs as part of a larger collaborative research effort aimed at examining and optimizing the OPase mechanism of PON1.5,8,17
II. Experimental and Computational Methods
II. A Enzyme Preparation and Kinetics
General
Substrate and inhibition kinetics were performed with recombinant PON1 (rePON1) variant G2E6 expressed as a 6×His-tagged thioredoxin fusion protein and purified as reported in the literature.7,18
Enzyme Kinetics
The enzymatic hydrolysis rate of all of the phosphoryl triester substrates of Groups I–V, were determined at pH 7.4, where the hydrolysis rate is known to be maximum,19 in 50 mM Tris•HCl, 50 mM NaCl, and 1 mM CaCl2 buffer. The enzyme was stored in 50 mM Tris•HCl, 50 mM NaCl, 1 mM CaCl2 buffer containing 0.1% tergitol and 10% glycerol. The presence of detergent and glycerol has been shown to keep the enzyme stable for a long period of time.20 A substrate stock was made in methanol as each substrate had limited solubility in aqueous solvent. We initially determined all values using a 0.1 M stock of each compound in methanol, with variable methanol concentration in the final reaction. Because it has been previously observed that increasing methanol results in apparently lower KM and often lower kcat values, we used a 0.26 M stock of each compound and determined the parameters again under constant 1% methanol conditions for the group I compounds. Only very small (less than 2-fold) changes in kcat and KM were observed, with virtually no effect on kcat/KM. However, due to instability of the compounds, we were unable to re-examine the group III phosphoryl azides under constant methanol conditions, and we did not re-examine compound 32, the only compound in groups II, IV or V with significant turnover. So the values for groups II-V must be regarded as apparent. The initial velocities from six different concentrations of each substrate were used to calculate kcat and KM. The concentration of substrate was generally varied in the range of 0.3 × KM up to (2–3) × KM, and the enzyme concentrations were varied for different substrates depending on the amount of activity (2 to 40 µM). The hydrolysis product was monitored spectrophotometrically on an Agilent 8453 UV-Vis spectrophotometer at 405 nm (for substrates with p-nitrophenolate leaving groups, Group I) or at 270 nm (for substrates with phenolate leaving groups, Group II-IV) in a 500 mL reaction volume. No significant background hydrolysis was observed for any substrates studied, within the conditions of the assay.
Inhibition
We carried out inhibition assays for molecules in Groups I–V by monitoring paraoxon hydrolytic activity with G2E6 (rePON1). The paraoxon hydrolysis product (p-nitrophenolate) was monitored at 405 nm at four different inhibitor concentrations (0.03–1 mM, depending on the inhibitor) and without an inhibitor, to calculate an apparent IC50. Substrate (paraoxon) concentration was kept constant at 0.065 mM, which is slightly more than 10-fold below the KM, with 1% co-solvent (methanol). Enzyme concentration was 13 µM. The group I compounds have the same leaving group as paraoxon, but we could still monitor inhibition for substrates that reacted much more slowly than paraoxon. The rest of the compounds in this study produce leaving groups with no significant absorbance at 405 nm.
Data Analysis
The kinetic parameters (KM, kcat) were obtained by fitting the Michaelis-Menten equation [ν=Vmax[S]/([S]+KM])] to the data using the program Kaleidagraph 4.0. In the cases of substrates where solubility limited higher substrate concentrations (compounds 27, 21 for example), data were fitted to the linear regime of the Michaelis-Menten model and Vmax/KM was derived from the slope. The kcat was then determined from Vmax using the known enzyme concentration. The averages of the three to five independent experiments were used in the calculations, and errors to the fits are reported.
II. B Computational Methods
Model Preparation
Human paraoxonase-1 (PON1) is a monomeric protein, with a 6-bladed β-propeller structure, which in serum is associated with high density lipoprotein (HDL). The human form of the protein is extremely insoluble, and obtaining crystal structures of the protein has been highly problematic. The sole crystal structure available at present is of a recombinant, gene-shuffled variant derived primarily from the rabbit paraoxonase; this variant, known as G2E6, shares 86% sequence homology with the human protein (PDB entry 1V04).7 The crystal structure of G2E6 was resolved at 2.2 Å resolution, with two fragments unresolved: the N-terminal 15 residues of the protein, as well as a flexible loop from residues 72–79. The center of the β-propeller contains two bound calcium ions, which have been designated ‘catalytic’ and ‘structural’ on the basis of mutagenesis studies,7,21–22 and a single phosphate ion was complexed to the ‘catalytic’ calcium ion in the crystal structure.
Preparation of a computational model of the G2E6 variant of PON1 was performed using the AMBER 10 suite of programs.23 The missing residues were added and the protonation states of titratable residues were assigned using the program pdb2pqr,24 with a solvent pH of 7.0. The FF0325 force field was employed for modeling of the protein models; simulations of ligands were performed using parameters assigned using the GAFF26,27 force field. To generate charges for the ligands, ChelpG28 partial atomic charges were generated at the B3LYP/6–311+G(d,p) level29,30 of theory using the Gaussian03 program.31 Parameters for the calcium ions were derived from published parameters.32
MD Simulations
There were only 340 amino acid residues resolved in the G2E6 crystal structure,7 along with the two calcium ions. A total of 24,074 TIP3P33 water molecules were added in a rectangular periodic box of 91 × 105 x 96 Å3. G2E6 contains a single disulfide bond, between C42 and C353, which was included in the computational model. Energy minimizations and molecular dynamics simulations were performed using the sander module within AMBER 9.23 A five-step equilibration procedure was employed in preparing the model for production MD simulations: First, a 1,000 step energy minimization was performed. This was followed by a four-step warming regime, where the protein was heated to 300 K over 4 ps of MD simulations, with a 1 fs time step in each case. No restraints were employed on the protein structure during this heating regime, and no instabilities were observed in the subsequent MD simulations. The temperature and pressure of the system were regulated using the Berendsen34 scheme of heat bath coupling, and a coupling time of 1.0 ps. While the Berendsen temperature bath has been demonstrated to result in instabilities for certain types of simulations,35 the use of explicit solvation and coupling to the temperature bath largely remove these concerns, and the method has been successfully used in the study of proteins.36,37 The SHAKE38 algorithm was employed to constrain bonds involving hydrogen atoms, and the particle-mesh Ewald (PME) method was used with a 12.0 Å cutoff for non-bonded interactions.
In order to understand the binding of the ligands screened in this study, we performed a series of molecular docking simulations into the computational model of G2E6 PON1 using AUTODOCK 4.0.39 In the absence of a crystal structure of a ligand bound in the active site of PON1, computational approaches, such as molecular docking, can aid in our understanding of the ligand’s binding in the active site.40 A total of one hundred poses were generated by AUTODOCK, clustered by root mean squared deviations (RMSD), and only clusters which had the P=O of the ligand oriented towards the catalytic calcium were defined as productive.17 These structures were pruned using several criteria, including proximity of the phosphoryl oxygen to the ‘catalytic’ calcium ion, to generate a series of plausible starting poses bound to the receptor. The resulting receptor-ligand complexes were subjected to 5 ns of MD simulations as described above for the G2E6 protein model. This protocol allowed for full receptor and ligand flexibility, and was followed by the estimation of free energies of binding using the molecular mechanics Poisson-Boltzmann surface area (MM-PBSA)41 and molecular mechanics Generalized Born surface area (MM-GBSA)42 methods. Following the MD simulations, the structural stability of the protein backbone was verified by plotting the RMSD of the protein-ligand complex over the course of the simulation. The RMSD was typically below 1–2 Å relative to the uncomplexed model system, and the simulations typically stabilized after 1–2 ns. Subsequently, predictions of the free energies of binding were made using42 the formula:
| (1) |
III. RESULTS
III. A Enzyme Catalysis and Binding Modes
Catalytic hydrolysis for each substrate by G2E6 PON1 was analyzed using a Michaelis-Menten model. When saturation by the substrate was possible, kcat and KM were determined separately; otherwise, only the catalytic efficiency (kcat/KM) was determined. Each variant was also examined for inhibition of paraoxon hydrolysis by determining the IC50 at a paraoxon concentration of 0.065 mM. Some of the molecules we studied were weak substrates or inhibitors for which we could not quantitatively determine the kinetic parameters. In the following tables, we report the qualitative kinetic parameters for such molecules as ‘weak’. (Structures for all compounds are provided in the supporting information, and substituents are noted in the subsequent tables.)
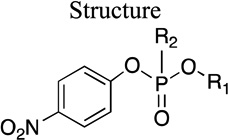
Group I
Paraoxon (1), the reference substrate, contains two O-ethyl groups and a p-nitrophenoxy substituent, which is the leaving group upon hydrolysis. As shown in Table 1, the KM of paraoxon (1) in the G2E6 variant is 0.9 mM, with a kcat of 2.1 s–1. (Note that the slight difference from the previously reported value8 is likely due to different buffer and cosolvent conditions.) Except for the dimethoxy analog (6), there were no significant changes in KM across this series. The kcat of the dipropyl analog of paraoxon (10) was significantly attenuated (0.07 s–1 for 10) relative to 1. The di-isopropyl analog (11) was not a substrate, and only a weak inhibitor, suggesting that any branching of the side chain affected either the binding or catalysis, or both. The dibutyl analog (13) was neither a substrate nor an inhibitor, suggesting that the symmetrical butyl side chains represent an upper limit on binding, at least for group I. The di-methylcyclopropyl analog (12) was a weak substrate, suggesting that restricting the torsional freedom of the side chains did improve the binding over the butyl analogs. On the other extreme of side chain size, the di-methyl analog (6) has a significantly higher KM (> 1.5 mM) than paraoxon, such that we could only determine the overall catalytic efficiency (kcat/KM of 100 M–1 s–1) due to solubility limits. We could still establish a lower limit on the kcat, however, using a KM of 1.5 mM as a lower limit, which leads to a minimum of 0.2 s–1. The trend across the series appears to be that increased size of the alkyl fragments reduces the rate of turnover (kcat) compared to paraoxon. Although paraoxon is not the natural substrate of PON1, surprisingly, paraoxon has an optimal size for overall catalytic efficiency when compared with other structurally similar analogs.
Table 1.
Kinetic parameters for hydrolysis of Group I paraoxon analogs by rePON1 G2E6
| Molecule ID |
 |
kcat s−1 |
KM mM |
kcat/KM M−1 s−1 |
IC50 mM |
|
|---|---|---|---|---|---|---|
| R1 | R2 | |||||
| 1 | Et | OEt | 2.1 ± 0.2 | 0.9 ± 0.2 | 2,300 ± 700 | |
| 6 | Me | OMe | > 0.2 | > 1.5 | 100 ± 10 a | |
| 7 | Et | OMe | 1.5 ± 0.1 | 0.6 ± 0.2 | 2,800 ± 800 | |
| 8 | Et | OPr | 0.63 ± 0.02 | 0.4 ± 0.1 | 1,700 ± 200 | |
| 9 | Et | Oi-Pr | 0.14 ± 0.01 | 0.6 ± 0.1 | 230 ± 50 | |
| 10 | Pr | OPr | 0.07 ± 0.01 | 0.3 ± 0.1 | 200 ± 100 | weak b |
| 11 | i-Pr | Oi-Pr | c | c | c | weak b |
| 12 | MeCyp | OMeCyp | weak | weak | weak | d |
| 13 | Bu | OBu | c | c | c | d |
| 14 | Et | OPentyl | 0.18 ± 0.04 | 1.3 ± 0.7 | 140 ± 80 | weak b |
| 15 | Et | OCyclohexyl | c | c | c | 0.5 b |
| 16 | Et | c | c | c | d | |
Saturation was not possible for this substrate under the experimental conditions; only kcat/KM could be determined via a linear fit.
Although the leaving group is the same as for paraoxon for all Group I compounds, we are able to observe inhibition of paraoxonase activity in substrates with turnover that is much slower than the turnover of paraoxon (1).
No observable activity.
No inhibition was observed.
Relative to paraoxon, when one of the two alkyl side chains is conserved as ethyl, the KM values are comparable with varying alkyl bulk from methyl through pentyl on the other alkyl chain. The results in this series indicate that the presence of one ethyl group was sufficient to allow for productive substrate binding in the active site, despite the increases in the other alkyl chain. However, the kcat values varied significantly over this series, indicating that while binding was not impaired, the increase in size of side-chains hindered catalysis, as in the symmetrically substituted cases above (1.5 s–1 for 7, 0.63 s–1 for 8, 0.14 s–1 for 9, and 0.18 s–1 for 14). It should also be noted that the ‘mixed’ substrates 7–9 and 14 have a chiral center, and the racemate was used in our study. Thus, it is possible that only one enantiomer in these cases is binding or being hydrolyzed; the experimental conditions did not allow for analysis of the stereospecificity of hydrolysis. It has been previously reported that the less-toxic RP isomer of cyclosarin is hydrolyzed more rapidly by PON1 compared to that of the SP isomer.43 It is also possible that one enantiomer is an inhibitor of the enzyme.
In order to examine the steric limits of the active site, we prepared two additional asymmetrically substituted paraoxon analogs, of which 15, which has an O-ethyl side-chain and an O-cyclohexyl side chain, was not turned over by G2E6; instead 15 was an inhibitor of paraoxon hydrolysis (IC50 ∼0.5 mM). However, 16, which has a N-morpholino side chain (making it a phosphoramidate) and an O-ethyl group, is neither a substrate nor an inhibitor. As the molecules are roughly isosteric, this result suggests that the linker atom (oxygen), which connects the phosphorus atom and the alkyl side-chain, may have played a role in differentiating the binding in the active site.
Overall, the results from this class of paraoxon analogs suggest that modifications of the retained O-alkyl groups can influence both the binding and turnover of substrates in PON1. A comparison of the number of carbon atoms present in the side chains against the kcat values shows that there is a strong inverse correlation with the carbon count (Figure 4); increased steric bulk of the retained groups reduces the rate of hydrolysis across the group I molecules with the exception of paraoxon (carbon count=2).
Figure 4.

Relationship between the number of carbons on the alkyl side chains versus KM (red squares) and kcat (blue diamonds) for group I ligands.
Group II
The molecules in group II possess modifications of the paraoxon scaffold where the p-nitrophenoxy group is replaced with a phenoxy group. Aside from this modification, 17 is identical to paraoxon; however (as also reported previously with serum PON115), 17 is a very weak substrate of G2E6 PON1, and it only very weakly inhibits paraoxon turnover. In order to probe whether alterations in the size of the alkyl side chains could compensate for the absence of the p-nitro group, we studied the dipropyl analog (18), which was also not a substrate of G2E6 PON1 but was a weak inhibitor of paraoxon turnover. Additional molecules in the group with larger side-chains did not show any improved activity (see Supporting Information). Because of the higher pKa of phenol versus p-nitrophenol, phenolate is a relatively poor leaving group, but it is also possible that some specific interaction with the p-nitro group occurs in the active site.
Group III
The molecules in group III (Table 2) are phosphoryl azides, which could potentially be employed as photoaffinity labels (PAL) for the direct identification of residues of the PON1 active site.14 In order to exploit the technique of photoaffinity labeling, it is important to identify photoaffinity labels that are good inhibitors of PON1. The intent in this set of compounds was accordingly to both develop such PALs and to study modifications of the retained groups on phosphorus for their effects on binding and turnover. We recently reported that having a p-nitrophenyl group is not suitable for the photochemistry of PALs, while the unsubstituted phenyl ring was favorable for the photochemical labeling to occur.44 As a result, we used the unsubstituted phenolate leaving group as a starting point and progressively explored a wider range of modifications. Note that the enzyme kinetics for groups II-V were done under variable methanol cosolvent conditions, and so the parameters should be regarded as apparent. For the group I compounds, little change in the parameters was observed between the constant and variable methanol conditions (see Methods).
Table 2.
Kinetic parameters for the hydrolysis of Group III phosphoryl azides by rePON1 G2E6
| Molecule ID |
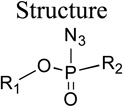 |
kcat s−1 |
KM mM |
kcat/KM M−1 s−1 |
IC50 mM |
|
|---|---|---|---|---|---|---|
| R1 | R2 | |||||
| 19 | Ph | OBu | 1.6 ± 0.1 | 0.5 ± 0.1 | 3,000 ± 800 | >1 |
| 20 | Ph | OPentyl | 0.73 ± 0.08 | 0.8 ± 0.2 | 1,000 ± 200 | 0.5–1 |
| 21 | Ph | OHexyl | 0.32 ± 0.02 | 0.40 ± 0.08 | 800 ± 200 | >1 |
| 22 | Ph | OOctyl | a | a | a | 0.5–1 |
| 23 | Ph | 1.2 ± 0.2 | 0.7 ± 0.2 | 1,900 ± 800 | >1 | |
| 24 | Ph | 0.60 ± 0.07 | 1.5 ± 0.2 | 390 ± 80 | 0.1–0.5 | |
| 25 | Ph | OPh | a | a | a | >1 |
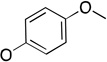
| 26 | Ph |  |
a | a | a | >1 |
| 27 | Ph |  |
a | a | a | 0.5–1 |
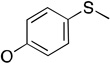
| 28 | 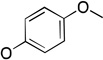 |
 |
a | a | a | b |
| 29 | 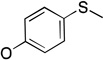 |
 |
a | a | a | 0.3 |
No observable activity.
No observable inhibition.
The first set of molecules in group III (19–27) are phosphoryl azides containing one phenyl substituent, with variations in the alkyl substituent either in terms of length/size or heteroatom linker, analogous to 7, 8, 9, and 14 from group I. Molecules 19–22 evaluate the length of the alkyl chain, and show decreasing kcat with increased size, in a fashion similar to the group I molecules above. Moreover, the results for KM and inhibition (IC50) do not show any significant trends with the bulk of the alkyl side chain. Alkynyl-substituted 23 has a higher kcat than the analogous alkyl-substituted 21, with overall catalytic efficiency between the smaller 19 and 20. Amino-substituted 24 has a lower kcat (0.60 s–1) when compared to the ether homologue 19 (1.6 s–1), perhaps due to the contribution of the resonance structures of 24 (Scheme 1), which could make the phosphorus atom less electrophilic. The most surprising result here is that several of these compounds demonstrated turnover, in contrast to the group II and other group III molecules (25–29).
Scheme 1.
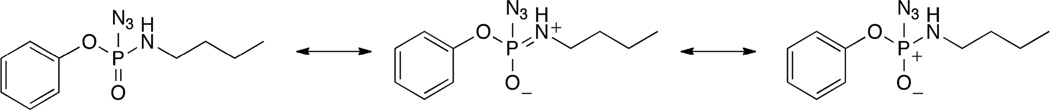
Selected resonance structures of 24
The lower kcat of 24 (0.60 s–1), compared to the similarly sized 19 (1.6 s–1), also suggests that differences in these compounds could be the result of the NH group in the former interacting with either the nucleophile in the active site or the calcium-binding residues such as E53 or D269. On the other hand, despite having comparable KM values, the p-nitrophenoxy substituted analog 10 (KM 0.3 mM), which has two propoxy groups connected to the phosphorus atom, and phenoxy-substituted phosphoryl azide 19 (KM 0.5 mM), which is roughly isosteric (a butoxy group and three azide nitrogen atoms), the kcat of 19 is considerably higher (1.6 s–1, as compared with 0.07 s–1 for 10). Not only is the azide compensating for the absence of the nitro group and enhancing binding to the protein, it appears to also improve the rate of hydrolysis beyond the simple steric aspect sampled in the group I molecules. The results suggest that there may be electrostatic interactions in the active site helping to anchor the azide for hydrolysis, which the O-alkyl substituted group I and II molecules were unable to participate.
Molecules 25–29 are phosphoryl azide analogs based upon a diphenoxy framework, which examine the effects of leaving group modifications on binding to PON1, and none of these molecules are substrates.45 Diphenoxy-substituted phosphoryl azide (25) (Table 2) demonstrates weak competitive inhibition of paraoxon turnover (IC50 > 1 mM). Symmetrical substitution of the phenyl rings at the para position with –SMe (29) results in improved inhibition (IC50 0.3 mM), while the corresponding p-OMe substituted analog (28) is not an inhibitor at all. Interestingly, the asymmetric, mono p-OMe substituted analog 26 was found to be a weak inhibitor (IC50 >1 mM), and the similar, propargyl-substituted 27 is an even better inhibitor of paraoxon hydrolysis (IC50 0.5–1.0 mM). From this series of compounds, it appears clear that modifications at the para position of the phenoxy rings in paraoxon analogs can significantly affect binding.
The SAR results suggest strong constraints for ligand binding to PON1, and additionally, more stringent constraints for turnover. Molecules 19, 21 and 23 (IC50 >1 mM for all three) are weaker inhibitors compared to 20 and 22 (IC50 0.5–1.0 mM for both) suggesting that there could be an appropriate size or volume for the ligand to cause effective inhibition. It is important to note that all of these substrates had only one aromatic ring, while those that had two aromatic rings (25–29) were not substrates, but were instead inhibitors; that is, turnover appears to be prevented with the presence of multiple bulky groups off of phosphorus.
Group IV
The molecules in group IV are paraoxon analogs (Table 3) with alterations in the electronics of the leaving group, similar to molecules 28 and 29 of group III, but which retain the two O-ethyl substituents. Analog 32, which has an electron donating p-SMe group on the phenyl ring, has slightly weaker binding (KM 1.5 mM) compared to paraoxon (KM 0.9 mM), with a comparable kcat (2.0 s–1 versus 2.1 s–1 for paraoxon), suggesting that the catalytic mechanism is not affected significantly by the difference in the leaving group abilities. To further probe these effects, molecules 30, 31, and 33 were studied, which consist of p-iPr, p-OMe, and p-Br substituents on the phenyl group, respectively. These molecules are neither substrates nor inhibitors of paraoxon hydrolysis. The pKa values for p-methoxyphenol, phenol and p-bromophenol are quite close (10.2, 10.0, 9.4), while the pKa of p-nitrophenol is much lower (7.2), meaning that p-nitrophenolate is expected to be a much better leaving group. Despite their electronic similarity, the absence of turnover in 31, in contrast to 32, is puzzling, although a similar trend was observed in the Group III molecules 28 and 29. It is surprising that none of the group IV compounds are inhibitors of paraoxon hydrolysis when the sole differences are on the distal portion of the leaving group. This may suggest that the lack of turnover of 30, 31, and 33 is not just because of a poorer leaving group than paraoxon, but because binding to the p-nitrophenoxy group is favorable.
Table 3.
Kinetic Parameters for Hydrolysis of Group IV by rePON1 G2E6
| Molecule ID |
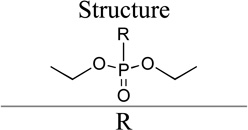 |
kcat s−1 |
KM mM |
kcat/KM M−1 s−1 |
IC50 mM |
|---|---|---|---|---|---|
| R | |||||
| 30 |  |
a | a | a | b |
| 31 |  |
a | a | a | b |
| 32 | 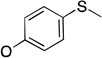 |
2.0 ± 0.3 | 1.5 ± 0.5 | 1,300 ± 400 | b |
| 33 | 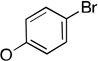 |
a | a | a | b |
No observable activity.
No observable inhibition.
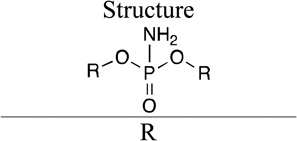
Group V
Phosphoramidates 34 and 35 (IC50 0.1–0.5 mM and 0.5–1 mM, respectively; Table 4) are known inhibitors of DFPase,16 a protein that is highly similar in structure to PON1. These molecules were tested for competitive inhibition of paraoxonase activity in G2E6 PON1, to assay whether the clear structural similarity of that protein would translate to similar kinetic trends. Both molecules did demonstrate inhibition of paraoxonase activity, with 34 being a better inhibitor than 35. These molecules were not tested as substrates due to the lack of a direct spectrophotometric assay. Interestingly, 36 with two phenoxy substituents, is both a substrate and good inhibitor (IC50 0.03 mM). Due to apparent substrate inhibition at higher concentrations (see Supporting Information), we could not directly fit a Michaelis-Menten model to the data, but we can estimate the kcat from the maximum rate (∼ 0.3 s−1) and a lower limit for the kcat/KM (300 M−1 s−1) from the rate at the lowest concentration tested. Molecule 36 is very similar in structure to phosphoryl azide 25. But 25 is not a substrate at all, and is a weak inhibitor (IC50 > 1 mM), suggesting that the NH2 group is favorable for binding.
Table 4.
Kinetic Parameters for Group V (phosphoryl amidates) hydrolysis by rePON1 G2E6
| Molecule ID |
 |
kcat s−1 |
KM mM |
kcat/KM M−1 s−1 |
IC50 mM |
|---|---|---|---|---|---|
| R | |||||
| 34 | ND | ND | ND | 0.1–0.5 | |
| 35 | ND | ND | ND | 0.5–1 | |
| 36 | ∼ 0.3 a | ND | > 300 a | 0.03 |
ND Not determined.
Due to substrate inhibition at higher concentrations, we could only estimate kcat and a kcat/KM (see text and Supporting Information).
The experimental results from our SAR studies suggest that the binding and turnover of paraoxon analogs in PON1 is governed by multiple factors including both steric and electronic properties of the leaving and retained groups. PON1 hydrolyzes a wide variety of different types of molecules, yet relatively small modifications in the leaving group result in dramatic differences in turnover while changes in the retained group have lesser influence in overall catalytic efficiency. In order to gain a molecular level understanding of the ligand–protein interactions behind this interesting result, we employed molecular modeling techniques that include molecular docking, MD simulations, and binding energy calculations to simulate potential binding modes of these substrates in the active site. We discuss our in silico observations of the interactions between ligand and receptor below, providing additional insights into the limits of the enzyme’s promiscuity.
III. B Computational Modeling
Based upon the structure activity relationships probed above as well as previous reports10 on PON1, it is clear that the active site of PON1, while generally tolerant of a range of different classes of substrates including aryl and alkyl esters, phosphoryl azides, as well as phosphoryl triesters, nonetheless has significant limits on the steric and electronic requirements of the leaving group and the retained portion of ligands. In a previous SAR study probing the native function of PON1, Tawfik et al.10 proposed that the protein in its native form is a lactonase, and further that nonspecific hydrophobic effects influenced the substrate binding modes for phosphoryl triesters. The weak inhibition or lack of inhibition of paraoxon hydrolysis by very similar compounds like 17, 30, 31, 32 and 33 suggests that productive interactions with the p-nitrophenoxy moiety (and presumably other peripheral groups) may also affect substrate binding.
The molecules in group I primarily explore the effects of increased steric congestion on the retained portion of the substrates, and generally show trends that can be justified in terms of nonspecific, hydrophobic interactions. With a conserved p-nitrophenolate leaving group, and the variations being only in the alkyl size and steric bulk, we observed two distinct trends across the series. For one, paraoxon is actually optimal in size for hydrolysis by PON1 compared to both larger and smaller near analogs. Above diethoxy, increase in the size and bulk reduced the kcat for turnover of substrates bound to the enzyme up to a point, after which binding was impeded entirely. For the two molecules, 6 and 7, which are smaller than paraoxon, the kcat values were also reduced compared to paraoxon. For the dipropyl analog 10, kcat values were significantly impaired, with KM values comparable with that of paraoxon, and only weak inhibition. The larger symmetrically substituted analogs 11–13 were very poor substrates and poor inhibitors, as they most likely exceeded the spatial requirements in the active site.
The other trend observed in the group I molecules involves the ability of the active site to tolerate asymmetrically substituted paraoxon analogs, and in particular those which have bulky substituents (e.g., O-pentyl-substituted 14). However, for the symmetric molecules, neither the di-O-iPr substituted 10 nor the di-O-butyl analog 12 demonstrated turnover, and only weak inhibition was observed for 10. Explanations for this differential tolerance include the possibility that the bulkier O-alkyl substituents in the symmetric molecules simply exceed the active site space, or that the hetero-substituted molecules reduce the overall size in a sufficient manner for binding and turnover to occur. However, the issue of stereochemistry arises in the ‘mixed’ molecules, and it is also possible that only one enantiomer of the racemate is able to productively bind for hydrolysis; that is, the steric constraint in the active site could be only at one of the two pockets interacting with the O-alkyl fragments, and the enantiomer with the smaller substitution at this position is able to productively bind. It is known that PON1 exhibits stereoselectivity in hydrolysis of some organophosphorus nerve agents, but to-date there have been no studies on the stereoselectivity of the enzyme for hydrolysis of paraoxon analogs.43 Efforts are ongoing to examine whether hydrolysis is proceeding to completion on the chiral analogs, or if turnover is exhibiting bimodal kinetics.
The trends in the group I molecules suggest that the productive binding of p-nitrophenoxy substituted paraoxon analogs in the active site is constrained in terms of retained group size, and may have a requirement for the presence of only two bulky groups off of the phosphorus center, including the leaving group. In the absence of experimentally determined stereospecificities for the enzyme, we can only speculate, based on the predicted mode of binding for the group I molecules in the active site, that the active site tolerance for bulk on one of the two chains is limited.
To attempt to understand these trends at the molecular level, paraoxon and analogs 6–8 and 14 were all docked into the active site of a computational model17 of the G2E6 variant (described previously) of PON1 using AUTODOCK 4.0.39 For paraoxon (1), seven of the ten lowest energy clusters met the basic criterion of having the phosphoryl oxygen bound to calcium, sampling a range of orientations for the p-nitrophenoxy group in the active site, including poses in which the nitro group interacted with residues K70 or K192 via electrostatic interactions, and poses with the p-nitrophenoxy oriented toward F292, without any polar or charged groups in proximity (Figure 5). In the predominantly identified binding modes, the p-nitrophenoxy group was observed either in the cavity between L240 and H285, or in proximity to H134 and K192 (Figure 6). Analyses of the MM-PBSA and MM-GBSA binding energies41,42 after 5 ns of molecular dynamics (MD) simulations suggest that the latter pose (near to H134 and K192) is generally lower in energy by 2–5 kcal mol−1. When the nitro group was in proximity to H134 and K192, the two O-ethyl substituents of paraoxon were in closest proximity to L69, V346, and H285, residues that border the active site. Similar poses were identified for the other molecules of Group I, with consistent energetic ordering, suggesting that such a pose could be a common mode of binding. This orientation of paraoxon analogs in the active site is consistent with other molecular modeling studies of the enzymes, which show a similar placement of paraoxon in a model of the WT variant of PON1.46
Figure 5.

Comparison of binding poses in the H115 and H285 pockets for (left) the SP stereoisomer of 8 and (right) paraoxon, respectively, using the structure from the final frame after 5 ns of MD simulations.
Figure 6.

Orientation of paraoxon (1) from MD simulations with coordinated water molecules near H285 and D269.
Based upon the predicted molecular modeling poses, we propose that the active site constraints across the group I molecules result largely from the hydrophobic pockets interacting with the O-alkyl fragments, and in particular, residues L69, V346, L240, and F222. These residues have been implicated as involved in modulating the selectivity of the enzyme,47 and their proximity and interactions with the bound substrates in our model provides some additional validation of the proposed binding modes. This pose is also consistent with the hypothesis that the nucleophile is in the vicinity of D269, as the p-nitrophenolate leaving group is in a roughly anti orientation (vide infra).
The group II molecules, generated from the removal of the p-nitro group from several group I analogs, showed dramatically reduced turnover and binding (inferred from KM and IC50 values) in PON1, relative to the group I molecules. As it is difficult, if not impossible, for modeling to provide a clear hypothesis in the absence of trends for calibration, we did not undertake detailed modeling studies for this group. The lack of turnover of the phenoxy analogs might simply be a function of the greater leaving group ability of p-nitrophenolate. In fact, G2E6 was produced by directed evolution toward increased activity against paraoxon, and it is possible that G2E6 is optimized for binding to the p-nitrophenoxy moiety.18a The basis of that binding is not clear, since nitro groups are electron-withdrawing but are not strongly polar and are not good hydrogen-bond acceptors. The pKa of a protonated nitrobenzene is about –11, and no binding is detected between nitrobenzene and substituted ureas in chloroform or DMSO.48 Nitrophenyl groups are common in haptens used to raise antibodies, and the nitro group has been shown to be important for binding relative to hydrogen, but comparable to iodo substitution.49 This suggests that the greatest effect of burial of the uncharged nitro group is in the liberation of water upon binding to a greasy active site. But it is also possible that specific interactions may occur, as was observed for nitrophenyl-binding mouse myeloma protein MOPC 315.50 G2E6, for example, has a Lys at 192 (as opposed to Gln or Arg in the human alloforms), and several docked poses of the nitrophenoxy compounds put the nitro group in the vicinity of that Lys. The lack of inhibition by the group IV variants (see below) is the strongest evidence for some specific interaction with the nitrophenoxy group.
The phosphoryl azides and phosphoramidates studied in groups III and V allowed us to probe the effects of modifications for both the retained and leaving groups of these paraoxon analogs. Despite lacking the p-nitrophenoxy group, the molecules in these groups did demonstrate quantifiable inhibition of paraoxonase activity, as well as detectable turnover, in contrast to the severely attenuated activity of the group II, phenolate substituted molecules. For example, paraoxon has a kcat of 2.1 s–1 and a KM of 0.9 mM, while the phenoxy-substituted analog 17 had extremely weak (but detectable) turnover and inhibition of paraoxonase activity. On the other hand, the phosphoryl azide analog closest in size to paraoxon is O-butyl and phenolate- substituted analog 19, which was found to have a kcat of 1.6 s–1 and a KM of 0.5 mM, nearly identical to the values for paraoxon. From these data, we can conclude that modifications of the retained portion of paraoxon analogs, and in particular the inclusion of a more polar azide group instead of the O-alkyl fragment, can compensate for removal of the nitro group from the leaving group portion of the substrates.
Based on our computational results for group III, the azide moiety can potentially play a significant role in the binding of the ligands in the active site: in most cases, the lowest energy pose (based on MM-PB/GBSA binding energies, see supporting information for additional details) was one in which the first nitrogen (N1) of the azide group participates in an electrostatic interaction with E53, which, in turn, is hydrogen bonded with H115 (Figure 7). Alternatively, in some low energy poses, the N1 of the azide is hydrogen bonded directly with H115. In the case of amino-substituted 24(R) for example, the NH group of the ligand is hydrogen-bonded with calcium binding residues N224 or D269 (Figure 8). Overall, the modeling results suggest that the heteroatom substitutions adjacent to phosphorus, and in particular the azide and amino groups, can participate in additional hydrogen bonding and/or electrostatic interactions in the active site relative to the simple O-alkyl substituents of group I. In conjunction with the loss of the p-nitrophenoxy group, it is possible that these interactions are vital to stabilize the orientation of substrate in a productive binding mode in the active site. Also, water molecules were observed interacting with H115, D269, H285, and E53.
Figure 7.

Orientation of 24(R) showing the first nitrogen of the azide group participating in an electrostatic interaction with E53, which is, in turn, hydrogen-bonded with H115.
Figure 8.

(a) Orientation of 24(R), for which the NH group of the ligand is hydrogen-bonded with the calcium coordinating residues N224 or D269. In the latter case, this could interfere with a potential catalytic dyad. (b) The alkyl side chain was often observed to be facing H285, and the oxygen of the aryloxy and alkoxy group is often hydrogen-bonded to N168, which, in turn, is hydrogen-bonded to D183.
It could be argued that heteroatom substitutions could affect the charges on the phosphorus atom, and thus its electrophilicity, as well as binding with the calcium through the phosphoryl oxygen. Calculations of atomic partial charges were performed using the ChelpG28 and natural population analysis (NPA)51 methods, and in the ChelpG charge calculations, the atomic charges on the phosphorus atoms of 19–23 (with O-alkyl substituents) were within the range of +1.36 and +1.39 e. The NPA charges were also quite similar across the series, in the range of +2.49 and +2.52 e, confirming that the variations in the length of alkyl chain did not affect the charges on the phosphorus atom. However, the positive charge on the phosphorus atom for the phosphoramidate 24 (+1.22 e ChelpG and +2.44 e NPA) was lower, consistent with the electronegativity differences between oxygen and nitrogen.
The results of the group IV series clearly demonstrate that the substitution at the para position affected the binding and turnover of paraoxon analogs. In general, the trend of these molecules was that the substitution of electron-donating groups at the para position destroys activity (as in the i-propyl and –OMe substituents). Weakly electron-withdrawing substitutions, such as p-Br also negated activity and inhibition. Interestingly, substitution with the electron-donating p-SMe, in 32, still allowed for the turnover by PON1 at a rate comparable to paraoxon.
The binding poses of 32 were similar to those identified for paraoxon, with similar leaving group orientations in the two main poses. That is, the leaving group was oriented toward either the H285 pocket or the H115/H134 pocket (Figure 9). Given the high structural similarity between paraoxon, 31, and 32, it is unlikely that computational modeling can explain the dramatic differences in activity, particularly without additional analogues, which do demonstrate turnover; this is an ongoing area of study.
Figure 9.

Two orientations of 32 from MD simulations, with the leaving group (a) toward the H285 pocket, and (b) anti to D269.
The last class of molecules, group V, are phosphoramidates that are either known DFPase inhibitors or derivatives thereof. We lacked a spectroscopic probe to monitor the potential hydrolysis of the cyclopropyl- and cyclopentyl-substituted analogs 34 and 35. Compound 36 is a relatively good substrate for G2E6, but its kinetics are complicated by substrate inhibition. Interestingly, 36 represents the only diphenoxy-substituted molecule which demonstrated turnover by PON1. More importantly, the IC50 values obtained for the group V molecules in general, and 36 in particular, were by far the lowest of any of the molecules we studied. In fact, 36 was the only compound with an IC50 value below the testing concentration of paraoxon, implying that it binds more tightly than paraoxon. While the mode of inhibition for the other groups was inconclusive due to solubility issues, the group V molecules were identified as competitive inhibitors (see Supporting Information), supporting the notion that binding in the active site is taking place. Comparison of inhibition between 36 and the azido analog 25 suggests that binding is strongly enhanced by the amino group.
When docking and molecular dynamics simulations were performed, poses similar to those for groups III and IV were identified for the phosphoramidate 36 following binding simulations. The two phenyl rings were oriented toward the H285 and H115 pockets, while the amine hydrogen bonded with N224, one of the calcium binding residues (Figure 10). Apart from clearly demonstrating that subtle modifications in the substrates lead to significant changes in the enzymatic activity against PON1, the results from this study also provide some interesting implications for the potential catalytic mechanism of the enzyme. Worek et al., on the basis of kinetic and mutagenesis data, in conjunction with a neutron scattering study of the related enzyme DFPase, have proposed a mechanism for the hydrolysis of OP compounds by PON1.16,52 Specifically, they proposed that D269 acts as a nucleophile and attacks the OP substrate at the electrophilic phosphorus atom, with the ‘catalytic’ calcium acting as an oxyanion hole to stabilize the developing negative charge on the phosphoryl oxygen, before the leaving group is expelled to form an OP–PON1 covalent complex (Scheme 2). As discussed above, a second hydrolysis step must necessarily follow to regenerate the free D269 for the protein to act in a catalytic fashion. However, both the crystal structure7 of G2E6 (which has a phosphate ion bound to the catalytic calcium) and our molecular modeling indicate that the oxygen atom of D269 might reside as far as ∼6 Å away from the phosphorus atom, which would require a large conformational change, both in terms of the coordination environment of the calcium ion and of the orientation of the substrate, for a nucleophilic attack to take place. Modeling by Wallqvist et al. suggests a separation of 4–5 Å between D269 and the phosphorus atom of paraoxon bound in the putative active site following docking and molecular dynamics simulations.46 Given that D269 also coordinates to the catalytic calcium, reducing its nucleophilicity, this mechanism, if operative, would likely involve, at a minimum, the transient loss of coordination between D269 and the calcium ion. Unfortunately, validation of this hypothesis through mutagenesis has been ambiguous: while mutants at D269 (including D269E)21 were inactive against OP compounds in prior studies, this is not necessarily indicative of the participation of this residue in catalysis, as the coordination of the ‘catalytic’ calcium coordination could also be disrupted.
Figure 10.

Pose from 5 ns MD simulation of 36 bound to PON1, showing that water molecules in the H115/134 pocket are not oriented anti (red arrow) to the leaving group for productive hydrolysis.
Scheme 2.

Mechanism of hydrolysis of OP compounds involving D269 as the nucleophile.52,56
The absence of either irreversible inhibition or aging (as in the case of acetylcholinesterase53,54) in the experimental data, which would likely occur along with formation of a covalent OP–PON1 adduct, further detracts from direct attack as a mechanistic hypothesis.55 Aging results in the inhibition of cholinesterases by OPs.53,54 The cholinesterases react with OPs via the formation of an acyl-enzyme intermediate, following nucleophilic attack of a catalytic triad of Glu-His-Ser onto the phosphorus of the OP. Following the formation of this ligated intermediate, aging occurs by the displacement of a second leaving group on the adduct via an activated water molecule, resulting in the generation of a negatively charged acyl-enzyme adduct, which is resistant to any known methods of reactivation.53,54 PON1 is not known to age, suggesting that even if a nucleophilic amino acid residue of the protein were involved in hydrolysis directly, the catalytic nature of the reaction would require regeneration of the free residue following a second hydrolytic step, most likely involving a water molecule, to regenerate the active protein.55 For carboxylesterases, which are known to form a covalent intermediate following reaction with OPs, the outcome is inhibition, followed in some instances by a spontaneous reactivation step.58 It is worth noting that the mixed anhydride that would form on phosphylation of D269 would be more labile to hydrolysis than the phosphylated Ser in AChE and carboxylesterase, which might explain the lack of aging in PON1 if a direct attack is occurring.
An alternative that is consistent with the experimental and computational studies we present here is a general-base catalytic mechanism, although the methodology employed herein does not allow for direct comparison of the several potential reaction pathways. For example, D269, possibly with assistance from E53 or another basic residue in proximity, could activate a coordinated water molecule to create hydroxide, which could subsequently attack the OP compound (Scheme 3). In many of our MD simulations, we observed water molecules close enough to D269 and E53 for this process to be plausible. Further, in most of the cases, the aryloxy leaving group is oriented towards the H115 pocket and the other substituents on the phosphorus center are oriented away from the H285/D269 pocket, providing a favorable anti orientation of the leaving group relative to an approaching nucleophile.
Scheme 3.

Proposed mechanism of hydrolysis of OP compounds involving activation of water by D269
In either of these proposed mechanisms, the anti and angular approach of the nucleophile to the phosphorus center would require displacement of the O-alkyl fragments, so the presence of larger alkyl retained groups could increase the kinetic barrier for reaction, or may completely prevent association and activation of the water molecules. In the MD simulations, the water molecules were fluxional, and only in some orientations was a stable coordination observed, such that a more detailed examination of the reaction pathway will be required to fully explore the mechanism. Such a process would require the use of hybrid QM/MM methods57 to fully map the potential energy surface for hydrolysis, and such detailed computational studies are underway.
This study certainly does not rule out a direct nucleophilic attack on OPs by D269 or some other residue, but activation of water is more consistent with our modeling data and the apparent lack of aging. Pre-steady state kinetics to look for a burst phase from formation of the putative mixed anhydride and mass spectrometry to look for small amounts of derivatized or even aged enzyme are required to shed more light on these alternative mechanisms. Such experiments are challenging, though, and lack of a burst phase or lack of detection of derivatized enzyme does not rigorously rule out direct attack. Further work toward this end is underway.
IV. CONCLUSIONS
The primary observation from our SAR studies is that while PON1 accepts a wide variety of substrates, subtle variations in the composition of paraoxon analogs led to significant changes in activity. In particular, alterations in the size and electronics of the peripheral groups on the phosphorus, as well as the leaving group, significantly alter both the binding and turnover of paraoxon analogs. We propose that these changes may be the result of a constrained substrate orientation for productive hydrolysis. However, with the nitro group as well as the modifications closer to the phosphorus atom in groups III and V of this study, the paraoxon analogs studied herein may well be capable of additional, specific interactions in the protein active site beyond those in molecules used earlier studies. Between the group I, II and IV molecules, dramatic differences in binding to the protein were observed following removal of the nitrophenoxy group, generally to the detriment of binding and turnover. However, when this was combined with alterations of the retained group, and in particular the inclusion of heteroatoms, productive binding to the protein was recovered.
Computational modeling provides a possible rationalization for these effects in the observation of electrostatic interactions between residues in the active site periphery and the nitro group or other heteroatom substituents. This could provide additional constraints on the orientation of ligands in the active site that contribute to the large differences in activity across sterically similar molecules. Closer to the phosphorus, several substrates varied in the heteroatom substitution of the side chain linkages, which could alter their interactions with the calcium binding pocket residues. Our modeling also suggests that several non-polar residues in the active site periphery (e.g. L69, F222, L240, V346) are most involved in hydrophobic interactions with the retained groups; thus, we predict that alterations of these residues modulate the steric constraints of the active site. Unfortunately, it will be difficult to make mutations to test this hypothesis, since such mutations may also alter the environment of the ‘catalytic’ calcium.
PAL labeling studies using molecules identified in this work are ongoing, and preliminary results suggest that labeling of active site residues has taken place. This will further improve our knowledge of the substrate binding modes, and may allow for direct identification of residues involved in specificity and activity of PON1.
Supplementary Material
Acknowledgments
We gratefully acknowledge very generous allocations of computational resources from the Ohio Supercomputer Center and funding from the National Institutes of Health via the Center of Excellence grant U54NS058183 (to TJM and CMH). We also appreciate many helpful discussions with Dr. David E. Lenz, Dr. Douglas M. Cerasoli and Dr. Tamara Otto (USAMRICD).
Footnotes
VI. Supporting Information Available
Additional experimental and computational information, including more detailed experimental procedures, results from analytical and spectral characterization, kinetic assays, complete ligand libraries, and additional results from modeling, are included in the supporting information.
References
- 1.(a) Furlong CE. In: Paraoxonases: An historical perspective. The Paraoxonases: Their role in disease, development and xenobiotic metabolism. Proteins and cell regulation. Mackness B, Mackness M, Aviram M, Paragh G, editors. NY: Springer; 2008. pp. 3–32. [Google Scholar]; (b) Gaidukov L, Tawfik DS. J. Lipid. Res. 2007;48:1637–1646. doi: 10.1194/jlr.D600045-JLR200. [DOI] [PubMed] [Google Scholar]
- 2.(a) Khersonsky O, Tawfik DS. ChemBioChem. 2006;7:49–53. doi: 10.1002/cbic.200500334. [DOI] [PubMed] [Google Scholar]; (b) Aharoni A, Gaidukov L, Khersonsky O, Gould SM, Roodveldt C, Tawfik DS. Nat. Genetics. 2005;37:73–76. doi: 10.1038/ng1482. [DOI] [PubMed] [Google Scholar]
- 3.(a) Yeung DT, Lenz DE, Cerasoli DM. Metabol. Clinic. Exptl. 2008:151–170. [Google Scholar]; (b) Cannard K. J. Neurol. Sci. 2006;249:86–94. doi: 10.1016/j.jns.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 4.Lenz DE, Yeung DT, Smith RJ, Sweeney RE, Lumley LA, Cerasoli DM. Toxicol. 2007;233:31–39. doi: 10.1016/j.tox.2006.11.066. [DOI] [PubMed] [Google Scholar]
- 5.(a) Costa LG, Vitalone A, Cole TB, Furlong CE. Biochem. Pharmacol. 2005;69:541–550. doi: 10.1016/j.bcp.2004.08.027. [DOI] [PubMed] [Google Scholar]; (b) Sorenson RC, Primo-Parmo SL, Kuo C, Adkins S, Lockridge O, La Du BN. Proc. Natl. Acad. Sci. USA. 1995;92:7187–719. doi: 10.1073/pnas.92.16.7187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rochu D, Chabrière E, Masson P. Toxicol. 2007;233:47–59. doi: 10.1016/j.tox.2006.08.037. [DOI] [PubMed] [Google Scholar]
- 7.Harel M, Aharoni A, Gaidukov L, Brumshtein B, Khersonsky O, Meged R, Dvir H, Ravelli RB, McCarthy A, Toker L, Silman I, Sussman JL, Tawfik DS. Nature Mol. Biol. 2004;11:412–419. doi: 10.1038/nsmb767. [DOI] [PubMed] [Google Scholar]
- 8.Otto TC, Harsch CK, Yeung DT, Magliery TJ, Cerasoli DM, Lenz DE. Biochem. 2009;48:10416–10422. doi: 10.1021/bi901161b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Ghamen E, Raushel FM. Toxicol. Appl. Pharmacol. 2005;207:S459–S470. doi: 10.1016/j.taap.2005.02.025. [DOI] [PubMed] [Google Scholar]; (b) Tsai PC, Fan YB, Kim J, Yang LJ, Almo SC, Gao YQ, Raushel FM. Biochemistry. 2010;49:7988–7997. doi: 10.1021/bi101058z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Khersonsky O, Tawfik DS. Biochemistry. 2005;44:6371–6382. doi: 10.1021/bi047440d. [DOI] [PubMed] [Google Scholar]; (b) Draganov DI, Teiber JF, Speelman A, Osawa Y, Sunahara R, La Du BN. J. Lipid. Res. 2005;46:1239–1247. doi: 10.1194/jlr.M400511-JLR200. [DOI] [PubMed] [Google Scholar]
- 11.(a) Chamers JE. Proc. Natl. Acad. Sci. USA. 2008;105:12639–12640. doi: 10.1073/pnas.0807062105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Draganov DI, La Du BN. Naunyn-Schmiedeberg's Arch. Pharmacol. 2004;369:78–88. doi: 10.1007/s00210-003-0833-1. [DOI] [PubMed] [Google Scholar]
- 12.Michel LP, Thierry D, Chapmann JM. Clinic. Chem. Med. 1998;36:431–441. [Google Scholar]
- 13.(a) Sklan EH, Lowenthal A, Kroner M, Ritov Y, Landers DM, Rankinen T, Bouchard C, Leon AS, Rice T, Rao DC, Wilmore JH, Skinner JS, Soreq H. Proc. Natl. Acad. Sci. USA. u2004;101:5512–5517. doi: 10.1073/pnas.0307659101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Aviram M, Rosenblat M. Paraoxonases (PON1, PON2, PON3) analyses in vitro and in vivo in relation to cardiovascular diseases. Methods in Molecular Biology (Totowa, NJ, United States) 2008 doi: 10.1007/978-1-60327-517-0_20. [DOI] [PubMed] [Google Scholar]
- 14.(a) Pinkse MWH, Rijkers DTS, Dostmann WR, Heck AJR. J. Biol. Chem. 2009;284:16354–16368. doi: 10.1074/jbc.M808521200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Voskresenska V, Wilson RM, Tarnovsky AN, Vyas S, Winter AH, Hadad CM. J. Am. Chem. Soc. 2009;131:11535–11547. doi: 10.1021/ja902224m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bargota R, Akhtar M, Biggadike K, Gani D, Allenmann RK. Bioorganic & Medicinal Chemistry Letters. 2003;13:1623–1626. doi: 10.1016/s0960-894x(03)00290-7. [DOI] [PubMed] [Google Scholar]
- 16.Blum M, Löhr F, Richardt A, Rüterjans H, Chen GC-H. J. Am. Chem. Soc. 2006;128:12750–12757. doi: 10.1021/ja061887n. [DOI] [PubMed] [Google Scholar]
- 17.Sanan TT, Muthukrishnan S, Beck JM, Tao P, Hayes CJ, Otto TC, Cerasoli DM, Lenz DE, Hadad CM. J. Phys. Org. Chem. 2010;23:357–369. doi: 10.1002/poc.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Aharoni A, Gaidukov L, Yagur S, Toker L, Silman I, Tawfik DS. Proc. Natl. Acad. Sci. USA. 2004;101:482–487. doi: 10.1073/pnas.2536901100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gan KN, Smolen A, Eckerson HW, La Du BN. Drug Met. Disposn. 1991;19:100–110. [PubMed] [Google Scholar]
- 19.Mieko KK, Yasuo S. Anal. Biochem. 2009;385:94–100. [Google Scholar]
- 20.Josse D, Ebel C, Stroebel D, Fontaine A, Borges F, Echalier A, Baud D, Renault F, Maire M, Chabrieres E, Masson P. J. Biol. Chem. 2002;277:33386–33397. doi: 10.1074/jbc.M200108200. [DOI] [PubMed] [Google Scholar]
- 21.Yeung DT, Josse D, Nicholson JD, Khanal A, McAndrew CW, Bahnson BJ, Lenz DE, Cerasoli DM. Biochimica et Biophysica Acta. 2004;1702:67–77. doi: 10.1016/j.bbapap.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 22.Yeung DT, Lenz DE, Cerasoli DM. FEBS J. 2005;272:2225–2230. doi: 10.1111/j.1742-4658.2005.04646.x. [DOI] [PubMed] [Google Scholar]
- 23.Case DA, Darden TA, Cheatham III TE, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Pearlman DA, Crowley M, Walker RC, Zhang W, Wang B, Hayik S, Roitberg A, Seabra G, Wong KF, Paesani F, Wu X, Brozell S, Tsui V, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Beroza P, Mathews DH, Schafmeister C, Ross WS, Kollman PA. AMBER 9. San Francisco: University of California; 2006. [Google Scholar]
- 24.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA, A N. Nucleic Acids Research. 2004;32:665–667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 26.Luo R, David L, Gilson MK. J. Comput. Chem. 2002;23:1244–1253. doi: 10.1002/jcc.10120. [DOI] [PubMed] [Google Scholar]
- 27.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, Kollman PA. J. Comput. Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 28.Breneman CM, Wiberg KB. J. Comput. Chem. 1990;11:361–373. [Google Scholar]
- 29.(a) Becke AD. J. Chem. Phys. 1993;98:5648–5656. [Google Scholar]; (b) Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 30.Labanowski J. Density Functional Methods in Chemistry. Heidelberg: Springer-Verlag; 1991. [Google Scholar]; (b) Parr RG, Y Weitao Y. Density-Functional Theory in Atoms and Molecules. New York: Oxford University Press; 1989. [Google Scholar]
- 31.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr., Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Revision C.02. Gaussian, Inc. Wallingford CT.: 2004. [Google Scholar]
- 32.Weiser J, Shenkin PS, Still WC. J. Comput. Chem. 1999;20:217–230. doi: 10.1002/(SICI)1096-987X(199905)20:7<688::AID-JCC4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 33.(a) Mahoney MW, Jorgensen WL. J. Chem. Phys. 2000;112:8910–8922. [Google Scholar]; (b) Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J. Chem. Phys. 1983;79:926–93. [Google Scholar]
- 34.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 35.Aqvist J. J. Chem. Phys. 1990;94:8021–8024. [Google Scholar]
- 36.Harvey SC, Tan RK, Cheatham TE., III J. Comput. Chem. 1998;19:726–740. [Google Scholar]
- 37.Chachra R, Rizzo RC. J. Chem. Theory. Comp. 2008;4:1526–1540. doi: 10.1021/ct800068v. [DOI] [PubMed] [Google Scholar]
- 38.Miyamoto S, Kollman PA. J. Comput. Chem. 1992;13:952–962. [Google Scholar]
- 39.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. J. Comput. Chem. 1998;19:1639–1662. [Google Scholar]
- 40.Bruice TC. Chem. Rev. 2006;106:3119–3139. doi: 10.1021/cr050283j. [DOI] [PubMed] [Google Scholar]
- 41.(a) Sharp SKA, Honig B. J. Phys. Chem. 1994;98:1978–1988. [Google Scholar]; (b) Luo R, David L, Gilson MK. J. Comput. Chem. 2002;23:1244–1253. doi: 10.1002/jcc.10120. [DOI] [PubMed] [Google Scholar]; (c) Brigo A, Molinaro H. Biophys. J. 2003;85:159–166. doi: 10.1016/S0006-3495(03)74462-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sigalov G, Onufriev P, Scheffel P. J. Chem. Phys. 2005;122:94511–9451. doi: 10.1063/1.1857811. [DOI] [PubMed] [Google Scholar]
- 43.Amitai G, Gaidukov L, Adani R, Yishay S, Yacov G, Kushnir M, Teitlboim S, Lindenbaum M, Bel P, Khersonsky O, Tawfik DS, Meshulam H. FEBS J. 2006;273:1906–1919. doi: 10.1111/j.1742-4658.2006.05198.x. [DOI] [PubMed] [Google Scholar]
- 44.(a) Vyas S, Muthukrishnan S, Kubicki J, McCulla RD, Burdzunski G, Sliwa M, Platz MS, Hadad CM. J. Am. Chem. Soc. 2010;132:16796–16804. doi: 10.1021/ja909327z. [DOI] [PubMed] [Google Scholar]; (b) Vyas S, Luk HL, Muthukrishnan S, Kubicki J, Platz MS, Hadad CM. (unpublished results) [Google Scholar]
- 45.We computed the thermochemistry for the hydrolysis of all the molecules in our library in the absence of the enzyme (see Supporting Information for additional details). It was observed that the enthalpy of hydrolysis for substrates with p-nitrophenolate leaving group (for example, 1, paraoxon) was only 2–3 kcal/mol more negative than those without the p-nitrophenolate leaving group (for example, 32) suggesting that the leaving group effects are minimal from a thermochemical view. For group III ligands, the possibility of the azide group being hydrolyzed can be excluded since such a reaction has an enthalpic penalty of >15 kcal/mol
- 46.Hu X, Jiang X, Lenz DE, Cerasoli DM, Wallqvist A. Proteins. 2009;75:486–498. doi: 10.1002/prot.22264. [DOI] [PubMed] [Google Scholar]
- 47.Gupta RD, Goldsmith M, Ashani Y, Simo Y, Mullokandov G, Bar H, Ben-Davis M, Leader H, Margalit R, Silman I, Tawfik DS. Nature Chem. Biol. 2011;7:120–125. doi: 10.1038/nchembio.510. [DOI] [PubMed] [Google Scholar]
- 48.Kelly TR, Kim MH. J. Am. Chem. Soc. 1994;116:7072–7080. [Google Scholar]
- 49.Nisonoff A, Shaw AR, Pressman D. J. A. Chem. Soc. 1959;81:1418–1423. [Google Scholar]
- 50.Gettins P, Givol D, Dwek RA. Biochem. J. 1978;173:713–722. doi: 10.1042/bj1730713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.(a) Reed AE, Weinhold RB. J. Chem. Phys. 1985;78:735–746. [Google Scholar]; (b) Roos G, Geerlings P, Messens J. J. Phys. Chem. A. 2009;113 doi: 10.1021/jp9034584. 13465-1347. [DOI] [PubMed] [Google Scholar]
- 52.(a) Blum M, Timperley CM, Williams GR, Thiermann H, Worek F. Biochem. 2008;47:5216–5224. doi: 10.1021/bi702222x. [DOI] [PubMed] [Google Scholar]; (b) Blum M, Koglin A, Rüterjans H, Schoenborn P, Langan P, Chen JCH. Acta Cryst. 2007;F63:42–4. doi: 10.1107/S1744309106052924. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Blum M, Mustyakimov M, Rüterjans H, Kehe K, Schoenborn BP, Langan P, Chen JCH. Proc. Natl. Acad. Sci. USA. 2009;106:713–718. doi: 10.1073/pnas.0807842106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ekstrom F, Akfur C, Tunemalm A, Lundberg S. Biochemistry. 2006;45:74–81. doi: 10.1021/bi051286t. [DOI] [PubMed] [Google Scholar]
- 54.Quinn DM. Chem. Rev. 1987;87:955–979. [Google Scholar]
- 55.Worek F, Eyer P, Szinicz L. Arch. Toxicol. 1998;72:996–998. doi: 10.1007/s002040050546. [DOI] [PubMed] [Google Scholar]
- 56.Khersonsky O, Tawfik DS. J. Biol. Chem. 2006;281:7649–7656. doi: 10.1074/jbc.M512594200. [DOI] [PubMed] [Google Scholar]
- 57.Ramos MJ, Fernandes PA. Acc. Chem. Res. 2008;41:689–698. doi: 10.1021/ar7001045. [DOI] [PubMed] [Google Scholar]
- 58.Fleming CD, Edwards CC, Kirby SD, Maxwell DM, Potter PM, Cerasoli DM, Redinbo MR. Biochemistry. 2007;46:5063–5071. doi: 10.1021/bi700246n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.