

Abstract
Impaired mitochondrial function represents an early manifestation of endothelial dysfunction and likely contributes to the development of cardiovascular diseases (CVD). The stimulation of mitochondrial function and/or biogenesis is seen as a means to improve the bioenergetic and metabolic status of cells and thus, reduce CVD. In this study we examined the capacity of the flavanol (−)-epicatechin and two novel derivatives to enhance mitochondrial function and protein levels in cultured bovine coronary artery endothelial cells. As nitric oxide production by endothelial cells is suspected in mediating mitochondria effects (including biogenesis), we also examined the dependence of responses on this molecule using an inhibitor of nitric oxide synthase. Results indicate that the flavanol (−)-epicatechin and derivatives are capable of stimulating mitochondria function as assessed by citrate synthase activity as well as induction of structural (porin, mitofilin) and oxidative phosporylation protein levels (complex I and II). Effects were blocked by the use of the chemical inhibitor of the synthase thus, evidencing a role for nitric oxide in mediating these effects. The results observed indicate that the three agents are effective in enhancing mitochondria function and protein content. The effects noted for (−)-epicatechin may serve to explain the healthy effects on cardiometabolic risk ascribed to the consumption of cocoa products.
Cardiovascular diseases (CVD) are a leading cause of mortality and morbidity worldwide. The health of the endothelium is central to the underlying pathophysiology of many CVD 1. Endothelial health is partly dependent on the physiological production of nitric oxide (NO) mainly by the endothelial nitric oxide synthase (eNOS) 2. Disruption in the bioavailability of NO facilitates a vasoconstricting and procoagulating state characteristic of many CVD. Impaired mitochondrial function represents an early manifestation of endothelial dysfunction and likely contributes to the development of CVD 3, 4. Physiological increases in NO have been linked with mitochondrial biogenesis and thus, mitochondrial function 5–8. Therefore, strategies that increase physiological NO production may enhance mitochondrial biogenesis in endothelial and other cell types and are promising for the preservation of cardiovascular health.
In vitro and in vivo studies suggest that the flavan-3-ol (−)-epicatechin (EPI) can exert CVD protection 9–11, potentially via improving mitochondrial structure/function 12–14. These effects have been partly ascribed to the capacity of EPI to activate eNOS and thereby increase NO production 15, 16. As previously reported by us, a possible means by which EPI (and possibly derivatives) triggers these responses can involve a putative receptor entity, as there is indirect evidence for its presence 11,15. Under physiological conditions, native EPI is prone to oxidation secondary to the reactivity of the phenolic hydroxyl groups (OH) 17. The pharmacological effects of native EPI are also limited by its susceptibility to phase II metabolism (sulfation and glucuronidation of the OH groups), which can lead to a short half-life 18. We propose that the development of more stable derivatives of EPI is a useful strategy to retain biological activity and enhance potency.
Two strategies may be useful in protecting EPI from oxidation. First, EPI in vitro and in vivo is mono-methylated by the endothelium in the -OH of the catechol ring (ring B, see figure 1), leading to a mixture of 3′- and 4′-O-methyl epicatechin 19, 20, which stabilizes the molecule against oxidation. Although, 3′-O-methyl epicatechin (3′-O-MeEPI) and EPI have different H-donating potentials, they appear to posses the same biological capacities. For example, both molecules inhibit cell death induced by H2O2, implying a mechanism of action independent of their antioxidant properties as Spencer et al proposed 21. Second, the acetylation of flavanols such as (−)-epigallocatechin gallate confers protection to all OH groups and improves the chemical stability of the compound under physiological conditions 22, which translates into enhanced biological actions 23.
Figure 1. Chemical structures of (−)-epicatechin and derivatives.
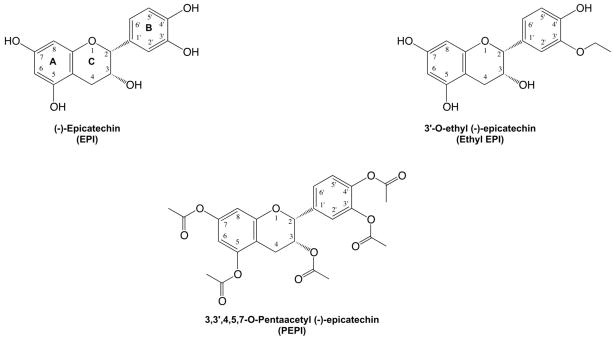
Bold letters and numbers correspond to the ring position and carbon atom numbering, respectively.
The goal of the current study was to rationally develop compounds derived from EPI with improved effects on NO production and thence on mitochondrial enzyme activity and protein content. We hypothesized that EPI acetylation or alkylation will confer stability to the molecule and thereby increase its efficacy on endothelial NO production and NO-mediated effects on mitochondrial endpoints. To test our hypothesis, we modified EPI via alkylation or acetylation of OH groups. For our first approach, we used an ethyl group as the alkylating agent, which mimics (but with an additional -CH2) the biotransformation of EPI into 3′-O-MeEPI, which should yield similar or enhanced biological activity as EPI, likely with improved stability. For our second approach, we acetylated all OH groups of EPI. The three compounds used were tested for biological activity using a cell culture model of bovine coronary artery endothelial cells (BCAEC).
For the acetylation modification, all OH groups of EPI were acetylated, leading to 3, 3′, 4′, 5, 7-O-penta-acetyl (−)-epicatechin (PEPI). The chemical structures of PEPI and Ethyl EPI are illustrated in figure 1. For the alkylation modification, EPI was ethylated at the position 3′ (ring B), leading to 3′-O-ethyl (−)-epicatechin (Ethyl EPI) (for synthesis procedures and spectra characterization see supplemental methods). In all experiments, EPI was used as a positive control. Administration of EPI to cultured endothelial cells stimulates NO synthesis secondary to the activation of eNOS; the effect peaks at 10 minutes and 1 μM EPI 15. As a prelude to determine the effects of NO on mitochondria-related endpoints, we generated concentration-response curves of NO production for EPI, PEPI, and Ethyl EPI (figure 2). The rank order of potency (EC50) derived from the curves was PEPI (0.006 nmol/L) > Ethyl EPI (1 nmol/L) > EPI (2 nmol/L) with respect to their capacities to increase NO production in BCAEC.
Figure 2. Dose response effects of EPI, Ethyl EPI and PEPI on nitric oxide (NO) levels.
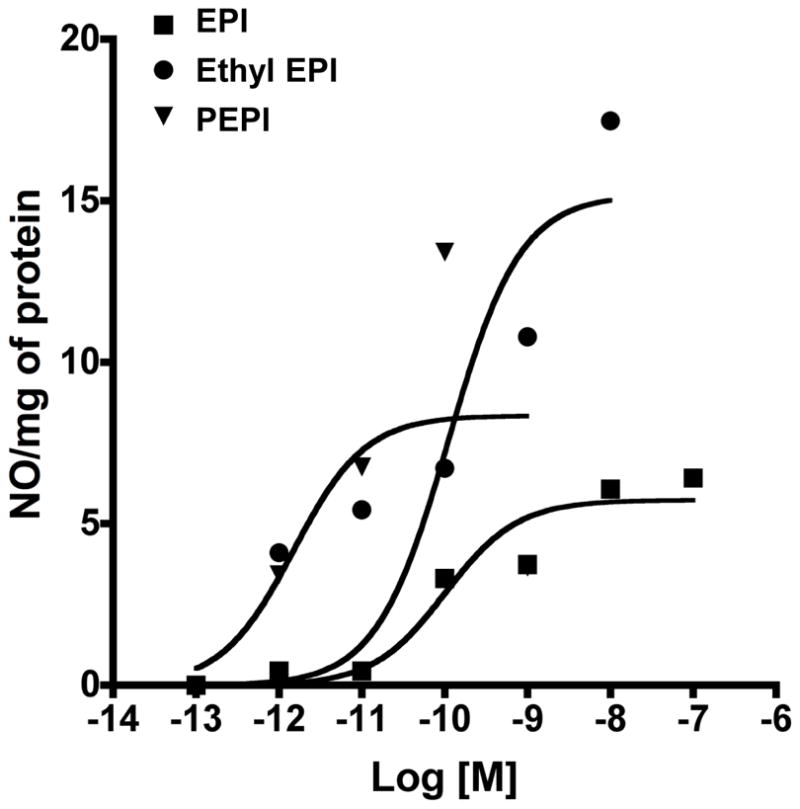
BCAEC (in triplicate) were treated at different doses with each compound and media collected 10 min after treatment. Nitric oxide (nitrite/nitrate) levels were assessed by kit (Cayman Inc.). Values represent means of triplicate assays and sigmoidal curves were generated by Prizm 5 (Graphpad Inc).
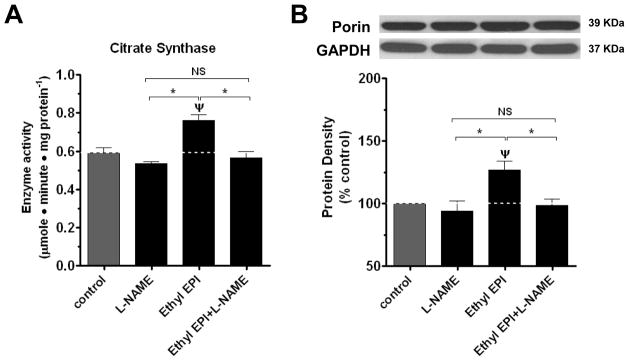
The regulatory role of NO on mitochondrial structure/function has been previously studied 24, 25. The sustained exposure of cells to NO can trigger mitochondrial biogenesis 5. However, mitochondrial biogenesis also appears to be only partly dependent on NO signaling. 5, 8, 26–29. We used the EC50 concentrations to evaluate effects of these compounds on mitochondrial endpoints over a 48 h treatment. We first examined citrate synthase activity (CS). Citrate synthase is the initial enzyme of the tricarboxylic acid (TCA) cycle 30. CS has been routinely used to assess the oxidative and respiratory capacity as well as mitochondrial volume and density in cells and tissues 31–33. All compounds significantly increased CS by ~ 30 % (figure 3A). This increase in CS suggests an augmented endothelial mitochondrial activity and/or density elicited by the epicatechin compounds. EC50 concentrations of PEPI, Ethyl EPI and EPI achieved similar increases in activity.
Figure 3. Effects of EPI, Ethyl EPI and PEPI on BCAEC citrate synthase activity and mitochondrial protein levels.

(A) CS was evaluated in BCAEC treated with either vehicle (control), 2 nmol/L EPI, 1 nmol/L Ethyl EPI, or 0.006 nmol/L PEPI for 48 h. BCAEC; Bovine coronary artery endothelial cells, CS; citrate synthase activity, EPI; (−)-epicatechin, Ethyl EPI; 3′-O-ethyl (−)-epicatechin, PEPI; 3, 3′, 4′, 5, 7-O-penta-acetyl (−)-epicatechin. Western blots were probed with specific antibodies against (B), Porin; (C), Mitofilin; (D), complex I, and (E), complex II. Values were normalized by arbitrarily setting the densitometry of control levels to 100. Values are mean ± SEM; n=3 per treatment. *p<0.05 vs. control, ψp<0.01 vs. control.
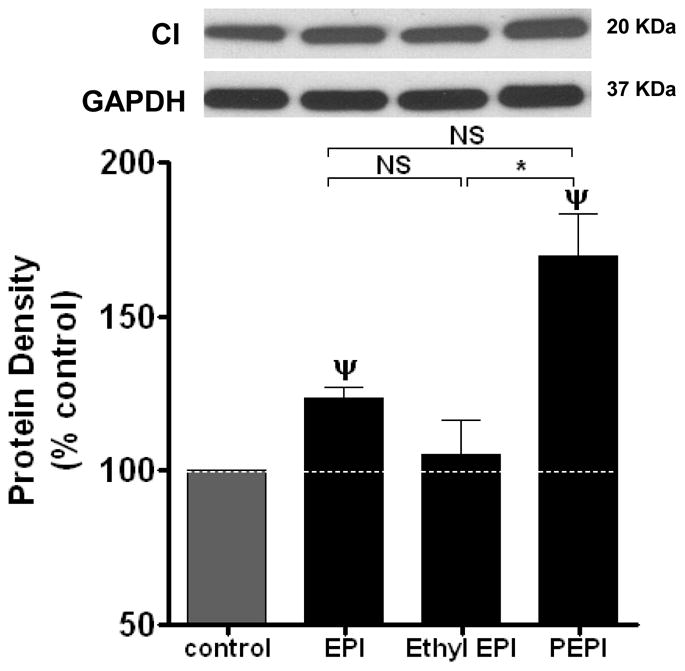
We then examined for changes in constitutive members of mitochondrial structure, specifically, porin, the most abundant outer mitochondrial membrane protein that often is used as a marker for cellular mitochondrial mass 34. PEPI and Ethyl EPI but not EPI significantly increased porin levels over control (figure 3B). We also examined changes in mitofilin, a protein from the inner mitochondrial membrane that promotes/induces cristae and cristae junction formation 35. EPI and PEPI significantly increase mitofilin protein levels (figure 3C), suggesting augmented inner mitochondrial mass. To assess changes in the oxidative phosphorylation system, we assessed levels of mitochondrial complex I (CI), the major entry point for electrons from NADH produced by the TCA cycle 36. EPI, and PEPI, stimulated CI by ~20 % and 60 % over control, respectively, while Ethyl EPI produced no significant effect (figure 3D). Complex II (CII) was also evaluated as a direct link between the TCA cycle and the respiratory chain 37, EPI and PEPI increased CII protein levels by ~25 % increase over control (figure 3E). Thus, PEPI treatment increased all the mitochondrial endpoints measured; EPI and Ethyl EPI appear to exert a semi-selective effect on specific mitochondrial proteins. Perhaps, a higher concentration is needed for EPI and Ethyl EPI to increase all the mitochondrial endpoints, as we only used the EC50 for NO production.
In order to determine the role of EPI and derivatives stimulated NO in mediating compound effects on mitochondrial endpoints, we used the nitric oxide synthase (NOS) inhibitor L-NG-Nitroarginine methyl ester (L-NAME). Treatment of BCAEC for 48 h with L-NAME by itself induced no significant change but blocked the effects of EPI, PEPI, and Ethyl EPI on CS and all assessed mitochondrial protein levels (porin, mitofilin, CI and CII) (figures 4, 5 and 6). To evidence the nature of the effects on a recognized mitochondria biogenesis related endpoint and the role played by NO, we also assessed the effects of treatments on a key regulator of mitochondria gene transcription, TFAM 38. Results indicate that EPI and PEPI induce significant increases in TFAM and effects are blocked by L-NAME (figure 7). The complete blockade of compound effects on mitochondrial endpoints by L-NAME strongly evidence the critical role that NO production may play in regulating mitochondrial biology in endothelial cells and thus, CVD. To further explore the relative role played by NO on mitochondrial protein (CI) levels induced by the compounds, we use a dose of each that yielded a similar induction in NO levels. As shown in figure 8, EPI and PEPI led to significant increases in CI levels. A possible means by which the compounds yield variable effects may be explained by different diffusion properties and/or selective metabolism.
Figure 4. Effects of L-NAME on EPI-induced increases in BCAEC citrate synthase activity and mitochondrial protein levels.

CS (A) and mitofilin (B), complex I (C), complex II (D) protein densities in BCAEC following exposure for 48 h to either vehicle (control), 2 nmol/L EPI, or 300 μmol/L L-NAME + 2 nmol/L EPI. BCAEC; Bovine coronary artery endothelial cells, CS; citrate synthase activity, EPI; (−)-epicatechin, L-NAME; L-NG-Nitroarginine methyl ester. Values were normalized by arbitrarily setting the densitometry of control levels to 100. Values are mean ± SEM; n=3 per treatment. *p<0.05 vs. control, ψp<0.05 EPI vs. EPI + L-NAME.
Figure 5. Effects of L-NAME on Ethyl EPI-induced changes on citrate synthase activity and mitochondrial protein levels.
CS (A) and porin (B) protein densities in BCAEC following exposure for 48 h to either vehicle (control), 1 nmol/L Ethyl EPI, or 300 μmol/L L-NAME + 1 nmol/L Ethyl EPI. BCAEC; Bovine coronary artery endothelial cells, CS; citrate synthase activity, Ethyl EPI; 3′-O-ethyl epicatechin, L-NAME; L-NG-Nitroarginine methyl ester. Values were normalized by arbitrarily setting the densitometry of control levels to 100. Values are mean ± SEM; n=3 per treatment. *p<0.05 vs. control, ψp<0.05 Ethyl-EPI vs. Ethyl-EPI + L-NAME.
Figure 6. Effects of L-NAME on PEPI-induced changes on citrate synthase activity and mitochondrial protein levels.

CS (A) and porin (B), mitofilin (C), complex I (D), complex II (E) protein densities in BCAEC following exposure for 48 h to either vehicle (control), 0.006 nmol/L PEPI, or 300 μmol/L L-NAME + 0.006 nmol/L PEPI. BCAEC; Bovine coronary artery endothelial cells, CS; citrate synthase activity, PEPI; 3, 3′, 4′, 5, 7-O-penta-acetyl (−)-epicatechin, L-NAME; L-NG-Nitroarginine methyl ester. Values were normalized by arbitrarily setting the densitometry of control levels to 100. Values are mean ± SEM; n=3 per treatment. *p<0.05 vs. control, ψp<0.05 EPI vs. EPI + L-NAME.
Figure 7. Effects of EPI, Ethyl EPI and PEPI and L-NAME on BCAEC TFAM levels.

BCAEC were treated with either vehicle (control), 2 nmol/L EPI, 1 nmol/L Ethyl EPI or 0.006 nmol/L PEPI for 48 h ± L-NAME (300 μM). Western blots were probed with specific antibodies against TFAM. (A) Effects of EPI, Ethyl-EPI and PEPI, (B) Effects of EPI ± L-NAME and (C) Effects of PEPI ± L-NAME. EPI; (−)-epicatechin, Ethyl EPI; 3′-O-ethyl (−)-epicatechin, PEPI; 3, 3′, 4′, 5, 7-O-penta-acetyl (−)-epicatechin. Values were normalized by arbitrarily setting the densitometry of control levels to 100. Values are mean ± SEM; n=3 per treatment. *p<0.01 ψp<0.01 vs. control.
Figure 8. Effects of EPI, Ethyl EPI and PEPI on BCAEC mitochondrial complex I protein levels.
BCAEC were treated with either vehicle (control), 100 nmol/L EPI, 0.01 nmol/L Ethyl EPI or 0.01 nmol/L PEPI for 48 h. Western blots were probed with specific antibodies against complex I (CI). EPI; (−)-epicatechin, Ethyl EPI; 3′-O-ethyl (−)-epicatechin, PEPI; 3, 3′, 4′, 5, 7-O-penta-acetyl (−)-epicatechin. Values were normalized by arbitrarily setting the densitometry of control levels to 100. Values are mean ± SEM; n=3 per treatment. *p<0.01, ψp<0.01 vs. control.
We provide evidence that ethyl O-alkylation of EPI’s B ring produces a more potent stimulant of NO production that can trigger significant increases in mitochondrial enzyme activity and protein levels. Surprisingly, the acetylation of all the EPI OH groups notably increased the potency on NO production vs. EPI and Ethyl EPI translating into significant increases in some but not all mitochondrial endpoints that we assessed. Thus, PEPI may represent a particular effective way of targeting NO production and mitochondrial activity at very low concentrations; however, more work is necessary to examine its in vivo metabolism since it could be rapidly hydrolyzed by intracellular and membrane-bound esterases, which may in turn release EPI. In other words, PEPI may represent a pro-drug form of EPI, which could be highly favorable for drug development. The testing of these compounds for mitochondrial targeting is warranted for diseases whose impaired cellular bioenergetics are an important part of the pathophysiology of endothelial dysfunction and/or CVD.
Supplementary Material
Acknowledgments
We would like to acknowledge the editorial help provided by Dr. Laurence Brunton. Study was supported by NIH R24 DK092154, R01 HL43617 and P60 MD00220 provided to Dr. F. Villarreal, CONACyT research grant #129889 and an unrestricted gift to Dr. Ceballos by Cardero Therapeutics Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S. Diabetes Care. 2009;32(Suppl 2):S314. doi: 10.2337/dc09-S330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Napoli C, Ignarro LJ. Arch Pharm Res. 2009;32:1103. doi: 10.1007/s12272-009-1801-1. [DOI] [PubMed] [Google Scholar]
- 3.Ballinger SW. Free Radic Biol Med. 2005;38:1278. doi: 10.1016/j.freeradbiomed.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 4.Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit C, Hamburg NM, Frame AA, Caiano TL, Kluge MA, Duess MA, Levit A, Kim B, Hartman ML, Joseph L, Shirihai OS, Vita JA. Circulation. 2011;124:444. doi: 10.1161/CIRCULATIONAHA.110.014506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, Carruba MO. Science. 2003;299:896. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- 6.Lira VA, Brown DL, Lira AK, Kavazis AN, Soltow QA, Zeanah EH, Criswell DS. J Physiol. 2010;588:3551. doi: 10.1113/jphysiol.2010.194035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tengan CH, Kiyomoto BH, Godinho RO, Gamba J, Neves AC, Schmidt B, Oliveira AS, Gabbai AA. Biochem Biophys Res Commun. 2007;359:771. doi: 10.1016/j.bbrc.2007.05.184. [DOI] [PubMed] [Google Scholar]
- 8.Nisoli E, Falcone S, Tonello C, Cozzi V, Palomba L, Fiorani M, Pisconti A, Brunelli S, Cardile A, Francolini M, Cantoni O, Carruba MO, Moncada S, Clementi E. Proc Natl Acad Sci U S A. 2004;101:16507. doi: 10.1073/pnas.0405432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamazaki KG, Romero-Perez D, Barraza-Hidalgo M, Cruz M, Rivas M, Cortez-Gomez B, Ceballos G, Villarreal F. Am J Physiol Heart Circ Physiol. 2008;295:H761. doi: 10.1152/ajpheart.00413.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamazaki KG, Taub PR, Barraza-Hidalgo M, Rivas MM, Zambon AC, Ceballos G, Villarreal FJ. J Am Coll Cardiol. 2010;55:2869. doi: 10.1016/j.jacc.2010.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramirez-Sanchez I, Nogueira L, Moreno A, Murphy A, Taub P, Perkins G, Ceballos GM, Hogan M, Malek M, Villarreal F. J Cardiovasc Pharmacol. 2012;60:429. doi: 10.1097/FJC.0b013e318269ae0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steffen Y, Schewe T, Sies H. Biochem Biophys Res Commun. 2005;331:1277. doi: 10.1016/j.bbrc.2005.04.035. [DOI] [PubMed] [Google Scholar]
- 13.Taub PR, Ramirez-Sanchez I, Ciaraldi TP, Perkins G, Murphy AN, Naviaux R, Hogan M, Maisel AS, Henry RR, Ceballos G, Villarreal F. Clin Transl Sci. 2012;5:43. doi: 10.1111/j.1752-8062.2011.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nogueira L, Ramirez-Sanchez I, Perkins GA, Murphy A, Taub PR, Ceballos G, Villarreal FJ, Hogan MC, Malek MH. J Physiol. 2011;589:4615. doi: 10.1113/jphysiol.2011.209924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramirez-Sanchez I, Maya L, Ceballos G, Villarreal F. Hypertension. 2010;55:1398. doi: 10.1161/HYPERTENSIONAHA.109.147892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brossette T, Hundsdorfer C, Kroncke KD, Sies H, Stahl W. Eur J Nutr. 2011;50:595. doi: 10.1007/s00394-011-0172-9. [DOI] [PubMed] [Google Scholar]
- 17.Zhu QY, Holt RR, Lazarus SA, Ensunsa JL, Hammerstone JF, Schmitz HH, Keen CL. J Agric Food Chem. 2002;50:1700. doi: 10.1021/jf011228o. [DOI] [PubMed] [Google Scholar]
- 18.Richelle M, Tavazzi I, Enslen M, Offord EA. Eur J Clin Nutr. 1999;53:22. doi: 10.1038/sj.ejcn.1600673. [DOI] [PubMed] [Google Scholar]
- 19.Steffen Y, Gruber C, Schewe T, Sies H. Arch Biochem Biophys. 2008;469:209. doi: 10.1016/j.abb.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 20.Ottaviani JI, Momma TY, Kuhnle GK, Keen CL, Schroeter H. Free Radic Biol Med. 2012;52:1403. doi: 10.1016/j.freeradbiomed.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 21.Spencer JP, Schroeter H, Kuhnle G, Srai SK, Tyrrell RM, Hahn U, Rice-Evans C. Biochem J. 2001;354:493. doi: 10.1042/0264-6021:3540493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huo C, Wan SB, Lam WH, Li L, Wang Z, Landis-Piwowar KR, Chen D, Dou QP, Chan TH. Inflammopharmacology. 2008;16:248. doi: 10.1007/s10787-008-8031-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Landis-Piwowar KR, Huo C, Chen D, Milacic V, Shi G, Chan TH, Dou QP. Cancer Res. 2007;67:4303. doi: 10.1158/0008-5472.CAN-06-4699. [DOI] [PubMed] [Google Scholar]
- 24.Erusalimsky JD, Moncada S. Arterioscler Thromb Vasc Biol. 2007;27:2524. doi: 10.1161/ATVBAHA.107.151167. [DOI] [PubMed] [Google Scholar]
- 25.Carreras MC, Franco MC, Peralta JG, Poderoso JJ. Mol Aspects Med. 2004;25:125. doi: 10.1016/j.mam.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 26.Chowanadisai W, Bauerly KA, Tchaparian E, Wong A, Cortopassi GA, Rucker RB. J Biol Chem. 2010;285:142. doi: 10.1074/jbc.M109.030130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Csiszar A, Labinskyy N, Pinto JT, Ballabh P, Zhang H, Losonczy G, Pearson K, de Cabo R, Pacher P, Zhang C, Ungvari Z. Am J Physiol Heart Circ Physiol. 2009;297:H13. doi: 10.1152/ajpheart.00368.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Funk JA, Odejinmi S, Schnellmann RG. J Pharmacol Exp Ther. 2010;333:593. doi: 10.1124/jpet.109.161992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rasbach KA, Schnellmann RG. J Pharmacol Exp Ther. 2008;325:536. doi: 10.1124/jpet.107.134882. [DOI] [PubMed] [Google Scholar]
- 30.Raimundo N, Baysal BE, Shadel GS. Trends Mol Med. 2011;17:641. doi: 10.1016/j.molmed.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoppeler H. Int J Sports Med. 1986;7:187. doi: 10.1055/s-2008-1025758. [DOI] [PubMed] [Google Scholar]
- 32.Spina RJ, Chi MM, Hopkins MG, Nemeth PM, Lowry OH, Holloszy JO. J Appl Physiol. 1996;80:2250. doi: 10.1152/jappl.1996.80.6.2250. [DOI] [PubMed] [Google Scholar]
- 33.Holloszy JO, Oscai LB, Don IJ, Mole PA. Biochem Biophys Res Commun. 1970;40:1368. doi: 10.1016/0006-291x(70)90017-3. [DOI] [PubMed] [Google Scholar]
- 34.Brinckmann A, Weiss C, Wilbert F, von Moers A, Zwirner A, Stoltenburg-Didinger G, Wilichowski E, Schuelke M. PLoS One. 2010;5:e13513. doi: 10.1371/journal.pone.0013513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.John GB, Shang Y, Li L, Renken C, Mannella CA, Selker JM, Rangell L, Bennett MJ, Zha J. Mol Biol Cell. 2005;16:1543. doi: 10.1091/mbc.E04-08-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fernie AR, Carrari F, Sweetlove LJ. Curr Opin Plant Biol. 2004;7:254. doi: 10.1016/j.pbi.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 37.Rutter J, Winge DR, Schiffman JD. Mitochondrion. 2010;10:393. doi: 10.1016/j.mito.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scarpulla RC. Phys Rev. 2008;88:611. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.