

Abstract
Purpose.
We hypothesized that loss of insulin-like growth factor binding protein 3 (IGFBP-3) signaling would produce neuronal changes in the retina similar to early diabetes.
Methods.
To understand better the role of IGFBP-3 in the retina, IGFBP-3 knockout (KO) mice were evaluated for neuronal, vascular, and functional changes compared to wild-type littermates. We also cultured retinal endothelial cells (REC) in normoglycemia or hyperglycemia to determine the interaction between IGFBP-3 and TNF-α, as data indicate that both proteins are regulated by β-adrenergic receptors and respond antagonistically. We also treated some cells with Compound 49b, a novel β-adrenergic receptor agonist we have reported previously to regulate IGFBP-3 and TNF-α.
Results.
Electroretinogram analyses showed decreased B-wave and oscillatory potential amplitudes in the IGFBP-3 KO mice, corresponding to increased apoptosis. Retinal thickness and cell numbers in the ganglion cell layer were reduced in the IGFBP-3 KO mice. As expected, loss of IGFBP-3 was associated with increased TNF-α levels. When TNF-α and IGFBP-3 were applied to REC, they worked antagonistically, with IGFBP-3 inhibiting apoptosis and TNF-α promoting apoptosis. Due to their antagonistic nature, results suggest that apoptosis of REC may depend upon which protein (IGFBP-3 versus TNF-α) is active.
Conclusions.
Taken together, loss of IGFBP-3 signaling results in a phenotype similar to neuronal changes observed in diabetic retinopathy in the early phases, including increased TNF-α levels.
Keywords: IGFBP-3, apoptosis, TNF-α
IGFBP-3 KO mice have the early characteristics of diabetic retinopathy noted in rodents.
Introduction
Early studies using sympathetic nerve ganglia suggested that diabetes could disrupt neurotransmitter release.1 Subsequently, we have found that surgical removal of the rat superior cervical ganglion, which provides sympathetic innervation to the eye, concomitantly produces retinal changes similar to those observed in diabetes, despite normal glucose levels.2 Loss of β-1–adrenergic receptor activity also elicits vascular and neuronal damage in the retina similar to diabetes.3 Importantly, restoration of β-adrenergic receptor signaling was able to abrogate these deleterious changes in the retina.4,5 Taken together, these data indicate that β-adrenergic receptors regulate cellular signaling in the retina that may maintain neuronal and vascular retinal homeostasis, and that loss of β-adrenergic signaling may lead to diabetes-like symptoms.
To determine if artificially enhancing β-adrenergic function could ameliorate diabetic retinal damage, we developed and tested a novel β-adrenergic receptor agonist, Compound 49b, using streptozotocin-induced diabetic rats. We found that, as predicted, treatment with Compound 49b reduced vascular, neuronal, and functional changes in the diabetic retina.5 One of the key diabetes-induced protein changes observed in these studies was a significant decrease in insulin-like growth factor binding protein 3 (IGFBP-3).5 IGFBP-3 was decreased in lysates from streptozotocin-treated diabetic retina as well as from primary human retinal endothelial cells exposed to high glucose conditions.5 Compound 49b reversed the decrease in both cases, suggesting that β-adrenergic stimulation of IGFBP-3 could trigger pathways that protect against diabetic damage in the retina.
While IGFBP-3 has been recognized as a regulator of apoptosis in several tissues, its exact actions vary with cell type. In primary retinal endothelial cells IGFBP-3 appears to be antiapoptotic, consistent with our studies of diabetic retina, whereas in immortalized cancer cell lines it has been well established that IGFBP-3 promotes apoptosis via several IGF-independent mechanisms.6–8 Interestingly, IGFBP-3 also has been shown to be antiapoptotic in retinal injury models.9 Thus, IGFBP-3 appears to promote apoptosis in cancer cells, but inhibit apoptosis in normal cells, such as retina.
In addition to its upregulation of IGFBP-3, Compound 49b also significantly reduced TNF-α levels in diabetic retina,5 suggesting the possibility that these two constituents may be linked in their response to diabetic conditions. Consistent with this notion, others have reported previously that TNF-α receptor knockout (KO) mice do not develop diabetic-like retinopathy.10 The exact interaction between IGFBP-3 and TNF-α in diabetic retina is unknown, and may be cell-type specific, as work in bronchial epithelial cells shows that IGFBP-3 can inhibit TNF-α levels,11 but TNF-α appears to increase IGFBP-3 in mammary epithelial cells,12 salivary gland tumor cells,13 and in mouse embryonic fibroblasts.14 Work in transgenic mice with TNF-α overexpression showed increased IGFBP-3 levels in the brain;15 however, this may be related to changes in IGF-1.
To examine further the actions of IGFBP-3 and TNF-α in the retina, we acquired IGFBP-3 KO mice to evaluate neuronal changes in the retina, as well as to investigate whether IGFBP-3 and TNF-α are linked antagonistically by pathways that, together, regulate apoptosis of neuronal elements that may be triggered in the diabetic retina.
Materials and Methods
Retinal Endothelial Cells
Primary human retinal microvascular endothelial cells (REC) were acquired from Cell System Corporation (Kirkland, WA). Cells were grown in M131 medium containing microvascular growth supplements (MVGS; Invitrogen, Carlsbad, CA), 10 μg/μL gentamicin and 0.25 μg/μL amphotericin B. In the high glucose condition, cells were transferred to high glucose (25 mM; Cell System Corporation) medium, supplemented with MVGS and antibiotics for 3 days. Only primary cells within passages 6 were used. Cells were quiesced by incubating in high or normal glucose medium without growth factors for 24 hours, and used to perform the experiments unless otherwise indicated. Treatment with Compound 49b was done for 30 minutes at 50 nM, while recombinant TNF-α was applied at 5 ng/mL for 30 minutes. We did not perform mannitol controls, as we have reported previously that cell death markers and IGFBP-3 levels are not altered by mannitol treatment.16
Transfection of Plasmid DNA and Small Interfering RNA (siRNA)
The cells were transfected with IGFBP-3 NB plasmid DNA at 1 μg/mL using Lipofectamine 2000, according to the manufacturer's instructions. The cells were used for experiment 24 hours after transfection.5 ON-TARGETplus SMARTpool (Dharmacon, Inc., Lafayette, CO) human IGFBP-3 siRNA were used at a final concentration of 20 nM using RNAiMAX transfection reagent according to the manufacturer's instructions. For control of siRNA experiments, nontargeting siRNA #1 (Dharmacon, Inc.) was used as a nonspecific control. REC were transfected with siRNA at a final concentration of 20 nM using RNAiMAX transfection reagent according to the manufacturer's instructions. The cells were used for experiments 24 hours after transfection.
IGFBP-3 KO Mice
IGFBP-3 KO mice were provided generously by Dr John Pintar (Rutgers University, New Brunswick, NJ). Confirmation of the knockout of the IGFBP-3 gene was completed using Southern blotting. C57/BL6 wild-type mice were used as controls, as this is the background of the IGFBP-3 KO mice. Wild-type mice were purchased from Charles River Laboratories, Inc. (Frederick, MD). All mice were sacrificed and ocular tissues collected at 2 months of age. All animal studies complied with ARVO Animal Statement for the Use of Animals in Ophthalmic and Vision Research.
Electroretinogram
ERG analyses were done to evaluate the changes in the electrical activity of the retina as we have reported previously.5,17 Briefly, mice were dark-adapted overnight. ERG responses were recorded from both eyes together using platinum wire corneal electrodes, forehead reference electrode, and ground electrode in the tail. Pupils were fully dilated using 1% tropicamide solution (Alcon Laboratories, Elkridge, MD). Methylcellulose (Celluvise; Allergan, Irvine, CA) drops were applied as well to maintain a good electrical connection, and body temperature was maintained at 37°C by a water-based heating pad. ERG waveforms were recorded with a bandwidth of 0.3 to 500 Hz and samples at 2 kHz by a digital acquisition system, and were analyzed with a custom-built program (MatLab; MathWorks, Natick, MA). Statistics was done on the mean ± SD amplitudes of the a- and b-wave of each treatment group.
Neuronal Analyses
Formalin-fixed paraffin sections were stained with hematoxylin and eosin for light microscopy and morphometry of retinal thickness as described previously.4 Photomicrographs were assessed for retinal thickness and the number of cells in the ganglion cell layer using methods described previously. Multiple images were taken throughout the retina and representative images are from the same region. The thickness of the retina and the cell count were measured using OpenLab software (Improvision, Lexington, MA).5
Vascular Analyses
Retinas from 1 eye of wild-type and IGFBP-3 KO mice (N = 5 mice in each group) were used to count degenerate capillaries. The eyes were enucleated, suspended in 10% buffered formalin for 5 days, and the retina was digested in elastase to remove the neuronal cells from the vasculature.18 The retinal vascular tree was dried onto a glass slide and stained with hematoxylin–periodic acid–Shiff. Degenerate capillaries were counted and identified as described previously.5
Western Blot Analysis
After appropriate treatments and rinsing with cold PBS, REC were lysed in the lysis buffer containing the protease and phosphatase inhibitors, and scraped into the tubes. Retinal extracts were prepared by sonication. Equal amounts of protein from the cell or tissue extracts were separated on the pre-cast tris-glycine gel (Invitrogen), blotted onto a nitrocellulose membrane. After blocking in TBST (10 mM Tris-HCl buffer, pH 8.0; 150 mM NaCl; 0.1% Tween 20) and 5% (wt/vol) BSA, the membrane was treated with appropriate primary antibodies followed by incubation with secondary antibodies labeled with horseradish peroxidase. Antigen–antibody complexes were detected by chemiluminescence reagent kit (Thermo Scientific, Waltham, MA). Primary antibodies used were phosphorylated Akt (Serine 473), Akt, Cytochrome C, Bax, Bcl-xL (all purchased from Cell Signaling, Danvers, MA), beta actin (Santa Cruz Biotechnology, Dallas, TX), and IGFBP-3 (GroPep Bioreagents Pty Ltd, Adelaide, Australia). Western blot analyses of proteins of interest were compared to beta actin levels, and a ratio is presented.
ELISA Analysis
A cleaved caspase 3 ELISA (Cell Signaling) was used to measure levels of the active apoptotic marker in whole retinal lysates, with equal protein loaded (50 μg) to allow for analyses using optical density measurements. TNF-α protein concentrations were measured using a TNF-α ELISA (ThermoFisher, Pittsburgh, PA). We confirmed IGFBP-3 levels using a mouse IGFBP-3 ELISA (R&D Systems, Minneapolis, MN). Cell Death was measured using the Roche Cell Death ELISA (Roche Diagnostics Corporation, Indianapolia, IN), which measures DNA fragmentation, and done according to manufacturer's instructions with equal protein loaded into all wells.
TUNEL Assay
TUNEL analyses were completed on C57/BL6 mice and IGFBP-3 KO mice at 2 months of age. TUNEL was done according to manufacturer's instructions using the ApopTag-Red kit (Millipore, Billerica, MA).
Results
IGFBP-3 KO Mice Have Increased Levels of Proapoptotic Factors
We have reported previously that streptozotocin-induced diabetes significantly reduced IGFBP-3 levels, which occurred concurrently with increased apoptotic protein levels in retinal endothelial cells and in vivo.5 To determine whether this was related directly to the reduced IGFBP-3 levels, we evaluated the same key apoptotic factors in IGFBP-3 KO mice. We found that the absence of IGFBP-3 increased bax, cleaved caspase 3, and cytochrome C levels, while significantly reducing the antiapoptotic proteins, Akt and Bcl-xL (Fig. 1). Results also showed that TNF-α levels were approximately 3-fold higher in the IGFBP-3 KO mice compared to the wild-type (Fig. 1). These findings, in concert with our previous results in REC cultured in high glucose, suggested that TNF-α-dependent proapoptotic actions may be countered by the antiapoptotic actions of IGFBP-3.
Figure 1. .

IGFBP-3 KO mice have increased proapoptotic protein levels, and increased TNF-α and caspase 8 compared to wild-type mice. Cleaved caspase 3 ELISA and Western blot results for wild-type mice and IGFBP3-KO mice for proapoptotic proteins (top panel – cleaved caspase 3, Bax, Cytochrome C) and antiapoptotic proteins (bottom panel – phosphorylated Akt, Bcl-xL). A representative Western blot is provided. *P < 0.05 versus wild-type. N = 6 mice in each group.
Increased TUNEL Labeling in IGFBP-3 KO Mice
Since we found increased protein levels of key apoptotic factors, we performed TUNEL analyses to localize the apoptotic cells. Based on the TUNEL labeling, the majority of apoptosis was occurring in the inner retina (Fig. 2E). Additionally, some labeling was noted in the outer plexiform layer, likely apoptotic cells on the blood vessels.
Figure 2. .
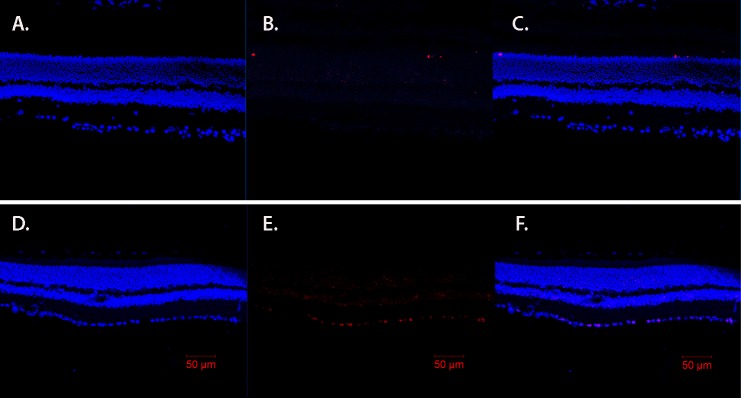
IGFBP-3 KO mice have increased TUNEL labeling of cells in the ganglion cell layer and light staining in outer plexiform layer. TUNEL results for wild-type mice (A–C) and IGFBP3-KO mice (D–F) showing DAPI staining (A, D), and TUNEL labeling (red, B, E) with overlay provided in (C) and (F). Scale bar: 50 μm.
B-wave and Oscillatory Potential Amplitudes Are Reduced in the IGFBP-3 KO Mice
To evaluate the function of the retina of IGFBP-3 KO and wild-type mice, we performed ERGs. While there were minimal changes in the a-wave amplitude, there was a significant decrease in the b-wave and oscillatory potential amplitudes of the IGFBP-3 KO mice compared to wild-type mice, which would be consistent with a functional loss of cells of the inner retina through apoptosis (Fig. 3). This functional loss mimics that observed in ERGs of diabetic animals,4,5,19 which also have reduced IGFBP-3 levels.
Figure 3. .
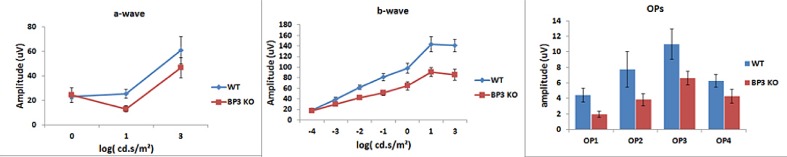
IGFBP-3 KO mice have reduced ERG responses (B-wave and oscillatory potentials) compared to wild-type. Mean ERG amplitudes for the IGFBP-3 KO and wild-type mice. Left is the A-wave, middle represents the B-wave, and the right is the amplitudes for the oscillatory potentials. N = 6 mice in each group.
Retinal Thickness and Cell Numbers in the Ganglion Cell Layer Are Reduced in IGFBP-3 KO Mice
To evaluate whether the IGFBP-3 KO mice have other retina changes similar to diabetic animals, we measured the retinal thickness and cell numbers in the ganglion cell layer. We found that in animals without IGFBP-3, the retina was significantly thinner than in wild-type mice (Fig. 4A). This alteration in retinal thickness correlated with the decreased cell numbers in the ganglion cell layer (Fig. 4B), as has been reported for diabetic animals.5,20,21
Figure 4. .
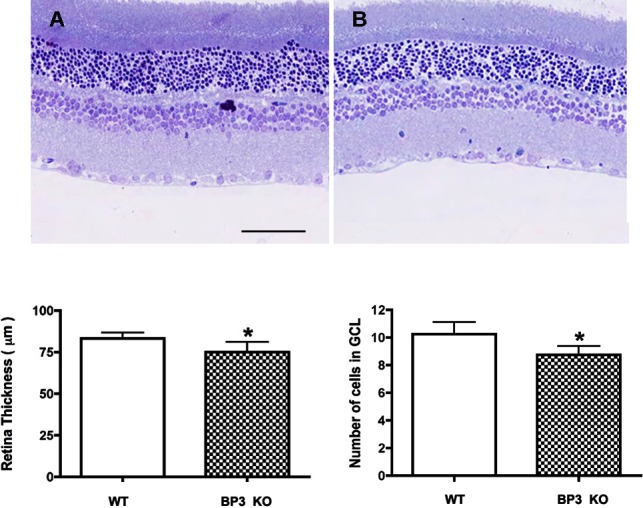
IGFPB-3 KO mice have thinner retinas and fewer cells in the ganglion cell layer. (C) Shows mean retinal thickness in IGFBP-3 KO and wild-type mice. (D) Shows the cell numbers in the ganglion cell layer. A representative image is provided (Wild-type is Image A and IGFBP-3 KO is Image B). *P < 0.05 versus wild-type mice. N = 6 mice in each group.
No Statistically Significant Differences Were Observed in Degenerate Capillaries in IGFBP-3 KO Mice
We measured the numbers of degenerate capillaries in the IGFBP-3 KO mice at 2 months of age, and found no significant changes in the KO versus wild-type animals (Fig. 5). Changes in degenerate capillaries are observed commonly at 6 to 8 months of age21,22; however, we have reported previously changes as early as 6 weeks after surgical sympathectomy in rats2 or by 4 months in beta-1-adrenergic receptor KO mice.3
Figure 5. .
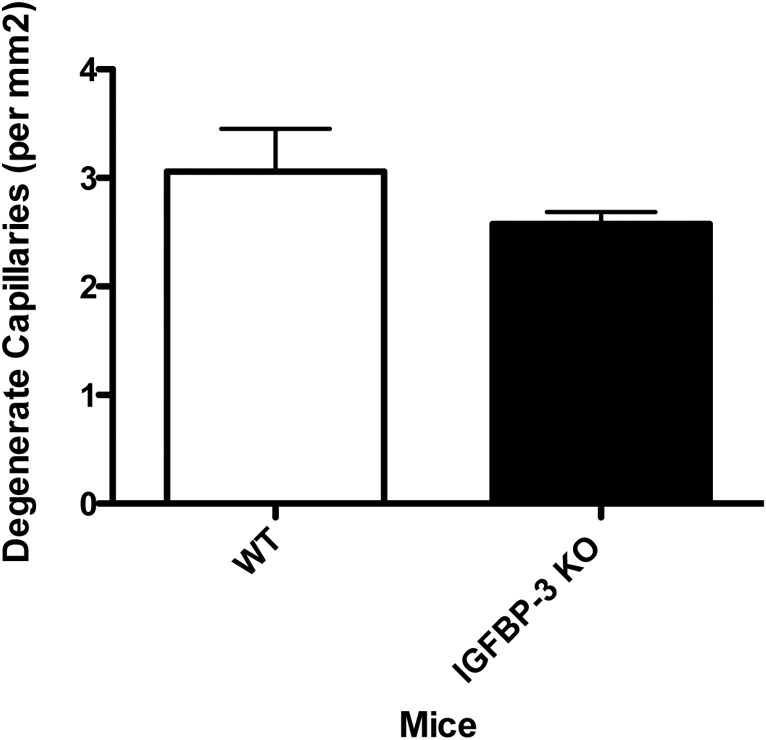
Vascular analyses in the IGFBP-3 KO mice show limited changes at 2 months of age versus wild-type mice. (A) Shows degenerate capillary numbers in IGFBP-3 KO and wild-type mice. N = 5 mice in each group.
Exogenous TNF-α Decreases IGFBP-3 Levels in Retinal Endothelial Cells Cultured in High Glucose
To verify the proposed antagonistic relationship between the levels, and resulting actions of IGFBP-3 and TNF-α, we analyzed further REC undergoing apoptosis in response to high glucose conditions. In this model, Compound 49b triggered increases in IGFBP-3 and, in confirmation of our previous observations, it also decreased apoptosis.4,5 When cells were treated concurrently with TNF-α and Compound 49b, cell death was increased significantly compared to cells treated with only Compound 49b, suggesting that Compound 49b-stimulated IGFBP-3 and TNF-α work in an antagonistic actions to regulate cell death (Fig. 6A). Interestingly, levels of IGFBP-3 were reduced significantly in cells treated with Compound 49b and TNF-α, suggesting that TNF-α can negatively regulate IGFBP-3 levels (Fig. 6B), so that Compound 49b can no longer increase IGFBP-3.
Figure 6. .
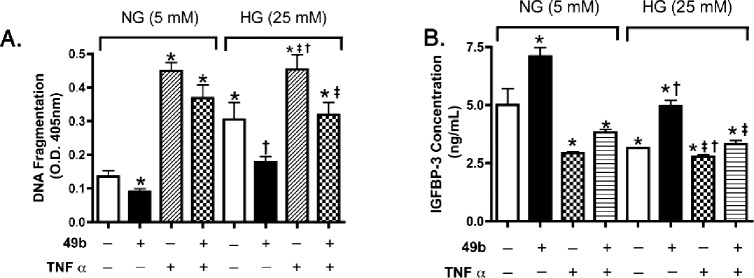
Apoptosis and IGFBP-3 levels in REC treated with a novel β-adrenergic receptor agonist, Compound 49b, or TNF-α. (A) Results from the Cell Death ELISA in REC cultured in normal (5 mM) and high glucose (HG, 25 mM), and treated with Compound 49b (50 nM, 30 minutes) or TNF-α (5 ng/mL; 30 minutes). (B) ELISA results for IGFBP-3 following Compound 49b and TNF-α treatments. *P < 0.05 versus NG no treatment (NT); †P < 0.05 versus HG NT; ‡P < 0.05 versus HG + 49b treatment. N = 3 for all groups.
Compound 49b Regulates Endogenous TNF-α Through IGFBP-3 Actions
While we observed that exogenous TNF-α blocked Compound 49-stimulated increases in IGFBP-3, we also found that Compound 49b significantly reduced endogenous TNF-α levels (Fig. 7A). Importantly, this response was blunted in cells pretreated with IGFBP-3 siRNA transfection (Fig. 7A), suggesting that Compound 49b requires IGFBP-3 for partial regulation of TNF-α. Figure 7B shows successful knockdown of IGFBP-3 with the siRNA.
Figure 7.
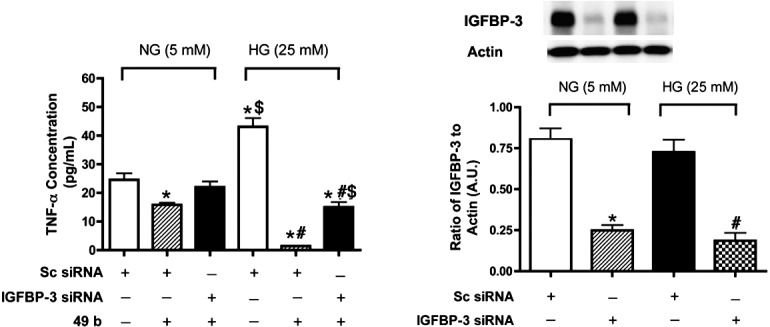
TNF-α levels after Compound 49b or IGFPB-3 siRNA. (A) ELISA results for the TNF-α ELISA in REC cultured in normal (5 mM) and high (25 mM) glucose, and treated with Compound 49b (50 nM, 30 minutes), scrambled siRNA (sc siRNA) or IGFBP-3 siRNA. (B) Western blot to verify successful knockdown using the IGFBP-3 siRNA. Data are presented as fold change from NG untreated. A representative blot is provided at the top. *P < 0.05 versus NG NT; #P < 0.05 versus HG NT; $P < 0.05 versus HG + 49b treatment. N = 3 for all groups.
IGFBP-3 Regulates TNF-α Through the IGFBP-3 Receptor
The receptor for IGFBP-3 actions was observed first in lung cells.11 We recently found that this receptor also is present in retinal endothelial cells.16 To determine whether IGFBP-3 regulates TNF-α through its receptor, we transfected REC with combinations of IGFBP-3 plasmid and IGFBP-3 receptor siRNA. When IGFBP-3 plasmid alone was added, TNF-α levels were reduced significantly. However, in cells transfected with IGFBP-3 plasmid and receptor siRNA, TNF-α levels were not reduced, suggesting that IGFBP-3 regulates TNF-α at least partially through activation of its receptor (Fig. 8).
Figure 8. .
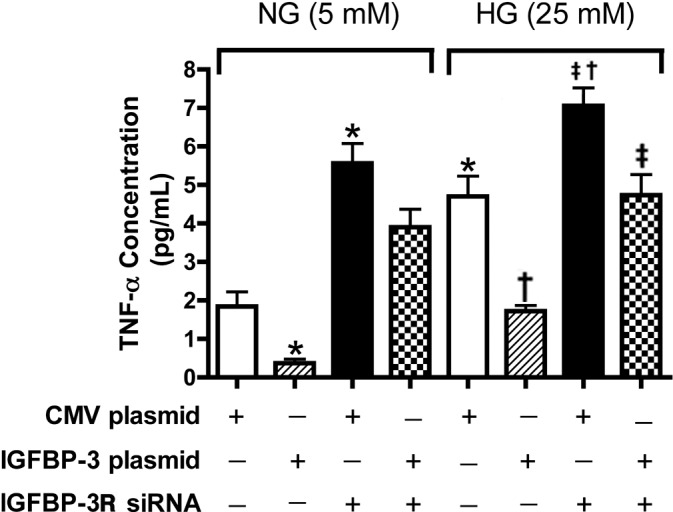
TNF-α concentration in REC showing that IGFBP-3 regulates TNF-α through IGFBP-3 receptor. ELISA results for the TNF-α ELISA in REC cultured in normal (5 mM) and high (25 mM) glucose, and treated with scrambled siRNA (sc siRNA), IGFBP-3 nonbinding plasmid, or IGFBP-3 receptor siRNA. *P < 0.05 versus NG NT; †P < 0.05 versus HG NT; ‡P < 0.05 versus HG + 49b treatment. N = 3 for all groups.
IGFBP-3 Plasmid Decreases Cell Death, Which Is Modulated by TNF-α
To substantiate further the functional link between IGFBP-3 and TNF-α, we transfected REC cultured in high glucose with IGFBP-3 plasmid. We observed a significant increase in cell death that was blocked partially by the addition of exogenous TNF-α (Fig. 9A). This suggested that IGFBP-3 and TNF-α counterbalance one another in REC cultured under hyperglycemic conditions to regulate apoptosis.
Figure 9. .
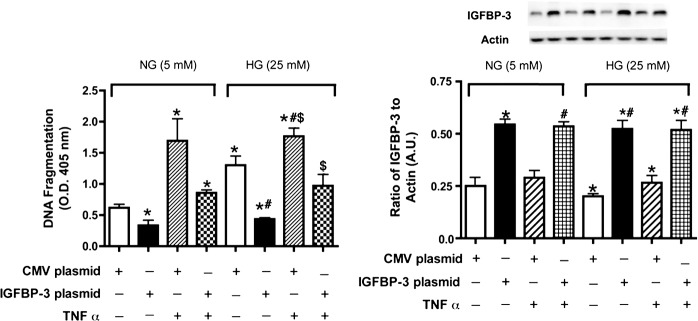
Cell death measurements in REC after TNF-α or IGFBP-3 plasmid transfection. (A) is ELISA results for the cell death ELISA in REC cultured in normal (5 mM) and high (25 mM) glucose, and TNF-α (5 ng/mL, 30 minutes) and/or transfected with CMV plasmid (control) or IGFBP-3 nonbinding plasmid. (B) Western blot results for IGFBP-3 following the same treatments. Data are presented as fold change from NG untreated. A representative blot is provided at the top. *P < 0.05 versus NG NT; #P < 0.05 versus HG NT; $P < 0.05 versus HG + 49b treatment. N = 3 for all groups.
Discussion
Two of the major findings of this study are that the IGFBP-3 KO mice have the neuronal markers observed in early diabetic retinopathy in rodents, and that TNF-α and IGFBP-3 contribute to the apoptotic actions observed in the IGFBP-3 KO mice. These results were confirmed further in cultured REC, suggesting these mice can be used for further examination of specific apoptotic pathways involved.
The three factors investigated in our study that regulate retinal apoptosis, that is beta-adrenergic stimulation, IGFBP-3, and TNF-α, likely have complex interactions. Data in REC demonstrate that β-adrenergic receptor agonists activate PKA, leading to enhanced IGFBP-3 levels.5,23 Occurring concurrently, hyperglycemia leads to reduced PKA activity, which allows for increased TNF-α levels promoting REC apoptosis. Using cells in culture, IGFBP-3 can reduce TNF-α levels directly, and TNF-α can reduce IGFBP-3 levels. Environmental factors (β-adrenergic receptor agonists, glucose levels) appear to influence strongly whether IGFBP-3 or TNF-α predominate in REC apoptosis. Similar findings have been observed in other systems, as discussed below.
IGFBP-3
IGFBP-3 has been reported to be a major regulator of apoptosis in a variety of tissues, able to act as either a pro- or antiapoptotic factor depending on the cell type.7,24 Our results and those of others suggest that, in the retina, IGFBP-3 is predominantly antiapoptotic.5,9,24 In IGFBP-3 KO mice and retinal endothelial cells, we found increased proapoptotic proteins that correspond to the intrinsic pathway (cytochrome C) and the classical death pathway, indicated by increased TNF-α and caspase 8 levels. In contrast, proapoptotic actions of IGFBP-3 have been reported in other tissues, including bronchial epithelial cells, in which IGFBP-3 regulates apoptosis through activation of the IGFBP-3 receptor, leading to caspase 3/7–induced apoptosis.11 Additionally using recombinant IGFBP-3 added to REC, Spoerri et al. found reduced cell proliferation and increased annexin V staining.25 While their findings disagree with the current work, it is potentially due to the use of a recombinant form of IGFBP-3, which is known to be extremely sticky, making effective transfer of the protein to the medium highly difficult. Additionally, in the work by Spoerri et al., recombinant IGFBP-3 also would have activated IGF-1 actions in the cells,25 which could produce different results from our findings using the transfection of an IGFBP-3 plasmid that cannot bind IGF-1. Taken together, our findings in REC in our previous work5 and these data suggest strongly that transfection of the IGFBP-3 plasmid reduces apoptosis of REC in an IGF-1–independent fashion.
IGFBP-3 and TNF-α Interactions
Literature strongly supports an interaction between IGFBP-3 and TNF-α in the regulation of apoptosis. In many cases, TNF-α has been shown to induce IGFBP-3.12–14,26 In contrast to these studies is the work in human fibroblasts, which demonstrated that TNF-α inhibits IGFBP-3, which led to an increase in IGF-1 levels.27 Additional studies also have shown that IGFBP-3 can regulate TNF-α. In prostate cancer cells (PC-3) IGFBP-3 mediates TNF-α–induced apoptosis.28 Similarly, work in colorectal carcinoma cells showed that IGFBP-3 potentiates TNF-α–mediated activation of TNF-related apoptosis–inducing ligand (TRAIL) to mediate apoptosis.29 In recent work on IGFBP-3 and its receptor, the investigators found that IGFBP-3 can inhibit TNF-α actions in bronchial epithelial cells.11 This inhibition of TNF-α occurred using a mutant IGFBP-3 that does not bind IGF-1, indicating that IGFBP-3 inhibited TNF-α in an IGF-1 independent manner,11 which is similar to what we observed in retinal endothelial cells
In contrast to much of the current literature, our findings are focused on IGFBP-3 actions that are IGF-1–independent. Our work in the retinal endothelial cells was all completed using a mutant form of IGFBP-3 that does not bind IGF-1.30 In the primary retinal endothelial cells, TNF-α does not induce IGFBP-3. When the IGFBP-3 plasmid and TNF-α are coadministered to cells, they have antagonistic actions, whereby TNF-α promotes apoptosis and IGFBP-3 inhibits apoptosis. When cells were treated with the IGFBP-3 plasmid and IGFBP-3 receptor siRNA, data suggested that IGFBP-3 inhibits TNF-α levels through activation of its receptor. Our novel β-adrenergic receptor agonist, Compound 49b, is able to reduce TNF-α and increase IGFBP-3,5 but exogenous TNF-α can eliminate the ability of Compound 49b to increase IGFBP-3. Compound 49b's regulation of TNF-α requires increased IGFBP-3 levels, as Compound 49b added with IGFBP-3 siRNA was not able to decrease TNF-α, as much as when Compound 49b is added alone. Therefore, the data show that IGF-1–independent actions of IGFBP-3 inhibit apoptosis of REC, while TNF-α promotes apoptosis in REC cultured in hyperglycemia.
IGFBP-3 KO Mice Phenotype
In IGFBP-3 KO mice we found that TNF-α levels are significantly higher in the IGFBP-3 KO mice compared to wild-type, suggesting that IGFBP-3 is inhibitory to TNF-α in the retina. This was confirmed by the increased caspase 8 levels in the retinal lysates from the IGFBP-3 KO mice. Additionally, we found significantly increased levels of proapoptotic proteins in the IGFBP-3 KO mice, which follows increased TNF-α, increased caspase 8 levels, and decreased IGFBP-3 actions in the retina.
In addition to evaluating the retinal proteins of the IGFBP-3 KO mice, we also investigated the neuronal changes in the retina of these mice. We have reported previously that Compound 49b prevented the diabetic retinopathy phenotype in the streptozotocin rat model.5 We proposed this occurred through increased IGFBP-3 levels, leading to decreased neuronal and vascular apoptosis. We found that retinal thickness is reduced significantly in the IGFBP-3 KO mice. Additionally, the IGFBP-3 KO mice have fewer cells in the ganglion cell layer. This corresponded well with our findings of decreased B-wave and oscillatory potential amplitudes in the electroretinogram of the KO mice. Taken together, these findings suggested that IGFBP-3 is involved in the neuronal changes associated with an early diabetic retinopathy phenotype in rodents, and restoration of IGFBP-3 levels may protect the retina against hyperglycemia-induced damage.
In conclusion, our work suggested that IGFBP-3 acts as an antiapoptotic agent in the retina. IGFBP-3 decreased TNF-α actions on retinal endothelial cells to reduce the apoptotic events by TNF-α. Use of the IGFBP-3 KO mice demonstrated that TNF-α levels are increased greatly in the retina when IGFBP-3 is absent. Furthermore, the IGFBP-3 KO mice have many of the neuronal characteristics of diabetic retinopathy in a rodent with normal glucose levels, including decreased ERG amplitudes, reduced retinal thickness and fewer cells in the ganglion cell layer, suggesting that IGFBP-3 is protective to the neural retina. Future work will evaluate retinal changes in a diabetic IGFBP-3 KO mice, as well as further dissect the cellular signaling involved in TNF-α/IGFBP-3 interplay in the retina. These studies can add greatly to our understanding of the inflammatory and apoptotic actions in the retina, specifically the role of IGFBP-3 in mediating the antiapoptotic and anti-inflammatory processes.
Acknowledgments
Supported by National Eye Institute Vision Grant R01EY022045 (JJS) and EY00300 (TSK), Juvenile Diabetes Research Foundation Grant 2-2011-597 (JJS), Oxnard Foundation (JJS), Research to Prevent Blindness Award (PI: Barrett Haik), NEI Vision Core Grant: PHS 3P30 EY013080 (PI: Dianna Johnson), a Merit Grant from the Veterans Administration (TSK), and a grant from the New Jersey Commission on Brain Injury Research (JP).
Disclosure: Q. Zhang, None; Y. Jiang, None; M.J. Miller, None; B. Peng, None; L. Liu, None; C. Soderland, Cell Systems Corp. (E), J. Tang, None; T.S. Kern, None; J. Pintar, None; J.J. Steinle, None
References
- 1. Burnstock G. Changes in expression of autonomic nerves in aging and disease. J Auton Nerv Syst. 1990; 30 (suppl): S25–S34 [DOI] [PubMed] [Google Scholar]
- 2. Wiley LA, Rupp GR, Steinle JJ. Sympathetic innervation regulates basement membrane thickening and pericyte number in rat retina. Invest Ophthalmol Vis Sci. 2005; 46: 744–748 [DOI] [PubMed] [Google Scholar]
- 3. Panjala SR, Jiang Y, Kern TS, Thomas SA, Steinle JJ. Increased tumor necrosis factor-alpha, cleaved caspase 3 levels and insulin receptor substrate-1 phosphorylation in the beta-adrenergic receptor knockout mouse. Mol Vis. 2011; 17: 1822–1828 [PMC free article] [PubMed] [Google Scholar]
- 4. Jiang Y, Walker RJ, Kern TS, Steinle JJ. Application of isoproterenol inhibits diabetic-like changes in the rat retina. Exp Eye Res. 2010; 91: 171–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang Q, Guy K, Pagadala J, et al. Compound 49b prevents diabetes-induced apoptosis through increased IGFBP-3 levels. Invest Ophthalmol Vis Sci. 2012; 53: 3004–3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ferry RJ Jr, Cerri RW, Cohen P. Insulin-like growth factor binding proteins: new proteins, new functions. Horm Res. 1999; 51: 53–67 [DOI] [PubMed] [Google Scholar]
- 7. Vasylyeva TL, Chen X, Ferry RJ Jr. Insulin-like growth factor binding protein-3 mediates cytokine-induced mesangial cell apoptosis. Growth Horm IGF Res. 2005; 15: 207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yamada PM, Lee KW. Perspectives in mammalian IGFBP-3 biology: local vs. systemic action. Am J Physiol Cell Physiol. 2009; 296: C954–C976 [DOI] [PubMed] [Google Scholar]
- 9. Kielczewski JL, Hu P, Shaw LC, et al. Novel protective properties of IGFBP-3 result in enhanced pericyte ensheathment, reduced microglial activation, increased microglial apoptosis, and neuronal protection after ischemic retinal injury. Am J Pathol. 2011; 178: 1517–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Joussen AM, Doehmen S, Le ML, et al. TNF-alpha mediated apoptosis plays an important role in the development of early diabetic retinopathy and long-term histopathological alterations. Mol Vis. 2009; 15: 1418–1428 [PMC free article] [PubMed] [Google Scholar]
- 11. Lee YC, Jogie-Brahim S, Lee DY, et al. Insulin-like growth factor-binding protein-3 (IGFBP-3) blocks the effects of asthma by negatively regulating NF-kappaB signaling through IGFBP-3R-mediated activation of caspases. J Biol Chem. 2011; 286: 17898–17909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leibowitz BJ, Cohick WS. Endogenous IGFBP-3 is required for both growth factor-stimulated cell proliferation and cytokine-induced apoptosis in mammary epithelial cells. J Cell Physiol. 2009; 220: 182–188 [DOI] [PubMed] [Google Scholar]
- 13. Katz J, Nasatzky E, Werner H, Le Roith D, Shemer J. Tumor necrosis factor alpha and interferon gamma--induced cell growth arrest is mediated via insulin-like growth factor binding protein-3. Growth Horm IGF Res. 1999; 9: 174–178 [DOI] [PubMed] [Google Scholar]
- 14. Zappala G, Rechler MM. IGFBP-3, hypoxia and TNF-alpha inhibit adiponectin transcription. Biochem Biophys Res Commun. 2009; 382: 785–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ye P, Price W, Kassiotis G, Kollias G, D'Ercole AJ. Tumor necrosis factor-alpha regulation of insulin-like growth factor-I, type 1 IGF receptor, and IGF binding protein expression in cerebellum of transgenic mice. J Neurosci Res. 2003; 71: 721–731 [DOI] [PubMed] [Google Scholar]
- 16. Zhang Q, Soderland C, Steinle JJ. Regulation of retinal endothelial cell apoptosis through activation of the IGFBP-3 receptor. Apoptosis. 2013; 18: 361–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jiang Y, Steinle JJ. Systemic propranolol reduces b-wave amplitude in the ERG and increases IGF-1 receptor phosphorylation in rat retina. Invest Ophthalmol Vis Sci. 2010; 51: 2730–2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Laver NM, Robison WG Jr, Pfeffer BA. Novel procedures for isolating intact retinal vascular beds from diabetic humans and animal models. Invest Ophthalmol Vis Sci. 1993; 34: 2097–2104 [PubMed] [Google Scholar]
- 19. Li Q, Zemel E, Miller B, Perlman I. Early retinal damage in experimental diabetes: electroretinographical and morphological observations. Exp Eye Res. 2002; 74: 615–625 [DOI] [PubMed] [Google Scholar]
- 20. Barber AJ, Antonetti DA, Kern TS, et al. The Ins2Akita mouse as a model of early retinal complications in diabetes. Invest Ophthalmol Vis Sci. 2005; 46: 2210–2218 [DOI] [PubMed] [Google Scholar]
- 21. Kern TS, Miller CM, Tang J, et al. Comparison of three strains of diabetic rats with respect to the rate at which retinopathy and tactile allodynia develop. Mol Vis. 2010; 16: 1629–1639 [PMC free article] [PubMed] [Google Scholar]
- 22. Engerman RL, Kern TS. Retinopathy in animal models of diabetes. Diabetes Metab Rev. 1995; 11: 109–120 [DOI] [PubMed] [Google Scholar]
- 23. Zhang Q, Steinle JJ. DNA-PK phosphorylation of IGFBP-3 is required to prevent apoptosis in retinal endothelial cells cultured in high glucose. Invest Ophthalmol Vis Sci. 2013; 54: 3052–3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lofqvist C, Chen J, Connor KM, et al. IGFBP3 suppresses retinopathy through suppression of oxygen-induced vessel loss and promotion of vascular regrowth. Proc Natl Acad Sci U S A. 2007; 104: 10589–10594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Spoerri PE, Caballero S, Wilson SH, Shaw LC, Grant MB. Expression of IGFBP-3 by human retinal endothelial cell cultures: IGFBP-3 involvement in growth inhibition and apoptosis. Invest Ophthalmol Vis Sci. 2003; 44: 365–369 [DOI] [PubMed] [Google Scholar]
- 26. Anwar A, Zahid AA, Scheidegger KJ, Brink M, Delafontaine P. Tumor necrosis factor-alpha regulates insulin-like growth factor-1 and insulin-like growth factor binding protein-3 expression in vascular smooth muscle. Circulation. 2002; 105: 1220–1225 [DOI] [PubMed] [Google Scholar]
- 27. Liu Y, Tsushima T, Miyakawa M, et al. Effect of cytokines on production of insulin-like growth factor binding proteins (IGFBPs) from human fibroblasts in culture. Endocr J. 1999; 46 (Suppl): S63–S66 [DOI] [PubMed] [Google Scholar]
- 28. Rajah R, Lee KW, Cohen P. Insulin-like growth factor binding protein-3 mediates tumor necrosis factor-alpha-induced apoptosis: role of Bcl-2 phosphorylation. Cell Growth Differ. 2002; 13: 163–171 [PubMed] [Google Scholar]
- 29. Williams AC, Smartt H, K-Zadeh AM, Macfarlane M, Paraskeva C, Collard TJ. Insulin-like growth factor binding protein 3 (IGFBP-3) potentiates TRAIL-induced apoptosis of human colorectal carcinoma cells through inhibition of NF-kappaB. Cell Death Differ. 2007; 14: 137–145 [DOI] [PubMed] [Google Scholar]
- 30. Kielczewski JL, Li Calzi S, Shaw LC, et al. Free insulin-like growth factor binding protein-3 (IGFBP-3) reduces retinal vascular permeability in association with a reduction of acid sphingomyelinase (ASMase). Invest Ophthalmol Vis Sci. 2011; 52: 8278–8286 [DOI] [PMC free article] [PubMed] [Google Scholar]