

Abstract
The ascomycin-producer strain Streptomyces ascomycinicus has been proven to be an extracellular poly(R)-3-hydroxybutyrate (PHB) degrader. The fkbU gene, encoding a PHB depolymerase (PhaZSa), has been cloned in E. coli and Rhodococcus sp. T104 strains for gene expression. Gram-positive host Rhodococcus sp. T104 was able to produce and secrete to the extracellular medium an active protein form. PhaZSa was purified by two hydrophobic interaction chromatographic steps, and afterwards was biochemically as well as structurally characterized. The enzyme was found to be a monomer with a molecular mass of 48.4 kDa, and displayed highest activity at 45°C and pH 6, thus being the first PHB depolymerase from a gram-positive bacterium presenting an acidic pH optimum. The PHB depolymerase activity of PhaZSa was increased in the presence of divalent cations due to non-essential activation, and also in the presence of methyl-β-cyclodextrin and PEG 3350. Protein structure was analyzed, revealing a globular shape with an alpha-beta hydrolase fold. The amino acids comprising the catalytic triad, Ser131-Asp209-His269, were identified by multiple sequence alignment, chemical modification of amino acids and site-directed mutagenesis. These structural results supported the proposal of a three-dimensional model for this depolymerase. PhaZSa was able to degrade PHB, but also demonstrated its ability to degrade films made of PHB, PHBV copolymers and a blend of PHB and starch (7∶3 proportion wt/wt). The features shown by PhaZSa make it an interesting candidate for industrial applications involving PHB degradation.
Introduction
Polyhydroxyalkanoates (PHAs) are intracellular polymers accumulated by a wide range of bacteria and archaea as a carbon and energy source when environmental conditions are not optimal for cell growth. Among these biopolymers poly(R)-3-hydroxybutyrate (PHB) is the best known and most common polyhydroxyalkanoate.
PHB has attracted much interest from the industry in the last two decades, since it is a natural thermoplastic that can be produced from renewable sources by different microorganisms or plants; it is biocompatible, nontoxic, biodegradable and presents some interesting chemical and mechanical properties, therefore PHB represents an interesting alternative to petroleum-derived plastics, despite its considerably higher production costs. Another important application of PHAs is related to the monomeric composition of this family of biopolymers, since all PHAs are enantiomerically pure polymers (all the monomers present R configuration). Thus, the degradation products of the PHAs are (R)-3-hydroxyalkanoic acids, valuable synthons for the chemical and pharmaceutical industries.
In the producer cell cytoplasm PHAs are stored in complex subcellular structures included under the term carbonosomes [1], which comprise amorphous PHA coated with a phospholipidic monolayer and different proteins involved in PHB production (polymerases), stabilization (phasins) or mobilization (depolymerases). These PHB carbonosomes are also known as native PHB or nPHB granules. When these granules are released into the extracellular medium as a consequence of cell lysis, PHAs become denatured, acquiring a semi-crystalline structure, known as denatured PHB or dPHB.
Insoluble dPHB granules are degraded in virtually every natural environment by extracellular PHB depolymerases produced by a wide variety of microorganisms, mainly bacteria and fungi. PHB depolymerases are specific for dPHB or nPHB, thus they are not able to degrade both types of PHB granules, except for the PHB depolymerase from Bacillus megaterium, classified as an intracellular nPHB depolymerase associated to PHB carbonosomes, but exhibiting dPHB depolymerase activity as well [2].
Many of PHB degrading microorganisms present in soil ecosystems are classified into the genus Streptomyces [3], which has been one of the most biotechnologically relevant genus during the last decades due to its metabolic versatility and the production of over 7000 natural antibiotics and other important bioactive compounds [4].
fkbU gene from S. ascomycinicus was described as a part of the FK520 gene cluster [5], responsible for the biosynthesis of ascomycin, a macrolide with immunosuppressive and antifungal activities. fkbU was proposed to encode a PHB depolymerase, but no experimental evidence regarding this enzyme was previously provided.
In this work, we demonstrate that S. ascomycinicus is able to degrade PHB and the identity of fkbU gene has been confirmed. In this sense, fkbU was cloned in the heterologous host Rhodococcus sp. T104, and its gene product, hereafter PhaZSa, was proven to be an extracellular dPHB depolymerase, which has been expressed in an active and extracellular form. Furthermore this depolymerase was purified as well as biochemically and structurally characterized, and a three-dimensional model was proposed for the tertiary structure of PhaZSa. Additionally, PhaZSa was used to perform film degradation tests employing pure PHB and PHB copolymers containing different monomeric contents of 3-hydroxyvalerate, as well as a blend of PHB and starch that has been reported to confer improved mechanical properties compared to PHB homopolymer, and also would allow industry to reduce the production costs of this kind of biodegradable plastics [6].
Materials and Methods
Chemicals
Cell culture medium reagents were from Difco (Becton Dickinson). All chemical reagents and polymers were purchased from Sigma-Aldrich.
Bacterial Strains, Media, and Growth Conditions
All strains used in this study are summarized in table 1. Streptomyces ascomycinicus sp. nov. DSMZ 40822 [8], (formerly known as S. hygroscopicus subsp. hygroscopicus or S. hygroscopicus subsp. ascomyceticus ATCC 14891), described as a putative extracellular PHB depolymerase producer, was used as chromosomal DNA source. Streptomyces exfoliatus DSMZ 41693 [9], [10], was used as positive control and Streptomyces coelicolor CECT 3243 as negative control for degradation of PHB. Escherichia coli DH5α was used as host for subcloning experiments, E. coli BL21(DE3) and wild type strain Rhodococcus sp. T104 KACC 21099 were used as hosts for gene expression [9], [11]. E. coli cells were grown in Luria–Bertani (LB) medium at 37°C, supplemented, when necessary, with 1 mM IPTG to induce overexpression of the cloned genes. For DNA purification, S. ascomycinicus cells were sporulated in solid SFM (Soya Flour Mannitol) medium and cultured aerobically under submerged conditions in S-YEME liquid medium (yeast extract/malt extract/0.5% glycine to allow dispersed growth) at 30°C and 250 rpm [12]. For PHB depolymerase extracellular activity detection, S. ascomycinicus spores previously collected and washed with 0.9% (wt/vol) NaCl were grown in solid basal mineral medium [13] supplemented with 1 mg/ml PHB as sole carbon source; plates were incubated for 120 hours at 30°C. Rhodococcus sp. T104 cells were grown in 2×YT (yeast extract/bactotriptone/NaCl) medium supplemented with glucose (5 g/l) [12].
Table 1. Bacterial strains, plasmids and constructs used in this study.
| Strain or plasmid | Relevant genotype or description | Reference |
| Strains | ||
| Escherichia coli DH5α | F− φ80dlacZΔM15 endA1 recA1 hsdR17 (rk −mK +) supE44 thi-1 gyrA96 relA1 Δ(lacZYA-argF)U169 λ− | [7] |
| Escherichia coli BL21(DE3) | F– ompT gal dcm lon hsdSB(rB − mB −) λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5]) | Invitrogen |
| Streptomyces ascomycinicusDSMZ 40822 | PHB depolymerase producer | [5], [8] |
| Streptomyces exfoliatusDSMZ 41693 | PHB and PHO depolymerase producer, used as positive control | [10] |
| Rhodococcus sp. T104KACC 21099 | Wild strain, suitable for gene cloning/expression with pNV19 vector | [11] |
| Plasmids | ||
| pET28 | Cloning/expression vector for E. coli strains. 5.4 kb | Novagen |
| pHPET | pET28 derivative containing fkbU gene | This work |
| pEM4 | Shuttle vector E. coli/Streptomyces. ApR TsrR permE* pUCori pWHM4ori 7.9 kb | [14] |
| pHPEM | pEM4 derivative containing fkbU gene | This work |
| pNV19 | Shuttle vector E. coli/Rhodococcus. KmR pAL5000ori lacZ CoE1ori. 4.4 kb | [9], [15] |
| pHPNV | pNV19 derivative containing fkbU gene under control of ermE* promoter | This work |
| pHPNV S131A/S131C/D209N/H269E/H269Q | pHPNV derivatives with mutated codons encoding catalytic amino acids | This work |
Plasmids, DNA Manipulation and Sequencing
All plasmids used in this study are summarized in table 1. pET28a(+) (KmR, T7 promoter, lacI) (Novagen) was used for gene expression in E. coli BL21(DE3). Bifunctional pEM4 (ApR, TsrR, permE*) [14] and pNV19 (KmR, lacZ) [15] plasmids were used to obtain the recombinant vectors for gene expression in Rhodococcus sp. T104. Chromosomal DNA from S. ascomycinicus DSMZ 40822 was purified according to the method described elsewhere [12]. Plasmid DNA preparations, restriction endonuclease digestions, ligations, and other DNA manipulations were carried out according to standard procedures for E. coli [16] and Streptomyces [12]. DNA sequences were determined by the dideoxy-chain-termination method [17] with an automated sequencer, DNA Analyzer 3730 (Applied Biosystems).
Construction of Strains Expressing the fkbU Gene
The putative PHB depolymerase encoding DNA sequence fkbU, (GenBank accession number: AF235504.1) was amplified by PCR using chromosomal DNA from S. ascomycinicus DSMZ 40822 as template. The PCR primers were designed according to the DNA sequence of fkbU [5]. Restriction sites NcoI, XbaI, and EcoRI were included in the primers to facilitate subcloning of PCR fragments. A Streptomyces RBS consensus sequence (GGAGG) was included in HPEM primer. PCR amplifications were performed in a Mastercycler Personal thermocycler (Eppendorf), employing Pfu DNA polymerase (Promega). The PCR products were purified by High Pure PCR Product Purification Kit (Roche), digested with endonucleases NcoI or XbaI and EcoRI, and cloned into the NcoI–EcoRI site of pET28 vector, resulting to recombinant plasmid pHPET or into the XbaI–EcoRI site of pEM4 vector, resulting to recombinant plasmid pHPEM. Recombinant plasmid pHPET was used to transform competent E. coli BL21(DE3) cells by heat shock. Recombinant pHPEM plasmid was digested with HindIII and EcoRI in order to obtain the fragments containing the ORF along with the strong ermE* promoter of the erythromycin resistance gene from Saccharopolyspora erythraea [18], [19] and transferred to the pNV19 vector, obtaining the recombinant plasmid pHPNV which was used to transform electrocompetent Rhodococcus sp. T104 cells, as previously described [9]. All resulting recombinant plasmids were purified by the High Pure Plasmid Isolation Kit (Roche) and sequenced to confirm the absence of mutations and the correct orientation.
Production and Purification of PhaZSa
Recombinant Rhodoccocus sp. T104 (pHPNV) cells were cultured aerobically under submerged conditions in 1 liter 2×YTG with 100 µg/ml kanamycin at 30°C for 72 h at 250 rpm orbital shaking. Ammonium sulfate was added to the cell-free culture broth at a final concentration of 0.5 M. Then, the solution was centrifuged at 10,000×g for 10 minutes and loaded onto a 100 ml Octyl FF sepharose column (GE Healthcare) equilibrated with 20 mM potassium phosphate buffer pH 7.0, 0.5 M ammonium sulfate (Buffer A) using a BioLogic LP chromatographic system (Bio-Rad). The column was extensively washed with 100 ml of the same buffer and the retained proteins were eluted with a linear decreasing gradient of 0.5 to 0 M ammonium sulfate in buffer A. The protein fractions containing PhaZSa were pooled and ammonium sulfate was added until final concentration of 0.5 M. After centrifugation at 10,000×g for 10 minutes, the supernatant was loaded onto a HiTrap Phenyl HP sepharose cartridge (GE Healthcare) (1 ml bed volume) equilibrated with buffer A. The cartridge was washed with 7 ml of the same buffer and a decreasing linear gradient from 0.5 to 0 M ammonium sulfate allowed elution of the bound proteins (purification table is shown in table S2). Purity of the fractions showing PHB depolymerase activity was analyzed by SDS-PAGE [20]. The amount of protein in the enzyme solutions was routinely determined by the Coomassie Blue method [21].
PHB Depolymerase Assays
Extracellular PHB depolymerase activity was measured by spot test or using the turbidimetric method, as described [9], [22] but using 50 mM MES buffer pH 6. One unit (U) of PHB depolymerase activity is the amount of enzyme needed to catalyze the decrease of 0.01 absorbance units (at 600 nm) per minute in the assay conditions described.
Effects of pH on PhaZSa stability were assessed by incubating 2 µg of pure PhaZSa for 45 minutes at 40°C and pH values from 4 to 9 in 20 mM phosphate/citrate/borate buffer with constant ionic strength of 120 mM, adjusted by addition of different amounts of NaCl.
Effects of temperature on PhaZSa stability were assessed by incubating 2 µg of pure PhaZSa for 45 minutes at temperatures ranging from 25 to 70°C in water bath with gentle shaking.
After the incubations, aliquots of enzyme were drawn at different times and placed on ice bath for five minutes. Remaining activity was immediately measured by the standard turbidimetric method. All assays were performed in triplicate.
PHB depolymerase activity of pure PhaZSa aliquots dialyzed against 50 mM MES buffer pH 6 was measured in the presence of different concentrations of divalent (MgCl2, CaCl2 and MnCl2) and monovalent (NaCl and KCl) cation chlorides by the standard turbidimetric assay. PhaZSa activity was also assessed by the standard turbidimetric assay in the presence of several concentrations of EDTA (with 2 mM MgCl2), methyl-β-cyclodextrin, polyethylenglycol 3350, reducing agents (DTT and 2-mercaptoethanol), corn starch and detergents (SDS, Tween 20 and Triton X-100), as well as in presence of twelve different organic solvents with 10% (vol/vol) concentration.
The apparent Km and Vmax values of PhaZSa for PHB hydrolysis were calculated by non-linear hyperbolic regression, using the starting values obtained by linear regression fitting of a Hanes-Woolf plot, [23], [24] with the Hyper32 software (freely available at http://homepage.ntlworld.com/john.easterby/hyper32.html). These parameters were calculated using the turbidimetric activity assay with PHB, the natural substrate of PhaZSa, and considering a PHB weight average molecular mass (MW) of 437 kDa, provided by the manufacturer.
The release of (R)-3-hydroxybutyrate by PhaZSa was measured using a spectrophotometric activity assay employing the β-hydroxybutyrate dehydrogenase from Pseudomonas lemoignei (Sigma) [22], [25]. Production of NADH as a result of (R)-3-hydroxybutyrate oxidation was measured at 340 nm after incubation of 100 µl samples for 30 minutes at 37°C with 9.5 mU of β-hydroxybutyrate dehydrogenase and 1 mM NAD+, in 75 mM Tris-HCl buffer pH 8, in a total volume of 500 µl. Reaction was stopped on ice and absorbance at 340 nm was immediately measured. Concentration of (R)-3-hydroxybutyrate was calculated by interpolating the absorbance values in a standard curve.
Degradation of PHB and PHBV films by PhaZSa
Thin solvent-cast films of pure PHB, PHBV with 5 or 12% (wt) 3-hydroxyvalerate and PHB-starch 7∶3 (wt) proportion were prepared by dissolving 100 mg of the polymer or blend in 20 ml of chloroform with heating and vigorous stirring. The solutions were poured on glass Petri dishes and then chloroform was evaporated at room temperature overnight. The films were subsequently submerged in 20 ml of 150 mM MES buffer pH 6 with 5 mM MgCl2 and 4 mM MβCD. Finally 250 µl of enzyme solution containing 30 µg of PhaZSa or 20 mM potassium phosphate buffer pH 7 (in the case of controls) were added to each plate. Plates were incubated at 37°C for 40 hours. Aliquots and pictures were taken at different times in order to monitor the degradation of the films; (R)-3-hydroxybutyrate concentration in the aliquots was quantified by the β-hydroxybutyrate dehydrogenase assay.
Chemical Modification of Recombinant PhaZSa
Modification by Phenylmethylsulfonyl fluoride (PMSF), diethylpyrocarbonate (DEPC), and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) were performed incubating 5 µg of recombinant PhaZSa with the appropriate amount of the suitable group-specific reagent as described [9]. PMSF solution was prepared in DMSO, DEPC in ethanol, and EDC was dissolved in 30 mM MES buffer pH 5. The concentration of DMSO or ethanol in the enzymatic assay did not exceed 0.1% (vol/vol) and was found to have no noticeable effect on the stability or activity of the enzyme. The remaining enzyme activity was determined by the standard turbidimetric assay.
Sulfhydryl groups were quantified spectrophotometrically with DTNB (5,5′-dithiobis-(2-nitrobenzoic acid) by Ellman’s method [26]. All experiments were carried out in triplicate, and mean values are shown in the tables. Controls for enzyme activity were carried out in all experiments.
Secondary Structure Elucidation
PhaZSa secondary structure was predicted using several bioinformatic tools or servers: PSIPRED (freely available at bioinf.cs.ucl.ac.uk/psipred) [27], Jpred 3 (freely available at www.compbio.dundee.ac.uk/www-jpred) [28] and PredictProtein, a collection of prediction tools available at www.predictprotein.org. Secondary structure content of PhaZSa was also experimentally obtained by circular dichroism spectrum deconvolution, using pure PhaZSa aliquots. Spectra were recorded with 102 µg/ml PhaZSa in 5 mM potassium phosphate buffer pH 7 at 25°C between 190 and 260 nm under thermostated conditions by using a JASCO J-715 spectropolarimeter. The CD readings were expressed as the mean residue molar ellipticity (degrees • cm2 • dmol−1), assuming a residue molecular mass of 104 Da according to the average amino acid molecular mass of PhaZSa. Secondary structure data was obtained from CD spectra deconvolution, using the CDNN V2.1 program [29]. Thermal unfolding of PhaZSa was analyzed by CD variation at 209 nm in 25–80°C range scanned at 20°C/h.
Identification of the PhaZSa Reaction Products
After 18 hours reaction with 1 µg PhaZSa and 300 µg/ml PHB in 20 mM MES buffer pH 6 at 40°C and 300 rpm orbital shaking, reaction products were derivatized with bromophenacyl bromide (BPB), and subsequently detected and identified by HPLC-MS as described [9], [30].
Site-directed Mutagenesis Studies
In order to ascertain the identity of the amino acids which constitute the catalytic triad of PhaZSa, the candidates chosen according to multiple sequence alignment were mutated using the Quikchange II XL site-directed mutagenesis kit (Stratagene). pHPNV plasmid was used as template for mutagenic PCR, using the primers listed in table S1. The resulting mutant constructions were sequenced to confirm the mutations and then transferred to Rhodococcus sp. T104 for protein expression. Serine 131 was exchanged for Alanine (mutant S131A) or Cystein (S131C); Aspartic acid 209 was exchanged for Asparagine (D209N), and Histidine 269 was exchanged for Glutamic acid (H269E) or Glutamine (H269Q).
Analytical Ultracentrifugation Analysis
Aliquots of pure PhaZSa with three different concentrations (65, 129 and 259 µg/ml) in 25 mM potassium phosphate pH 7 with 100 mM NaCl were subjected to sedimentation velocity experiments with a Beckman Coulter XL-I analytical ultracentrifuge equipped with absorbance optics, at 48,000×g and 20°C, using an An-60Ti rotor and standard (12 mm optical path) double sector centerpieces of Epon-charcoal. Baseline offsets were measured afterwards at 200,000×g. The apparent sedimentation coefficient of distribution, c(s), and sedimentation coefficient s were calculated from the sedimentation velocity data using the SEDFIT software [31].
Results
Multiple Sequence Alignment of PhaZSa Amino Acid Sequence
Complete amino acid sequence of the putative PHB depolymerase PhaZSa (519 amino acids, accession number AAF86381.1) deduced from fkbU gene from Streptomyces ascomycinicus sp. nov. DSMZ 40822 [5], was analyzed and aligned with a selection of homologous PHB depolymerase sequences from different Gram-positive (Catenulispora acidiphila, Streptomyces flavogriseus, Nocardiopsis dassonvillei, Thermobifida fusca and Janibacter sp.) and Gram-negative (Haliangium ochraceum, Paucimonas lemoignei and Ralstonia pickettii) microorganisms (Figure 1). All chosen sequences present a maximum identity below 58% among each other, in order to minimize redundancy; sequence similarity/identity matrix was calculated using MatGAT v 2.01 [32]. Multiple sequence alignment was performed with T-Coffee software, freely available at www.tcoffee.org [33]. In this alignment some highly conserved regions can be found, including the putative oxyanion hole and lipase box (Gly-Leu-Ser-Ala-Gly). Putative catalytic amino acids (Ser131-Asp209-His269) are strictly conserved, as well as the putative histidine in the oxyanion hole (His48). PhaZSa shows the typical arrangement of extracellular dPHB depolymerases with secretion signal peptide, catalytic domain, fibronectin type III linking domain and a C-terminal substrate binding domain [34]. According to the PHA Depolymerase Engineering Database [35] available at http://www.ded.uni-stuttgart.de, PhaZSa belongs to the dPHAscl homologous family 11.
Figure 1. Multiple sequence alignment of PhaZSa with other PHB depolymerase sequences.
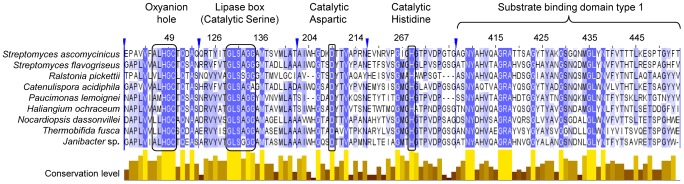
Only regions with conserved amino acids are shown. All shown sequences present an identity below 58% among each other to avoid redundancy. Main catalytic amino acids are marked with black boxes. Conservation level of each position can be observed in the yellow bars below. Vertical blue lines represent gaps in the complete sequence.
Detection of PHB Depolymerase Activity in S. ascomycinicus
The putative gene fkbU encoding PHB depolymerase from S. ascomycinicus was previously described [5], however no activity has been reported so far. In order to determine the extracellular PHB depolymerase activity of S. ascomycinicus, fresh spores were grown on solid basal mineral medium with PHB as sole carbon source, as described in materials and methods section. After 120 hours of incubation at 30°C a clear zone around the streak could be observed (Figure 2) demonstrating that S. ascomycinicus is able to degrade extracellular denatured PHB. This result shows the ability of this microorganism to produce an extracellular dPHB depolymerase.
Figure 2. Detection of extracellular PHB depolymerase activity in Streptomyces ascomycinicus.
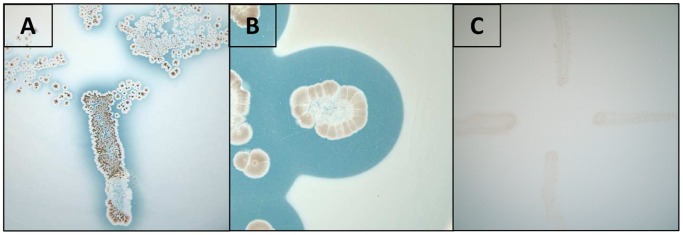
Basal mineral medium plates supplemented with PHB were inoculated with fresh spores of Streptomyces ascomycinicus (A), Streptomyces exfoliatus (positive control) (B) or Streptomyces coelicolor (negative control) (C), and incubated for 120 hours at 30°C. The clear halos surrounding the microbial growth indicate the degradation of PHB.
Construction of Strains Expressing the fkbU Gene and Detection of PhaZSa
The putative PHB depolymerase-coding gene fkbU was amplified by PCR and cloned into different vectors with the aim of expressing an active form of this enzyme. The pET28 derivative plasmid pHPET was used to clone and overexpress fkbU in E. coli BL21(DE3), but after inducing its expression with IPTG this recombinant strain was not able to produce an active form of PhaZSa, leading to the formation of insoluble inclusion bodies of PhaZSa.
In order to express fkbU gene in a homologous host, pHPEM plasmid was constructed, this pEM4 derivative plasmid contains the strong and constitutive ermE* promoter upstream the fkbU ORF, enabling the expression of this gene in gram positive hosts.
pHPEM was digested, and the HindIII-EcoRI fragment was cloned into pNV19, giving rise to the expression plasmid pHPNV, which was used to transform electrocompetent Rhodococcus sp. T104 cells. The recombinant Rhodococcus sp. T104 pHPNV cells were grown in 2×YTG medium and the PHB depolymerase activity of intracellular cell extract, insoluble cell debris and cell-free culture broth was assayed by the spot test method (Figure S1–B). Most of the activity was located in the fermentation broth, and a protein band of around 48 kDa corresponding to PhaZSa was detected by SDS-PAGE analysis of the fermentation broth, demonstrating that this enzyme was being secreted to the extracellular medium (Figure S1–A).
The N-terminal sequence of this extracellular protein obtained by Edman sequential degradation [36] was Ala-Ala-Gly-Leu-Ala-Lys-Pro-Gly-Leu-Thr-Lys-Ala-Asp-Leu-Thr-Glu-Val. Therefore, mature PhaZSa consists of 461 amino acids, with a theoretical mass of 48 kDa. It is noteworthy that the signal peptide is correctly recognized and cleaved by Rhodococcus sp. T104.
Purification and Analysis of Recombinant PhaZSa
Recombinant PhaZSa produced by Rhodococcus sp. T104 pHPNV was purified by only two consecutive hydrophobic interaction chromatographic (HIC) steps (Figure 3) MALDI-TOF analysis of pure enzyme showed a main peak of 48.4 kDa which fits the theoretical value deduced from the sequence (48.0 kDa), and several minor peaks corresponding to different protein aggregation states (Figure S2), the peak of 24.2 kDa corresponds to the same form with double electric charge. In addition, aliquots of pure PhaZSa with three different concentrations (65, 129 and 269 µg/ml) were subjected to sedimentation velocity analysis to ascertain the expected monomeric nature of this enzyme. The experiments with all these preparations showed a single peak with an apparent molecular mass between 40 and 46 kDa, corresponding to the active monomeric form of PhaZSa, and also showed characteristic values of a globular shaped protein.
Figure 3. Analysis of recombinant PhaZSa purification steps.
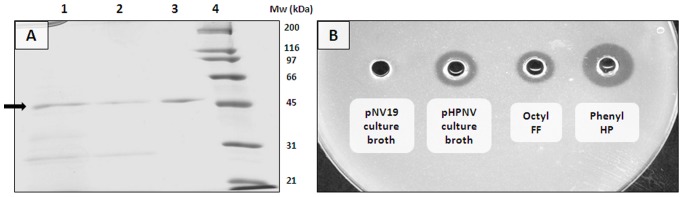
A) SDS-PAGE analysis: Lane 1: Rhodococcus T104 pHPNV culture broth; lane 2∶1 µg protein after octyl FF sepharose purification step; lane 3∶0.89 µg of purified protein after phenyl HP sepharose purification step; lane 4: Bio-Rad broad range molecular weight standards. Bands corresponding to PhaZSa are marked with an arrow. B) Spot test activity assay: Rhodococcus T104 pNV19 culture broth (negative control), Rhodococcus T104 pHPNV culture broth, pooled fractions after octyl FF sepharose purification step and pooled fractions after phenyl HP sepharose.
The secondary structure content of PhaZSa was experimentally calculated by CD spectrum deconvolution (Figure 4), and was also predicted according to the amino acid sequence with the online servers Jpred 3, PSIPRED and PredictProtein. Experimental and theoretical structural data were compared (Table 2). Experimentally obtained structural percentages differ slightly from the predicted values. However, all the programs employed for the prediction of secondary structure provide similar values, pointing to an alpha-beta structure.
Figure 4. Far UV circular dichroism spectrum of pure recombinant PhaZSa.
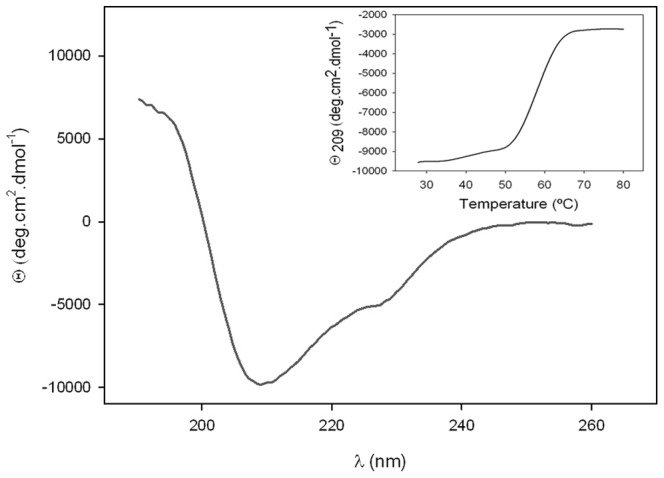
Inset: thermal unfolding of PhaZSa, studied by CD variation at 209.0 nm in 25–80°C range.
Table 2. Structural composition percentages of PhaZSa.
| Structure type | CD | PSIPRED | Jpred 3 | PredictProtein |
| α-helix | 16.8 | 12.4 | 11.5 | 12.8 |
| β-sheet | 32.2 | 27.1 | 29.9 | 26.5 |
| Other | 51.2 | 60.5 | 58.6 | 60.7 |
Comparison between the values deduced by CD spectrum deconvolution and those obtained by different secondary structure prediction servers.
Thermal denaturation of PhaZSa was also monitored by CD, measuring the change of ellipticity at 209 nm in a temperature range from 25 to 80°C (Figure 4 inset), showing a single melting temperature (T m) of 58.4°C.
Biochemical Characterization of Recombinant PhaZSa
Recombinant PhaZSa was functionally characterized, showing highest activity at pH 6 and 45°C. It showed full stability at the entire range of pH values (4 to 9) and temperatures up to 50°C.
In addition, the effect of different cations on activity of dialyzed PhaZSa was also studied, (Figure 5), revealing a strong dependence on the presence of divalent cations such as magnesium, calcium or manganese. Monovalent cations did not exert evident effects on activity of PhaZSa at concentrations up to 30 mM, although the increase of ionic strength gradually inhibited the PHB depolymerase activity leading to its complete inactivation at 2.5 M NaCl. The activity of PhaZSa was also assayed in the presence of several organic solvents at 10% (vol/vol) (Table 3), as well as in the presence of different additives (Table 4). It is remarkable the slight effect of reducing agents on PhaZSa activity compared to other PHB depolymerases, [10], [37], [38], [39] and the notable inhibitory effect of EDTA, which was expected due to the high dependence of PhaZSa activity on divalent cations.
Figure 5. Activity of dialyzed PhaZSa in presence of different concentrations of Magnesium (▴), Calcium (•), Manganese (▪), Sodium (×) or Potassium (★) chlorides.
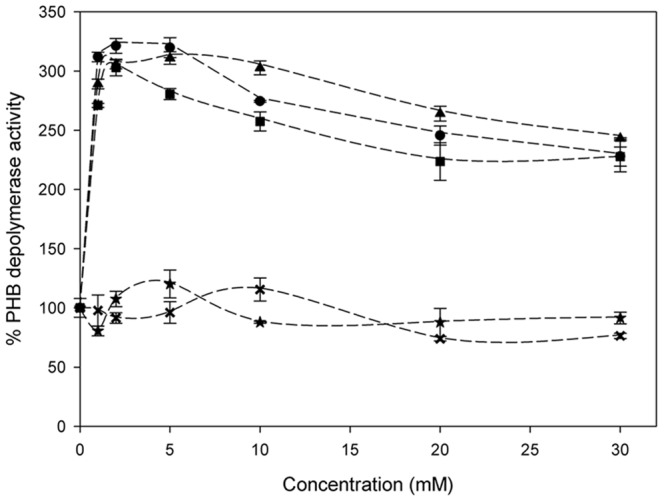
Table 3. PHB depolymerase activity of PhaZSa in the presence of 10% (vol/vol) of different organic solvents.
| Solvent | Relative activity (%) |
| Control | 100 |
| Glycerol | 97 |
| Ethylene glycol | 72 |
| THF | 49 |
| Acetonitrile | 30 |
| Diethylene glycol | 29 |
| 2-propanol | 29 |
| Triethylene glycol | 24 |
| Acetone | 18 |
| Ethanol | 16 |
| DMF | 15 |
| DMSO | 14 |
| Methanol | 9 |
Table 4. Effect of different concentrations of several reagents on the PHB depolymerase activity of PhaZSa.
| Reagent | Concentration | Relative activity (%) |
| Control | – | 100 |
| EDTA (+2 mM MgCl2) | 1 mM | 88 |
| 10 mM | 18 | |
| 20 mM | 14 | |
| Methyl-β-cyclodextrin | 1 mM | 126 |
| 5 mM | 146 | |
| 10 mM | 146 | |
| PEG 3350 | 1 mM | 112 |
| 5 mM | 106 | |
| 10 mM | 98 | |
| DTT | 1 mM | 90 |
| 5 mM | 44 | |
| 10 mM | 27 | |
| 2-Mercaptoethanol | 1 mM | 100 |
| 5 mM | 97 | |
| 10 mM | 91 | |
| Corn starch | 50 µg/ml | 103 |
| 200 µg/ml | 99 | |
| 400 µg/ml | 98 | |
| 1000 µg/ml | 104 | |
| SDS | 0.1% | Not detected |
| 1% | ||
| Tween-20 | 0.1% | |
| 1% | ||
| Triton X-100 | 0.1% | |
| 1% |
Furthermore, the kinetic parameters of recombinant PhaZSa for PHB hydrolysis were also determined. The apparent Km and Vmax values were 0.61±0.11 µM (269±48 µg/ml) and 9796.8±186.8 U/mg of enzyme, respectively.
Finally, the products released after PHB hydrolysis catalyzed by recombinant PhaZSa were derivatized with bromophenacyl bromide (BPB) and analyzed by HPLC-MS. As it can be observed in figure 6, derivatized monomers and dimers of (R)-3-hydroxybutyrate could be detected and identified in the reaction medium, suggesting that PhaZSa displays an exo-type hydrolysis mechanism.
Figure 6. Identification of PhaZSa reaction products by HPLC–MS.

The insets on the spectrum show the structure of the BPB derivatized compounds and their corresponding molecular weights. Double picks with similar intensity and a mass difference of 2 Da correspond to bromine-containing molecules, due to the isotopic abundance of this element (50.69% for Br79 and 49.31% for Br81).
Degradation of PHB and PHBV Films by PhaZSa
The degradation by recombinant PhaZSa of films made of PHB, PHBV with 5 or 12% (wt) content of 3-hydroxyvalerate and PHB-starch 7∶3 (wt) proportion was assessed for 40 hours at 37°C and pH 6. Images of the films at different times are shown in figure 7. Release of (R)-3-hydroxybutyrate to the reaction medium was quantified by the β-hydroxybutyrate dehydrogenase method. As observed in Figure 8, PhaZSa was able to degrade all the films tested. However, depolymerization was slower in the case of PHBV 12% due to its high 3-hydroxyvalerate content. Films made of pure PHB, PHBV 5% and PHB-starch yielded a similar amount of product, and were completely disintegrated within the first 24 hours. Control films were incubated in the same conditions without enzyme to evaluate the possible spontaneous degradation of the polymers. In such cases, concentration of (R)-3-hydroxybutyrate was undetectable during the first 40 hours.
Figure 7. Degradation of different PHB films by PhaZSa.

Solvent cast films of PHB, PHBV 5%, PHBV 12% and PHB-starch 7∶3 were incubated with 30 µg of PhaZSa at 37°C without shaking. Control films were incubated in the same conditions without enzyme. Pictures were taken at different times.
Figure 8. Release of (R)-3-hydroxybutyrate from different PHB films catalyzed by PhaZSa.
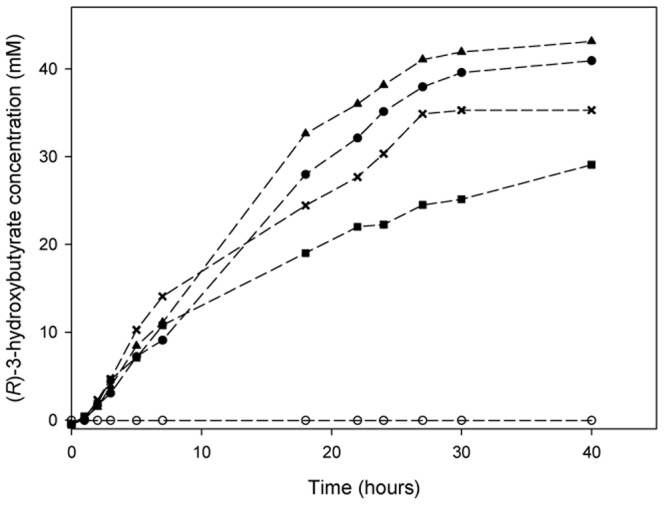
Concentration of (R)-3-hydroxybutyrate, released from the films by PhaZSa, was monitored during the first 40 hours of reaction by the β-hydroxybutyrate dehydrogenase method. The films used were PHB (•), PHBV 5% (▴), PHBV 12% (▪), and PHB-starch 7∶3 proportion (×). All the controls without enzyme (O) presented undetectable concentrations of (R)-3-hydroxybutyrate.
Identification of Catalytic Amino Acids of PhaZSa
The presence of a catalytic triad involving a serine, a histidine and an aspartic acid was clearly suggested by previously described PHB depolymerases with known tertiary structure or catalytic mechanisms [34]. This fact is also supported by the clearly conserved amino acids found when performing a multiple sequence alignment (Figure 1). With the purpose of identifying the essential catalytic residues of PhaZSa, responsible for the hydrolysis of PHB, three specific amino acid-modifying reagents were used in this study (PMSF for serines, DEPC for histidines and glycinamide-EDC for carboxyl residues).
The residual PHB depolymerase activity after chemical modification of PhaZSa with different concentrations of modifying reagents can be observed in table 5; PMSF abolishes activity whereas DEPC and glycinamide-EDC exerted an important inhibition of the activity; these results support the presence of essential serine, histidine and carboxyl-residues for the catalytic activity of PhaZSa. Titration of sulfhydryl groups with Ellman’s reagent [26] revealed two free cysteine residues per enzyme molecule. Since PhaZSa has a total number of eight cysteine residues in its sequence, it means that two of these cysteines are in sulfhydryl form, and the other six are involved in the formation of three disulfide bonds, stabilizing the tertiary structure of the protein.
Table 5. Residual PHB depolymerase activity after chemical modification of PhaZSa by several amino acid-specific reagents.
| Reagent | Concentration (mM) | Relative activity (%) |
| Control | – | 100 |
| PMSF | 0.5 | 4 |
| 1 | 0 | |
| DEPC | 5 | 60 |
| 20 | 10 | |
| EDC+Glycinamide | 1 | 85 |
| 5 | 67 |
In order to identify the serine, histidine and carboxyl-residues involved in the hydrolysis of PHB catalyzed by PhaZSa, site-directed mutagenesis of the enzyme was performed. Based on multiple sequence alignment, Ser131, Asp209 and His269 were strongly suggested as candidates for constituting the catalytic triad since they are strictly conserved. To demonstrate the importance of these putative catalytic amino acids, several mutant forms of the enzyme have been engineered by site-directed mutagenesis experiments. Five mutants of PhaZSa (namely S131A, S131C, D209N, H269E and H269Q) were expressed in Rhodococcus sp. T104; the correct secretion of these mutant enzymes to the extracellular medium was confirmed by SDS-PAGE (Figure S3–A). PHB depolymerase activity of cell-free culture broths was qualitatively checked by spot test activity assay. All mutant forms were correctly secreted by Rhodococcus sp. T104, and the PHB depolymerase activity was undetectable in every case (Figure S3-B). These results demonstrate the presence and identity of the catalytic triad in the active site of PhaZSa.
Discussion
PHB depolymerases from many Gram-negative bacteria have been purified and are well-characterized in contrast to the limited knowledge of PHB depolymerases from Gram-positive bacteria. With this work, Streptomyces ascomycinicus has been proven to produce an extracellular PHB depolymerase (PhaZSa) specific for degradation of denatured and partially crystalline granules of PHB, present in the environment as a product of cell lysis of PHB-producer bacteria. Therefore, the role of this enzyme is not the mobilization of intracellular PHB, but the use of extracellular denatured PHB, in contrast to the hypothesis proposed by Wu et al. (5); in that work fkbU gene was proposed to encode a PHB depolymerase that could be responsible for maintaining the intracellular levels of butyryl-CoA during the stationary phase of growth, using the accumulated PHB as the carbon storage for this biosynthetic pathway. Since fkbU encodes an extracellular depolymerase, it cannot be directly related to the FK520 synthetic pathway, and despite its adjacent location, this gene should not be considered as a member of this cluster.
In addition, fkbU gene, encoding PhaZSa, has been cloned into pNV19 vector, expressed under control of the strong and constitutive ermE* promoter and secreted to extracellular medium by Rhodococcus sp. T104. The success of this expression system confirms the suitability of Rhodococcus sp. T104 for the heterologous expression of Streptomyces enzymes [9], and also demonstrates that this Gram-positive host recognizes genetic elements from Streptomyces genes, such as the ermE* promoter, and the consensus ribosome binding site, as well as secretory signal peptides. According to the predicted amino acid sequence of PhaZSa (accession number AAF86381.1) it presents an unusually long secretion putative signal peptide (58 residues), which predicted cleavage point was confirmed by N-terminal sequencing of the mature protein. However, fkbU could be expressed using an alternative translation start codon (GTG, residue number 33 in the predicted sequence) [12], preceded by a potential ribosome binding site (GGACG) located seven bp upstream, which would yield a 26 amino acids signal peptide, same length as reported for the PHB depolymerase from S. exfoliatus [9], [10]. This short signal peptide would fit the typical structure of secretory signal peptides from Streptomyces strains [40]. The analysis of PhaZSa secondary structure (Table 2) supports an alpha/beta hydrolase fold, present in many hydrolytic enzymes such as lipases. A catalytic domain type 1 according to the position of the putative oxyanion hole histidine (closer to the N-terminus than the catalytic amino acids) has been identified, as well as a substrate binding/recognizing domain type 1, with some clearly conserved amino acids (Figure 1) that might play an important role in the establishment of hydrophilic, hydrophobic or electrostatic interactions with the polymeric substrate [41], [42]. The catalytic triad (Ser131-Asp209-His269) which was previously postulated by multiple sequence alignment [34], is also present, as well as a lipase box pentapeptide (Gly-Leu-Ser-Ala-Gly), which includes the catalytic serine (Ser131). In this work, the identity of the catalytic triad has been confirmed by chemical modification and site-directed mutagenesis of specific amino acid residues.
The homology modeling of the tertiary structure of PhaZSa using Phyre2 server [43] (Figure 9), suggested that this enzyme lacks a lid domain similarly to other described extracellular PHA depolymerases [44]. In this model the spatial arrangement of the catalytic amino acids can be observed, as well as the proximity of His48, the putative oxyanion hole histidine that stabilizes the tetrahedral transition state. The structure of the substrate binding domain has not been reliably established yet, due to the lack of homologous proteins with known three-dimensional folding, however, ab initio modeling of this domain provides an outline of its structure, which would comprise coiled regions and several β-sheets.
Figure 9. Three-dimensional structure model of PhaZSa.

A) Complete model. This structure was modeled by the Phyre2 server (Protein Homology/analogY Recognition Engine V 2.0) [43] available at www.sbg.bio.ic.ac.uk/phyre2, which combines homology and ab initio modeling algorithms. These figures were rendered using Discovery Studio 3.1 software (Accelrys software Inc.) B) Detail of the active site, catalytic amino acids (Ser131-Asp209-His269) and oxyanion hole histidine (His48).
The extracellular expression of PhaZSa has facilitated its purification from the fermentation broth by only two hydrophobic interaction chromatography (HIC) steps. A good level of separation is achieved in the first HIC step (octyl sepharose), nevertheless a second HIC step (butyl HP sepharose) is required to reach a higher degree of purity and concentration, in order to characterize this enzyme.
Regarding the optimum reaction conditions, PhaZSa shows highest activity at 45°C, which is a very common optimum temperature among PHB depolymerases from soil microorganisms. However, the optimum pH of this enzyme is 6, this acidic pH is quite usual among fungal PHB depolymerases, but it is very uncommon in bacterial PHB depolymerases. Only two enzymes of this group, both from the Gram-negative Ralstonia pickettii have been reported to show acidic optimum pH values of 5.5 and 6 [45]. Hence, to the best of our knowledge PhaZSa is the first reported PHB depolymerase from a Gram-positive bacterium with an acidic pH optimum. This singular feature makes PhaZSa an interesting biocatalyst, suitable for PHB-derived residues degradation in acidic media.
Hydrolytic activity of PhaZSa is strongly enhanced (over 300%) in presence of low concentrations of divalent cations such as calcium, magnesium or zinc. However, PhaZSa remains slightly active even when it is thoroughly dialyzed in the presence of EDTA. These results indicate that PhaZSa shows a non-essential activation by divalent cations. Likewise, methyl-β-cyclodextrin (MβCD) exerts an important activating effect (up to 146%) at concentrations as low as 5 mM. This effect was previously described for PHB depolymerase from Streptomyces exfoliatus [9], but it remains unclear whether the effect of MβCD is produced directly on the enzyme, on the polymeric substrate (e.g. facilitating the access to PHB particles), or even associating with the reaction products, enhancing its solubility in the medium or shifting the equilibrium of the reaction towards the products.
Regarding kinetic parameters of PhaZSa, it is a difficult task to compare our data with those previously reported from other PHB depolymerases, since different measurement methods were employed in literature; moreover, the polymeric nature of the substrate is a problem when substrate concentration should be expressed in molarity terms. In this work, this problem was overcome by using the weight average molecular mass/MW of the PHB, estimated by the manufacturer (437,000 g/mol), to calculate the moles of polymer degraded in activity assays. Using this approach, PhaZSa parameters (KM 0.61 µM or 268 µg/ml and Vmax 9,797 U/mg) can only be compared to those of PhaZSex from S. exfoliatus [9], which were obtained by the same procedure. In this sense, PhaZSa shows a 2-fold higher KM value and 3.5-fold higher Vmax value than PhaZSex, what means a better catalytic efficiency (1.75-fold higher). In general terms, when comparing KM value of PhaZSa with those previously reported for other PHB depolymerases, several enzymes with higher affinity (lower KM) for PHB were found, such as PHB depolymerases from Leptothrix sp. HS [46], Thermus thermophilus HB8 [47] or Paecilomyces lilacinus D218 [48]. However PHB depolymerases with acidic pH optima (predominantly fungal) generally had a higher KM value than PhaZSa, ranging from 0.69 to 14 mg/ml [49], [50], [51], [52]. This result supports PhaZSa as a suitable biocatalyst for PHB degradation in slightly acidic conditions.
Finally, PhaZSa has been successfully employed for degradation of PHB solvent cast films. Two PHBV copolymers with 5 or 12% hydroxyvalerate have been tested, as well as a blend of PHB and starch in 7∶3 (wt/wt) proportion. PHBV polymers containing both hydroxybutyrate and hydroxyvalerate monomers have many important advantages regarding their thermomechanical properties in comparison to the PHB homopolymer: they are more ductile, flexible, less crystalline and present better tensile strength and lower melting points [53], [54]. Likewise, blends of PHB with starch has been reported to confer improved mechanical properties as well, giving these films more tensile strength and increasing the extension needed to break them. Additionally, blending virgin PHB with starch would reduce considerably the high production costs of PHB without affecting its biodegradability [6]. In this work, PhaZSa has been proven to degrade all types of PHB films tested (Figure 7); the presence of starch or a moderate content of hydroxyvalerate in the film did not affect enzymatic depolymerization, although degradation of PHBV containing 12% hydroxyvalerate is relatively slower than the other tested bioplastics. During polyester degradation, (R)-3-hydroxybutyrate was released and quantified using a specific enzymatic assay, reaching approximately 40 mM concentration in the first 30 hours of incubation. Production of (R)-3-hydroxybutyrate by PhaZSa under mild conditions is an interesting feature, since this enantiomerically pure hydroxyalkanoic acid has a wide range of industrial and medical applications, serving as building block for synthesis of many fine chemicals or tailor-made plastics with controlled properties [55], [56].
To conclude, PhaZSa has very interesting properties for its industrial implementation, since it is an extracellular hydrolytic enzyme, stable up to 50°C and within a broad pH range (from 4 to 9), and is able to degrade different PHB copolymers. Moreover, PhaZSa can resist freezing and lyophilization with no need to add any protective additive, and its PHB depolymerase activity can be recovered even after denaturation with 6 M guanidinium chloride. These features make PhaZSa a promising candidate for the development of a robust biocatalyst able to degrade PHB-derived residues present in urban solid wastes.
Supporting Information
Production of recombinant PhaZ Sa in Rhodococcus T104. A) SDS-PAGE analysis; Lanes 1–3: Rhodococcus T104 pNV19 (control strain). Lanes 5–7: Rhodococcus T104 pHPNV (fkbU clone). Lanes 1 and 5: culture broths, lanes 2 and 6: cell extracts, lanes 3 and 7: cellular debris, lane 4: Sigma wide range molecular weight standards. A protein band of about 50 kDa not present in the control strain is marked with an arrow in the culture broth of pHPNV clone. B) Spot test activity assay of different fractions of these strains. Wells are marked with their corresponding lane number in SDS-PAGE analysis from panel A.
(TIF)
MALDI-TOF mass spectrum of pure recombinant PhaZ Sa expressed by Rhodococcus T104 pHPNV.
(TIF)
Site-directed mutagenesis of the catalytic triad residues of PhaZ Sa . A) SDS-PAGE of the fermentation broths from the Rhodococcus T104 strains carrying the mutant pHPNV plasmids. Lane 1: S131A; lane 2: S131C; lane 3: D209N; lane 4: H269E; lane 5: H269Q; lane 6: pNV19 negative control; lane7; pHPNV positive control; lane 8: Bio-Rad broad range molecular weight standards. Band corresponding to PhaZSa or its mutant forms is marked with an arrow. B) Spot test PHB depolymerase activity assay of the fermentation broths containing the mutant forms of PhaZSa, the native PhaZSa and the negative control pNV19.
(TIF)
PCR primers used in this work for cloning of fkbU and site-directed mutagenesis of PhaZ Sa .
(DOCX)
Purification table of PhaZ Sa .
(DOCX)
Acknowledgments
The authors wish to acknowledge the collaboration of Dr. Gerben Zylstra from Rutgers University (NJ, USA) for kindly providing Rhodococcus sp. T104 KACC 21099 and the plasmid pNV19, and Dr. J. A. Salas from the University of Oviedo for providing the pEM4 expression vector.
Funding Statement
This work was carried out in the framework of the IP-Project “Sustainable Microbial and Biocatalytic Production of Advanced Functional Materials” (BIOPRODUCTION/NMP-2-CT- 2007-026515) funded by the European Commission. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Jendrossek D (2009) Polyhydroxyalkanoate granules are complex subcellular organelles (carbonosomes). J. Bacteriol. 191: 3195–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen HJ, Pan SC, Shaw GC (2009) Identification and characterization of a novel intracellular poly(3-Hydroxybutyrate) depolymerase from Bacillus megaterium. Appl. Environ. Microbiol. 75: 5290–5299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mergaert J, Webb A, Anderson C, Wouters A, Swings J (1993) Microbial degradation of poly(3-hydroxybutyrate) and poly(3-hydroxybutyrate-co-3-hydroxyvalerate) in soils. Appl. Environ. Microbiol. 59: 3233–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berdy J (2005) Bioactive Microbial Metabolites. J. Antibiot. 58: 1–26. [DOI] [PubMed] [Google Scholar]
- 5. Wu K, Chung L, Revill WP, Katz L, Reeves CD (2000) The FK520 gene cluster of Streptomyces hygroscopicus var. ascomyceticus (ATCC 14891) contains genes for biosynthesis of unusual polyketide extender units. Gene 251: 81–90. [DOI] [PubMed] [Google Scholar]
- 6. Godbole S, Gote S, Latkar M, Chakrabarti T (2003) Preparation and characterization of biodegradable poly-3-hydroxybutyrate-starch blend films. Bioresour. Technol. 86: 33–37. [DOI] [PubMed] [Google Scholar]
- 7. Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166: 557–580. [DOI] [PubMed] [Google Scholar]
- 8. Kumar Y, Goodfellow M (2010) Reclassification of Streptomyces hygroscopicus strains as Streptomyces aldersoniae sp. nov., Streptomyces angustmyceticus sp. nov., comb. nov., Streptomyces ascomycinicus sp. nov., Streptomyces decoyicus sp. nov., comb. nov., Streptomyces milbemycinicus sp. nov. and Streptomyces wellingtoniae sp. nov. Int. J. Syst. Evol. Microbiol. 60: 769–775. [DOI] [PubMed] [Google Scholar]
- 9. García-Hidalgo J, Hormigo D, Prieto MA, Arroyo M, de la Mata I (2011) Extracellular production of Streptomyces exfoliatus poly(3-hydroxybutyrate) depolymerase in Rhodococcus sp. T104: determination of optimal biocatalyst conditions. Appl. Microbiol. Biotechnol. 93: 1975–1988. [DOI] [PubMed] [Google Scholar]
- 10. Klingbeil B, Kroppenstedt RM, Jendrossek D (1996) Taxonomic identification of Streptomyces exfoliatus K10 and characterization of its poly(3-hydroxybutyrate) depolymerase gene. FEMS Microbiol. Lett. 142: 215–221. [DOI] [PubMed] [Google Scholar]
- 11. Hernández BS, Koh SC, Chial M, Focht DD (1997) Terpene-utilizing isolates and their relevance to enhanced biotransformation of polychlorinated biphenyls in soil. Biodegradation 8: 153–158. [Google Scholar]
- 12.Kieser T, Bibb M, Buttner M, Chater K, Hopwood D (2000) Practical Streptomyces genetics. The John Innes Foundation, Norwich.
- 13. Pridham TG, Gottlieb D (1948) The utilization of carbon compounds by some actinomycetales as an aid for species determination. J. Bacteriol. 56: 107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Quirós LM, Aguirrezabalaga I, Olano C, Méndez C, Salas JA (1998) Two glycosyltransferases and a glycosidase are involved in oleandomycin modification during its biosynthesis by Streptomyces antibioticus. Mol. Microbiol. 28: 1177–1185. [DOI] [PubMed] [Google Scholar]
- 15. Chiba K, Hoshino Y, Ishino K, Kogure T, Mikami Y, et al. (2007) Construction of a pair of practical Nocardia-Escherichia coli shuttle vectors. Jpn. J. Infect. Dis. 60: 45–47. [PubMed] [Google Scholar]
- 16.Sambrook J, Russell DW (2001) Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 17. Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 74: 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bibb MJ, Janssen GR, Ward JM (1985) Cloning and analysis of the promoter region of the erythromycin resistance gene (ermE) of Streptomyces erythraeus. . Gene 38: 215–226. [DOI] [PubMed] [Google Scholar]
- 19. Bibb MJ, White J, Ward JM, Janssen GR (1994) The messenger RNA for the 23S ribosomal RNA methylase encoded by the ermE gene of Saccharopolyspora erythraea is translated in the absence of a conventional ribosome-binding site. Mol. Microbiol. 14: 533–545. [DOI] [PubMed] [Google Scholar]
- 20. Laemmli UK (1970) Cleavage of structural proteins during assembly of head of bacteriophage T4. Nature 227: 680–685. [DOI] [PubMed] [Google Scholar]
- 21. Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248–254. [DOI] [PubMed] [Google Scholar]
- 22. Jendrossek D (2007) Peculiarities of PHA granules preparation and PHA depolymerase activity determination. Appl. Microbiol. Biotechnol. 74: 1186–96. [DOI] [PubMed] [Google Scholar]
- 23. Wilkinson GN (1961) Statistical estimations in enzyme kinetics. Biochem. J. 80: 324–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Duggleby RG (1981) A nonlinear regression program for small computers. Anal. Biochem. 110: 9–18. [DOI] [PubMed] [Google Scholar]
- 25. Shirakura Y, Fukui T, Tanio T, Nakayama K, Matsuno R, et al. (1983) An extracellular D(-)-3-hydroxybutyrate oligomer hydrolase from Alcaligenes faecalis. Biochim. Biophys. Acta 748: 331–339. [DOI] [PubMed] [Google Scholar]
- 26. Ellman GL (1959) Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82: 70–77. [DOI] [PubMed] [Google Scholar]
- 27. McGuffin L, Bryson K, Jones DT (1999) The PSIPRED protein structure prediction server. Bioinformatics 16: 404–405. [DOI] [PubMed] [Google Scholar]
- 28. Cole C, Barber JD, Barton GJ (2008) The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36: 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Böhm G, Muhr R, Jaenicke R (1992) Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 5: 191–195. [DOI] [PubMed] [Google Scholar]
- 30. Gebauer B, Jendrossek D (2006) Assay of poly(3-hydroxybutyrate) depolymerase activity and product determination. Appl. Environ. Microbiol. 72: 6094–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brown PH, Schuck P (2006) Macromolecular size-and-shape distributions by sedimentation velocity analytical ultracentrifugation. Biophys. J. 90: 4651–4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Campanella JJ, Bitincka L, Smalley J (2003) MatGAT: an application that generates similarity/identity matrices using protein or DNA sequences. BMC Bioinformatics 4: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Notredame C, Higgins DG, Heringa J (2000) T-coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302: 205–217. [DOI] [PubMed] [Google Scholar]
- 34. Jendrossek D, Handrick R (2002) Microbial degradation of polyhydroxyalkanoates. Annu. Rev. Microbiol. 56: 403–432. [DOI] [PubMed] [Google Scholar]
- 35. Knoll M, Hamm TM, Wagner F, Martinez V, Pleiss J (2009) The PHA Depolymerase Engineering Database: A systematic analysis tool for the diverse family of polyhydroxyalkanoate (PHA) depolymerases. BMC Bioinformatics 10: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Edman P, Begg G (1967) A protein sequenator. Eur. J. Biochem. 1: 80–91. [DOI] [PubMed] [Google Scholar]
- 37. Nakayama K, Saito T, Fukui T, Shirakura Y, Tomita K (1985) Purification and properties of extracellular poly(3-hydroxybutyrate) depolymerases from Pseudomonas lemoignei. Biochim. Biophys. Acta 827: 63–72. [DOI] [PubMed] [Google Scholar]
- 38. Kim DY, Yun JH, Kim HW, Bae KS, Rhee YH (2002) Purification and characterization of poly(3-hydroxybutyrate) depolymerase from a fungal isolate, Emericellopsis minima W2. J. Microbiol. 40: 129–133. [Google Scholar]
- 39. Sznajder A, Jendrossek D (2011) Biochemical characterization of a new type of intracellular PHB depolymerase from Rhodospirillum rubrum with high hydrolytic activity on native PHB granules. Appl. Microbiol. Biotechnol. 89: 1487–1495. [DOI] [PubMed] [Google Scholar]
- 40. Gilbert M, Morosoli R, Shareck Fo, Kluepfel D (1995) Production and secretion of proteins by Streptomycetes. Crit. Rev. Biotechnol. 15: 13–39. [DOI] [PubMed] [Google Scholar]
- 41. Hiraishi T, Hirahara Y, Doi Y, Maeda M, Taguchi S (2006) Effects of mutations in the substrate-binding domain of poly[(R)-3-hydroxybutyrate] (PHB) depolymerase from Ralstonia pickettii T1 on PHB degradation. Appl. Environ. Microbiol. 72: 7331–7338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hermawan S, Jendrossek D (2010) Tyrosine 105 of Paucimonas lemoignei PHB depolymerase PhaZ7 is essential for polymer binding. Polym. Degrad. Stab. 95: 1429–1435. [Google Scholar]
- 43. Kelley LA, Sternberg MJE (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protocols 4: 363–371. [DOI] [PubMed] [Google Scholar]
- 44. Gangoiti J, Santos M, Prieto MA, de la Mata I, Serra JL, et al. (2012) Characterization of a novel subgroup of extracellular medium-chain-length polyhydroxyalkanoate depolymerases from actinobacteria. Appl. Environ. Microbiol. 78: 7229–7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kurusu Y, Kohama K, Uchida Y, Saito T, Yukawa H (1994) In Doi Y, Fukuda K (ed) Biodegradable plastics and polymers. Elsevier Science B V, Amsterdam: 357–361.
- 46. Takeda M, Koizumi J, Yabe K, Adachi K (1998) Thermostable poly(3-hydroxybutyrate) depolymerase of a thermophilic strain of Leptothrix sp. isolated from a hot spring. J. Ferment. Bioeng. 85: 375–380. [Google Scholar]
- 47. Papaneophytou CP, Pantazaki AA, Kyriakidis DA (2009) An extracellular polyhydroxybutyrate depolymerase in Thermus thermophilus HB8. Appl. Microbiol. Biotechnol. 83: 659–668. [DOI] [PubMed] [Google Scholar]
- 48. Oda Y, Osaka H, Urakami T, Tonomura K (1997) Purification and properties of poly(3-hydroxybutyrate) depolymerase from the fungus Paecilomyces lilacinus D218. Curr. Microbiol. 34: 230–232. [DOI] [PubMed] [Google Scholar]
- 49. Brucato CL, Wong SS (1991) Extracellular poly(3-hydroxybutyrate) depolymerase from Penicillium funiculosum: general characteristics and active site studies. Arch. Biochem. Biophys. 290: 497–502. [DOI] [PubMed] [Google Scholar]
- 50. Liu HY, Zhang H, Chen S, Liu DB, Xia HM (2006) Purification and properties of a poly (beta-hydroxybutyrate) depolymerase from Penicillium sp. J. Polym. Environ. 14: 419–426. [Google Scholar]
- 51. Zhou HL, Wang ZY, Chen S, Liu DB, Xia HM (2009) Purification and characterization of extracellular poly(-hydroxybutyrate) depolymerase from Penicillium sp DS9701-D2. Polym.-Plast. Technol. Eng. 48: 58–63. [Google Scholar]
- 52. Miyazaki K, Takahashi K, Shiraki M, Saito T, Tezuka Y, et al. (2000) Properties of a poly(3-hydroxybutyrate) depolymerase from Penicillium funiculosum. J. Polym. Environ. 8: 175–182. [Google Scholar]
- 53. Luzier WD (1992) Materials derived from biomass/biodegradable materials. Proc. Natl. Acad. Sci. U. S. A. 89: 839–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Braunegg G, Lefebvre G, Genser KF (1998) Polyhydroxyalkanoates, biopolyesters from renewable resources: Physiological and engineering aspects. J. Biotechnol. 65: 127–161. [DOI] [PubMed] [Google Scholar]
- 55. Tokiwa Y, Ugwu CU (2007) Biotechnological production of (R)-3-hydroxybutyric acid monomer. J. Biotechnol. 132: 264–272. [DOI] [PubMed] [Google Scholar]
- 56. Park SJ, Kim TW, Kim MK, Lee SY, Lim S-C (2012) Advanced bacterial polyhydroxyalkanoates: Towards a versatile and sustainable platform for unnatural tailor-made polyesters. Biotechnology Advances 30: 1196–1206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Production of recombinant PhaZ Sa in Rhodococcus T104. A) SDS-PAGE analysis; Lanes 1–3: Rhodococcus T104 pNV19 (control strain). Lanes 5–7: Rhodococcus T104 pHPNV (fkbU clone). Lanes 1 and 5: culture broths, lanes 2 and 6: cell extracts, lanes 3 and 7: cellular debris, lane 4: Sigma wide range molecular weight standards. A protein band of about 50 kDa not present in the control strain is marked with an arrow in the culture broth of pHPNV clone. B) Spot test activity assay of different fractions of these strains. Wells are marked with their corresponding lane number in SDS-PAGE analysis from panel A.
(TIF)
MALDI-TOF mass spectrum of pure recombinant PhaZ Sa expressed by Rhodococcus T104 pHPNV.
(TIF)
Site-directed mutagenesis of the catalytic triad residues of PhaZ Sa . A) SDS-PAGE of the fermentation broths from the Rhodococcus T104 strains carrying the mutant pHPNV plasmids. Lane 1: S131A; lane 2: S131C; lane 3: D209N; lane 4: H269E; lane 5: H269Q; lane 6: pNV19 negative control; lane7; pHPNV positive control; lane 8: Bio-Rad broad range molecular weight standards. Band corresponding to PhaZSa or its mutant forms is marked with an arrow. B) Spot test PHB depolymerase activity assay of the fermentation broths containing the mutant forms of PhaZSa, the native PhaZSa and the negative control pNV19.
(TIF)
PCR primers used in this work for cloning of fkbU and site-directed mutagenesis of PhaZ Sa .
(DOCX)
Purification table of PhaZ Sa .
(DOCX)