

Abstract
Holocarboxylase synthetase (HLCS) is a chromatin protein that facilitates the creation of histone H3 lysine 9-methylation (H3K9me) gene repression marks through physical interactions with the histone methyltransferase EHMT-1. HLCS knockdown causes a depletion of H3K9me marks in mammalian cell cultures and severe phenotypes such as short lifespan and low stress resistance in Drosophila melanogaster. HLCS displays a punctuate distribution pattern in chromatin despite lacking a strong DNA-binding domain. Previous studies suggest that the binding of HLCS to chromatin depends on DNA methylation. We tested the hypothesis that HLCS interacts physically with the DNA methyltransferase DNMT1 and the methyl CpG binding protein MeCP2 to facilitate the binding of HLCS to chromatin, and that these interactions contribute toward the repression of long-terminal repeats (LTRs) by H3K9me marks. Co-immunoprecipitation and limited proteolysis assays provided evidence suggesting that HLCS interacts physically with both DNMT1 and MeCP2. The abundance of H3K9me marks was 207% greater in the LTR15 locus in HLCS overexpression human embryonic kidney HEK293 cells compared with controls. This gain in H3K9me was inversely linked with a 87% decrease in mRNA coding for LTRs. Effects of HLCS abundance on LTR expression were abolished when DNA methylation marks were erased by treating cells with 5-azacytidine. We conclude that interactions between DNA methylation and HLCS are crucial for mediating gene repression by H3K9me, thereby providing evidence for epigenetic synergies between the protein biotin ligase HLCS and dietary methyl donors.
Keywords: Biotin, DNA methyl transferase, holocarboxylase synthase, methyl CpG binding protein 2, methylation, repression, long terminal repeats
Introduction
HLCS is the sole protein biotin ligase in the human proteome1 and has translational start sites in methionine-1, methionine-7 and methionine-58.2 The classical role of HLCS is to catalyze the covalent binding of biotin to five distinct carboxylases in cytoplasm and mitochondria.3 More recently, it became evident that HLCS also enters the cell nucleus4,5 where it binds to chromatin in a punctuate, locus-specific pattern.6,7 Unambiguous evidence suggests that recombinant HLCS catalyzes biotinylation of histone H3 in vitro and that HLCS knockdown causes a loss in histone H3 and H4 biotinylation marks in vivo.8-11 HLCS-dependent histone biotinylation marks are enriched in repressed genomic loci such as long-terminal repeats (LTRs) suggesting a role of HLCS in gene repression.9-13 Biotinylation of histones H3 and H4 is a natural modification, but the abundance of biotinylation marks is too low (< 0.001%) to account for the severe phenotypes seen in HLCS knockdown cells and flies.14-16 These phenotypes include a de-repression of LTRs in human and murine cell lines and in Drosophila melanogaster, and short life span and low stress resistance in Drosophila6,9 Collectively, these observations point toward a role of HLCS in gene repression at the epigenetic level that is independent of histone biotinylation.
We recently proposed that the roles of HLCS in gene repression are caused by interactions between HLCS and other chromatin proteins (Fig. 1).16 This theory integrates the discoveries described above with the following observations into a coherent model. First, HLCS binds to chromatin despite lacking a strong DNA-binding domain.6,7 Second, two HLCS-binding proteins have been identified, namely histone H3 and the histone H3 lysine-9 methyltransferase EHMT-1.8,17 Lysine-9 methylated histone H3 (H3K9me) is an abundant gene repression mark and EHMT-1 is one of the enzymes capable of creating this mark.18 HLCS physically interacts with EHMT-1 and catalyzes the biotinylation of lysine (K) residues in EHMT-1, which strengthens the interactions between the proteins in vitro.17 Both biotin depletion and loss of HLCS cause a decrease of H3K9me marks.9-11,19 Third, when DNA methylation marks are erased by 5-azacytidine treatment in human cell cultures, the abundance of histone biotinylation marks decreases considerably in LTRs; this is an uni-directional effect and biotin depletion has no effect on the abundance of DNA methylation marks.9
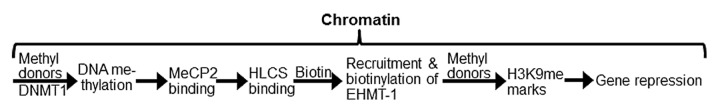
Figure 1. HLCS facilitates methylation events in the epigenome through physical interactions with DNA methyltransferase 1 (DNMT1), methyl CpG binding protein 2 (MeCP2) and eukaryotic histone methyltransferase (EHMT-1).
Here we tested the hypothesis that HLCS interacts with DNA methyltransferases (DNMT) and methyl-CpG-binding proteins (MeCPs), and that the binding of HLCS to loci rich in methylated DNA increases the local abundance of H3K9me marks, thereby contributing toward the repression of LTRs. Our rationale for testing DNMT1, as opposed to DNMT3a and DNMT3b, was that the maintenance methyltransferase DNMT1 mediates DNA methylation in all somatic cells, whereas the de novo methyltransferases DNMT3a and DNMT3b mediate DNA methylation only in embryonic cells.20,21 However, DNMT3a and DNMT3b were tested as specificity controls. Our rationale for testing MeCP2, as opposed to other methylated DNA binding proteins such as MBD1, MBD2 and MBD421,22 was that MeCP2 binds exclusively to methylated DNA to cause gene repression, whereas MBD1 can bind to non-methylated DNA and MBD4 does not cause gene repression; also MeCP2 is more abundant and widely expressed compared with MBD1.22,23 Our rationale for assessing the effects of DNA methylation on HLCS-mediated and H3K9me-dependent repression of LTRs, as opposed to other loci, was that DNA methylation, HLCS activity, biotin availability and H3K9me have been implicated in LTR repression.9,10,24,25 At least 54 LTRs in the human genome are transcriptionally active,26 and pose a threat to genome stability, as their mobilization facilitates recombination between non-homologous loci, leading to chromosomal deletions and translocations.25,27 Drug-induced hypomethylation of DNA is associated with activation of LTRs in mice28-30 and with chromosomal instability and tumors.31,32 Mobilization of LTR transposons is associated with 10% of all spontaneous mutations in mice.33 Pericentromeric α satellite repeats on chromosomes 1 (Chr1alpha) and 4 (Chr4alpha) were tested as specificity controls because HLCS-dependent biotinylation events are implicated in their repression.12
Results
HLCS interacts with DNMT1 and MeCP2
HLCS interacts physically with both DNMT1 and MeCP2, based on the following lines of evidence. Human embryonic kidney HEK293 cells were co-transfected with pCMV-Myc-HLCS and pCMV-HA-DNMT1. When cell lysates were precipitated with anti-Myc and probed with anti-DNMT1, a distinct signal was obtained for DNMT1 (Fig. 2A, first lane). Likewise, when cell lysates were precipitated with anti-HA and probed with anti-HLCS, a distinct signal was obtained for HLCS (Fig. 2B, first lane). When empty vectors (pCMV-Myc or pCMV-HA) were substituted for pCMV-Myc-HLCS and pCMV-HA-DNMT1, respectively, no signals were obtained (negative controls, second lane in both gels). Overexpression of DNMT1 and HLCS was confirmed by probing whole cell extracts with anti-DNMT1 and anti-HLCS (input control, third and fourth lane in both gels). In contrast, no signal was obtained when HEK293 cells were co-transfected with pCMV-Myc-HLCS and pCMV-HA-DNMT3a or pCMV-HA-DNMT3b, followed by probing anti-Myc precipitates with anti-HA, or when anti-HA precipitates were probed with anti HLCS (specificity control, data not shown).

Figure 2. HLCS interacts with both MeCP2 and DNMT1 in vivo, judged by co-immunoprecipitation assay. Myc-HLCS and HA-DNMT1 were overexpressed in HEK293 cells and protein extracts were immunoprecipitated using anti-Myc (A) and anti-HA (B). Proteins were resolved by electrophoresis and probed with anti-DNMT1 (A) and anti-HLCS ( B).Panels C and D are similar to A and B. But HA-MeCP2 was substituted for HA-DNMT1 in overexpression experiments and anti-MeCP2 (C) and anti-Myc (D) were substituted for anti-DNMT1 and anti-HLCS, respectively. Transfections with empty vectors were used as negative controls (second lane in each gel), and whole cell lysates were used as input controls (third and fourth lane in each gel). Gels were electronically re-arranged to facilitate comparisons.
Similar experiments were conducted for MeCP2. HEK293 cells were co-transfected with pCMV-Myc-HLCS and pCMV-HA-MeCP2. When cell lysates were precipitated with anti-Myc and probed with anti-MeCP2, a distinct signal was obtained for MeCP2 (Fig. 2C, first lane). Likewise, when cell lysates were precipitated with anti-HA and probed with anti-Myc, a distinct signal was obtained for HLCS (Fig. 2D, first lane). When empty vectors (pCMV-Myc or pCMV-HA) were substituted for pCMV-Myc-HLCS and pCMV-HA-MeCP2, respectively, no signals were obtained (second lane in both gels). Overexpression of MeCP2 and HLCS was confirmed by probing whole cell extracts with anti-MeCP2 and anti-Myc (input control, third and fourth lane in both gels).
Next, limited proteolysis assays were conducted using recombinant HLCS, DNMT1 and MeCP2. Limited proteolysis assays are based on the principle that physical interactions between proteins delay their degradation by dilute solutions of proteases such as trypsin.34 When His-tagged DNMT1 was mixed with GST-tagged HLCS prior to treatment with trypsin, the proteolytic digestion of DNMT1 and HLCS was slow, as evidenced by staining gels with blue stain (Fig. 3A, left panel). Meaningful amounts of protein were detected even after 90 min of incubation with trypsin. In contrast, when DNMT1 was incubated with GST tag alone, DNMT1 was degraded within less than 5 min, and GST started to degrade within 5 min of incubation (Fig. 3A, right panel). Likewise, when His-tagged MeCP2 was mixed with GST-tagged HLCS prior to treatment with trypsin, the proteolytic digestion of HLCS and MeCP2 was slow. Meaningful amounts of protein were detected even after 45 min of incubation with trypsin. (Fig. 3B, left panel). In contrast, when MeCP2 was incubated with GST tag alone, both MeCP2 and GST were degraded within 10 to 30 min of incubation (Fig. 3B, right panel).
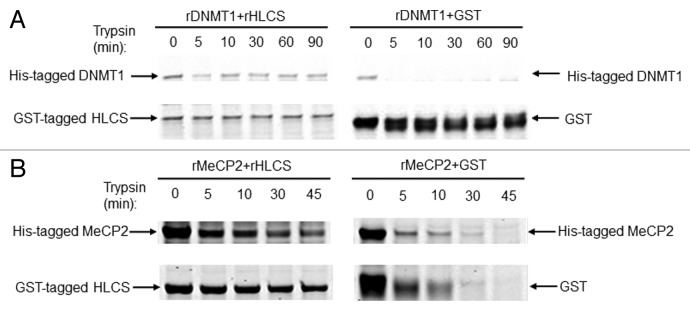
Figure 3. HLCS interacts with both MeCP2 and DNMT1 in vitro, judged by limited proteolysis assay. (A) Purified recombinant GST-HLCS protected His-DNMT1 from trypsin digestion (left panel) compared with DNMT1 that was pre-incubated with GST (right panel). (B) Purified recombinant GST-HLCS protected His-MeCP2 from trypsin digestion (left panel) compared with MeCP2 that was pre-incubated with GST (right panel).
Synergies between DNA methylation events and HLCS in the repression of LTRs
Our central hypothesis is that DNMT1 and MeCP2 facilitate the binding of HLCS to distinct loci in chromatin, and that HLCS recruits the histone methyltransferase EHMT-1 to repress genes through creating H3K9me marks. This hypothesis is partly based on previous observations suggesting that HLCS knockdown causes a loss of H3K9me mark in LTRs.9 Here we substantiated these previous observations by conducting HLCS overexpression experiments. HLCS was stably overexpressed in HEK293 cells. The abundance of mRNA coding for HLCS was 69 ± 4.6 times greater in overexpression cells compared with non-transfected controls; values were normalized by the abundance of mRNA coding for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The differences were similar when HLCS was probed with anti-HLCS or anti-Myc in western blots (Fig. 4).
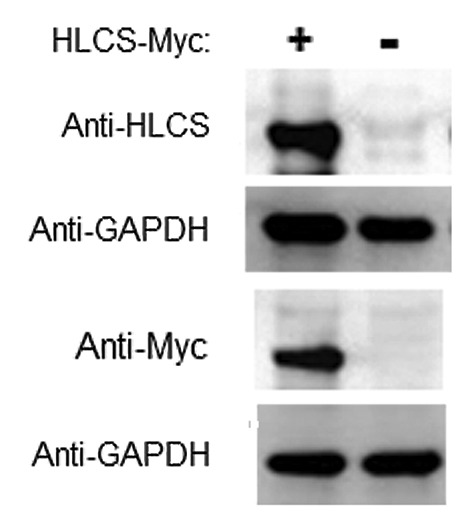
Figure 4. Transfection with plasmid FLAG/Myc-HLCS produced a stable overexpression of HLCS compared with non-transfected controls. The Myc-tagged HLCS in cell extracts was probed with anti-HLCS and anti-Myc. GAPDH was probed as loading control.
Consistent with our central hypothesis, HLCS overexpression caused a 207% increase of H3K9me marks in the LTR15 locus in HEK293 cell compared with non-transfected controls (Fig. 5A). A similar HLCS-dependent increase (33%) of H3K9me marks was also seen in the LTR22 locus but the difference was not statistically different between HLCS overexpression cells and controls. The HLCS-dependent enrichment of H3K9me marks was not specific for LTRs but was also seen in Chr1alpha and Chr4alpha repeats (Fig. 5A). In contrast, HLCS overexpression had no effect on the enrichment of H3K9me marks in promoter of GAPDH gene in euchromatin and the promoter of the transcriptionally repressed, colon-specific Apobec1 gene.
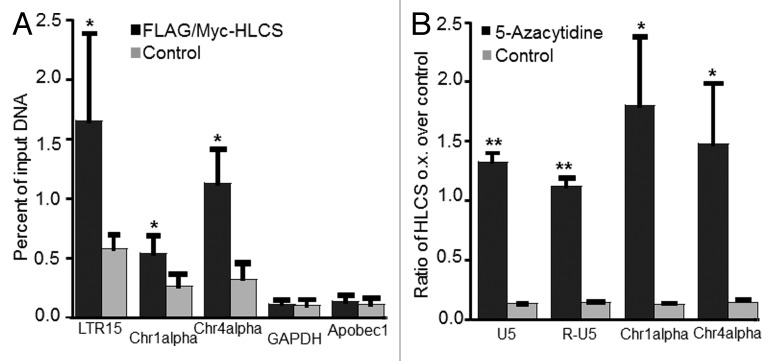
Figure 5. The effects of HLCS on LTR repression depend on DNA methylation. (A) The enrichment of H3K9me3 marks in loci coding for LTR15, Chr1alpha, Chr4alpha, GAPDH and Apobec1 was quantified by chromatin immunoprecipitation assay/qRT-PCR in HEK293 HLCS overexpression cells and control cells (H = 6.9018, 1 d.f., *p-value = 0.0086 HLCS overexpression vs. control; H = 4.8402, 1 d.f., *p-value = 0.0278 for Chr1alpha; H = 6.8598, 1 d.f., p-value = 0.0088 for Chr4alpha; H = 0.1756, 1 d.f., p-value = 0.6752 for GAPDH; H = 0.7245, 1 d.f., p-value = 0.3947 for Apobec1). (B) The transcription of LTRs, Chr1alpha and Chr4alpha was repressed in HLCS overexpression HEK293 cells compared with controls. Treatment of 5-azacytidine (1 µM) abrogated the effects of HLCS on repeat repression (**p-value < 0.0001, *p-value < 0.05, 5-azacytidine treatment vs. control).
The HLCS-dependent enrichment of H3K9me marks in LTRs caused an 87% decrease in the abundance of mRNA coding for LTRs (Fig. 5B). Importantly, this effect was abrogated when DNA methylation marks were erased by treating HEK-293 cells with 5-azacytidine. Note that transcripts coding for distinct LTRs are nearly identical and cannot be distinguished by regular quantitative real-time PCR. Therefore the mRNA values reported for LTRs represents the global expression of LTRs, as opposed to representing individual LTRs. Also note that LTRs may produce two distinct transcripts, U5 and R-U5, and that both transcripts depended on HLCS expression and DNA methylation. Again, effects of HLCS overexpression and 5-azacytidine treatment on transcriptional activity were not specific for LTRs, but were also seen in Chr1alpha and Chr4alpha repeats (Fig. 5B).
Discussion
This is the first report offering a potential mechanism for recruiting HLCS to distinct loci in chromatin. Based on this report, physical interactions between HLCS and DNMT1 and MeCP2 contribute toward the positioning of HLCS in human chromatin. This observation goes beyond our previous studies, which suggest that HLCS interacts physically with the core histones H3 and H4 and the histone methyltransferase EHMT-1.8,17 The interactions between HLCS and histones may account for the binding of HLCS to chromatin, but does not explain the punctuate pattern of HLCS localization that was observed in previous studies,6,7 simply because core histones can be found in all nucleosomes in chromatin. Likewise, the interactions between HLCS and EHMT-1 are unlikely to mediate the initial positioning of HLCS, based on previous observations that HLCS knockdown causes a loss of EHMT-1 dependent H3K9me marks.9,10,19 This study provides compelling evidence that HLCS interacts physically with DNMT1 and MeCP2 and that DNA methylation marks are important for subsequent HLCS/EHMT-1 interactions leading to the creation of H3K9me marks.
This paper is also the first report to identify epigenetic synergies between dietary methyl donors such as folate, methionine and choline, which are essential one-carbon donors in DNA and histone methylation events,35-37 and the micronutrient biotin, which appears to be essential for the creation of H3K9me marks.9,10,19 It remains to be determined whether methyl donors and biotin can compensate for each other’s deficiency or whether the deficiency of one of the two can precipitate the other’s deficiency symptoms. We do not propose that biotinylation of histones per se contributes in quantitatively meaningful ways to gene repression, because histone biotinylation marks are too rare to elicit such effects.14-16
Our model that HLCS is member of a multiprotein gene repression complex is more complicated than shown in Figure 1. For example, we have developed an algorithm to predict HLCS-interacting proteins and that algorithm identified not only EHMT-1 but also the nuclear co-repressor N-CoR2 as an HLCS interacting protein.17 N-CoR participates in the recruitment of histone deacetylases to human chromatin.38 Histone deacetylases catalyze the removal of acetylation marks from histones, thereby causing gene repression.39 Ongoing studies in our laboratory suggest that HLCS interacts physically with both N-CoR and histone deacetylases (D. Liu and J. Zempleni, unpublished). Depending on the outcome of these studies, the model shown in Figure 1 will need to be amended in the future.
The chromatin HLCS/multiprotein complex constitutes a plausible machinery contributing toward transcriptional repression of LTRs. Transcriptionally competent LTRs are enriched with H3K9me3 marks and the CpGs located in LTRs are generally hypermethylated.40,41 Evidence suggests that LTRs are also docking sites for HLCS, judged by the local enrichment of HLCS-dependent biotinylation marks.9 Both LTR15 and LTR22 are transcriptionally active and were chosen as representative examples of abundant 3′ proviral and solitary LTRs respectively, in the human genome.26 Considering the importance of repressing LTRs for maintaining genome stability, this paper may have implications for methyl/biotin and HLCS/protein synergies in gene regulation and disease risk. Importantly, our studies suggest that these synergies are not limited to the repression of LTRs, but also play roles in the repression of other repeats including pericentromeric α satellite repeats.
Some uncertainties remain. One might ask why HLCS participates in the creation of repression marks in the LTR15 locus, but to a much small extent in the LTR22 locus, and at non-detectable levels in the Apobec1 locus. A possible explanation could be that LTR15 is a proviral LTR, whereas LTR22 is a solitary LTR.26 Redundancies among histone K9-methyltransferases might also contribute to site specificity. The human genome encodes for H3K9 methyltransferases other than EHMT-1, e.g., SETDB1 and ESET.42,43 Histone methyltransferases other than EHMT-1 might not interact with HLCS. This issue will need to be addressed in future studies demonstrating co-localization of HCS and EHMT-1 in the same loci by using chromatin immunoprecipitation studies, or by using transgenic cell lines in which histone methyltransferases other than EHMT-1 have been knocked down. Likewise, methyl-CpG-binding proteins other than MeCP2 may have distinct effects in regard to recruiting HLCS to target loci. The absence of effect in the Apobec1 locus might be due to the fact that HLCS containing multiprotein complexes specifically target repeat regions such as LTRs and pericentromeric α satellite repeats, but that is untested speculation.
We conclude that HLCS exerts some of its roles in gene regulation through the formation of multiprotein gene repression complexes in human chromatin. Possible members of this complex include proteins involved in DNA methylation, EHMT-1, N-CoR2 and histone deacetylases. This theory would integrate previous observations that HLCS and biotin are important for gene repression, the low abundance of biotinylated histones in chromatin, and the known roles of members of the putative multiprotein complex in mediating gene repression into a coherent model.
Materials and Methods
Plasmids
Full-length human HLCS (NM_000411) was subcloned from plasmid pET41a (+)-HLCS44 into vector p3XFLAG-CMV-26 (Sigma-Aldrich, Cat# E7283) using NotI and XbaI, thereby creating plasmid FLAG/Myc-HLCS for studies of HLCS overexpression and its effects on H3K9me marks and LTR transcriptional activity. Plasmid pCMV-myc-HLCS was created as described previously17 and was used for overexpression of Myc-tagged HLCS in co-immunoprecipitation studies. Full-length human MeCP2 (GenBank Accession:NM_004992) was subcloned from plasmid pGADT7-MeCP2 (unpublished) into vector pCMV-HA (Clontech, Cat# 635690) using Sfil and XhoI for overexpression of HA-tagged MeCP2. Full-length DNMT1 (GenBank Accession:NM_001130823.1) was subcloned from pBluescript SK (+)-DNMT1 (unpublished) into pCMV-HA vector using NdeI and SalI for overexpression of HA-tagged DNMT1. Plasmid pET41a (+)-HLCS was used to prepare GST-tagged recombinant full-length HLCS as described previously.8 Full-length MeCP2 was subcloned from plasmid pET41a (+)-MeCP2 (unpublished) into vector pET28a+ (Novagen, Cat# 698646) using EcoRI and XhoI (creating pET28a-MeCP2), and full-length DNMT1 was subcloned from pET41a (+)-DNMT1 (unpublished) into pET28a+ vector using EcoRI and SalI (creating pET28a-DNMT1) for the preparation of recombinant proteins. As specificity controls, full-length human DNMT3a and DNMT3b were subcloned from plasmids pCMV-sport6-DNMT3A (Invitrogen, cat#6150112) and pENTR223.1-DNMT3B (Invitrogen, cat#40080762) into vector pCMV-HA using EcoRI and SalI to create plasmids pCMV-HA-DNMT3a and pCMV-HA-DNMT3b, respectively, for overexpression of HA-tagged DNMT3a and DNMT3b. The identities of all plasmids were verified by sequencing.
Cell lines
Human embryonic kidney HEK293 cells (American Type Culture Collection, CRL-1573) were cultured following the vendor’s recommendations. Cells were transfected with plasmid FLAG/Myc-HLCS by using electroporation and the transfectants were selected using 1 mM G418 for 14 d, and maintained in medium containing 0.4 mM G418. The expression of HLCS and GAPDH (control) was assessed by quantitative real-time PCR (qRT-PCR; see below) and by western blot analysis using anti-Myc (Abcam, cat# ab9106) and anti-HLCS serum.45 For co-immunoprecipitation assays, HLCS, DNMT1 and MeCP2 were transiently overexpressed as described below.
Co-immunoprecipitation assays
HEK293 cells were seeded in 25-cm2 flasks (2.5 x 106 cells/flask) 24 h before transient transfection with 8 μg of plasmids (4 µg each for Myc- and HA-tagged proteins) in TurboFect reagent (Fermentas, Cat# R0531) and Opti-MEM I medium (Invitrogen, Cat# 31985). Cells from six flasks were collected 48 h after transfection and lysed in 20 mM TRIS-HCl buffer (pH 8.0), containing 137 mM NaCl, 10% glycerol, 1% CA-630, 2 mM EDTA, phenylmethylsulfonyl fluoride and protease inhibitor cocktail (Sigma-Aldrich, Cat# P2714). Samples were pre-cleared using rabbit IgG before precipitation of proteins with rabbit polyclonal anti-HA (Abcam, Cat# ab9110), anti-Myc (Abcam, Cat# ab9106), and protein A resin (Thermo Scientific, Cat#20338). Western blots were performed using anti-MeCP2 (Abcam, Cat# ab2028), anti-DNMT1 (Abcam, Cat# ab16632), anti-Myc, anti-HLCS,45 and appropriate fluorophore conjugated secondary antibodies.10
Limited-proteolysis assays
Recombinant proteins were overexpressed in ArcticExpress (DE3) E. coli and purified using a Glutathione resin (Genscript, Cat# L00206) or a HisTrap FF column (GE Healthcare, Cat# 17–5319–01) as described previously.8 Limited proteolysis was performed at 37°C in 75 mM TRIS-acetate buffer (pH 7.5), containing 0.3 mM dithiothreitol and 45 mM MgCl2. One aliquot (time-zero sample) was collected before adding trypsin (Sigma-Aldrich, Cat# T6567), and equal volumes of aliquots were collected at timed intervals after initiation of digestion. Proteins were separated by gel electrophoresis and stained using Coomassie blue.17 In control experiments MeCP2 and DNMT1 were incubated with GST peptide to formally eliminate the possibility that the GST tag protected the proteins against proteolytic digestion.
5-Azacytidine treatment
5-Azacytidine solutions were prepared freshly immediately prior to use. Pilot tests suggested that 1 μM 5-azacytidine does not affect HEK293 cell viability. HEK293 cells were seeded in T25 flasks in fresh growth medium at day 0 and 5-azacytidine was added on day 1 to produce a final concentration of 1 μM. Media were changed daily maintaining the concentration of 5-azacytidine at 1 μM. On day 5, the cells were washed with phosphate-buffered saline and collected for analysis of gene expression by qRT-PCR.
Micro chromatin immunoprecipitation assay
The enrichment of H3K9me in distinct loci in human chromatin was assessed by micro chromatin immunoprecipitation (μChIP) assay as described before, using 2.5 × 106 HEK293 cells.10,46 ChIP-grade anti-H3K9me was purchased from Abcam (ab8898). Quantification of amplicons, calculation of relative enrichment of histone marks and normalization of nucleosomal occupancy using (nonmodified) C-terminus in histone H3 (Abcam, Cat# ab1791) were conducted as described previously.9,11 Data are expressed as percent of input DNA.
qRT-PCR
Power SYBR Green PCR Master Mix (Applied Biosystems, Cat# 4309155) was used to quantify the abundance of transcripts by qRT-PCR. Perfecta SYBR Green FastMix (Quanta Biosciences, Cat# 95073) was used to quantify the abundance of amplicons in immunoprecipitated chromatin.10 PCR primers are listed in Table 1. Pericentromeric α satellite repeats are repressed through HLCS- and methylation-dependent epigenetic events and were used as specificity controls.12,25 Apobec1 is specifically expressed in colon and its expression is silenced in kidney tissues as confirmed by searching against the BioGPS database.47
Table 1. PCR primers.
|
Target Sequence F/Ra Templateb |
| Apobec1 5′-TCCCATAACTGCCTGAGATG-3′ F DNA |
| 5′-TTGTTCCTGGACTTTGTTGC-3′ R DNA |
| Chr1alpha 5′-TCATTCCCACAAACTGCGTTG-3′ F DNA/Transcript |
| 5′-TCCAACGAAGGCCACAAGA-3′ R DNA/Transcript |
| Chr4alpha 5′-CTGCACTACCTGAAGAGGAC-3′ F DNA/Transcript |
| 5′-GATGGTTCAACACTCTTACA-3′ R DNA/Transcript |
| GAPDH 5′-CGTAGCTCAGGCCTCAAGA-3′ F DNA |
| 5′-GCTGCGGGCTCAATTTATAG-3′ R DNA |
| GAPDH 5′-TCCACTGGCGTCTTCACC-3′ F Transcript |
| 5′-GGCAGAGATGATGACCCTTT-3′ R Transcript |
| HLCS 5′-TGAGACCTGATCCTTAACTTCC-3′ F Transcript |
| 5′-ATGGAAGATAGACTCCACAT-3′ R Transcript |
| LTR15 5′-TTGTCATTGGTTCTGTGTAGGG-3′ F DNA |
| 5′-CCTCCATATGCTGAACGCTG-3′ R DNA |
| LTR22 5′- GACCATTTGCATGGACAAATC-3′ F DNA |
| 5′- CCTCCATATGCTGAACGCTG-3′ R DNA |
| LTR, R-U5 5′-GCGGGCAGCAATACTGCTTTGTAA-3′ F Transcript |
| 5′-ACCAGCGTTCAGCATATGGAGGAT-3′ R Transcript |
| LTR, U5 5′-AACTCAGAGGCTGGCG-3′ F Transcript |
| 5′-AGACACAGAGACAAAGTATAGAGA-3′ R Transcript |
Apobec1,apolipoprotein B mRNA editing enzyme, catalytic polypeptide 1 (GenBank NC_000012.11); Chr1alpha, chromosome 1 α satellite repeats (GenBank M26919); Chr4alpha, chromosome 4 α satellite repeats (GenBank M38467); GAPDH, glyceraldehyde-3-phosphate dehydrogenase (GenBank NG_007073.2 for genomic DNA, NM_002046.4 for transcript); HLCS, Holocarboxylase Synthetase (GenBank NM_000411.6); LTR15, intact LTR (GenBank AC116309)26; LTR22, solitary LTR (GenBank AL451165)26; LTR R-U5, region between R and U5 of LTR of HERV-K families50; LTR U5, U5 region of LTR of HERV-K families.50
a f, Forward; R, Reverse.
b DNA, genomic DNA for ChIP assay; Transcript, cDNA for qRT-PCR.
Statistical analysis
Data from μChIP assays were tested for normality of distribution and homogeneity of variances by constructing the probability plot of the residues, the stem-and-leaf plot of the residuals, and the normal probability plot and performing the Shapiro-Wilk test48 using the SAS 9.2 software package (SAS Institute, USA). After log transformation, the data were still not normally distributed and, therefore, were analyzed by the Kruskal-Wallis test as a robust substitute for one-way Anova.49 Calculations are based on five repeats. Differences were considered statistically significant if p-value was < 0.01. The data for quantification of LTR transcripts were analyzed by student’s t-test. Calculations were based on three repeats. Differences were considered statistically significant if p-value was < 0.05. Data are expressed as mean ± SD.
Acknowledgments
This work was supported in part by the University of Nebraska Agricultural Research Division with funds provided through the Hatch Act. Additional support was provided by NIH DK063945 and DK077816.
Glossary
Abbreviations:
- Holocarboxylase synthetase
HLCS
- histone H3 K9-methylation
H3K9me
- euchromatic histone-lysine N-methyltransferase 1
EHMT-1
- DNA methyltransferase 1
DNMT1
- methyl CpG binding protein 2
MeCP2
- long terminal repeats
LTRs
- pericentromeric alpha satellite repeats on chromosomes 1 and 4
Chr1alpha and Chr4alpha
- quantitative real-time PCR
qRT-PCR
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/24449
References
- 1.Suzuki Y, Aoki Y, Ishida Y, Chiba Y, Iwamatsu A, Kishino T, et al. Isolation and characterization of mutations in the human holocarboxylase synthetase cDNA. Nat Genet. 1994;8:122–8. doi: 10.1038/ng1094-122. [DOI] [PubMed] [Google Scholar]
- 2.Hiratsuka M, Sakamoto O, Li X, Suzuki Y, Aoki Y, Narisawa K. Identification of holocarboxylase synthetase (HCS) proteins in human placenta. Biochim Biophys Acta. 1998;1385:165–71. doi: 10.1016/S0167-4838(98)00032-6. [DOI] [PubMed] [Google Scholar]
- 3.Zempleni J, Wijeratne SSK. Kuroishi, T. (2012) Biotin, In Present Knowledge in Nutrition (Erdman, JW, Jr., Macdonald, I Zeisel, SH, Eds.), pp 587-609, International Life Sciences Institute, Washington, D.C. [Google Scholar]
- 4.Bailey LM, Wallace JC, Polyak SW. Holocarboxylase synthetase: correlation of protein localisation with biological function. Arch Biochem Biophys. 2010;496:45–52. doi: 10.1016/j.abb.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 5.Bao B, Wijeratne SS, Rodriguez-Melendez R, Zempleni J. Human holocarboxylase synthetase with a start site at methionine-58 is the predominant nuclear variant of this protein and has catalytic activity. Biochem Biophys Res Commun. 2011;412:115–20. doi: 10.1016/j.bbrc.2011.07.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camporeale G, Giordano E, Rendina R, Zempleni J, Eissenberg JC. Drosophila melanogaster holocarboxylase synthetase is a chromosomal protein required for normal histone biotinylation, gene transcription patterns, lifespan, and heat tolerance. J Nutr. 2006;136:2735–42. doi: 10.1093/jn/136.11.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh D, Pannier AK. Zempleni, J. Identification of holocarboxylase synthetase chromatin binding sites using the DamID technology. Anal Biochem. 2011;413:55–9. doi: 10.1016/j.ab.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bao B, Pestinger V, Hassan YI, Borgstahl GE, Kolar C, Zempleni J. Holocarboxylase synthetase is a chromatin protein and interacts directly with histone H3 to mediate biotinylation of K9 and K18. J Nutr Biochem. 2011;22:470–5. doi: 10.1016/j.jnutbio.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chew YC, West JT, Kratzer SJ, Ilvarsonn AM, Eissenberg JC, Dave BJ, et al. Biotinylation of histones represses transposable elements in human and mouse cells and cell lines and in Drosophila melanogaster. J Nutr. 2008;138:2316–22. doi: 10.3945/jn.108.098673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pestinger V, Wijeratne SSK, Rodriguez-Melendez R, Zempleni J. Novel histone biotinylation marks are enriched in repeat regions and participate in repression of transcriptionally competent genes. J Nutr Biochem. 2011;22:328–33. doi: 10.1016/j.jnutbio.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rios-Avila L, Pestinger V, Zempleni J. K16-biotinylated histone H4 is overrepresented in repeat regions and participates in the repression of transcriptionally competent genes in human Jurkat lymphoid cells. J Nutr Biochem. 2012;23:1559–64. doi: 10.1016/j.jnutbio.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Camporeale G, Oommen AM, Griffin JB, Sarath G, Zempleni J. K12-biotinylated histone H4 marks heterochromatin in human lymphoblastoma cells. J Nutr Biochem. 2007;18:760–8. doi: 10.1016/j.jnutbio.2006.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wijeratne SS, Camporeale G, Zempleni J. K12-biotinylated histone H4 is enriched in telomeric repeats from human lung IMR-90 fibroblasts. J Nutr Biochem. 2010;21:310–6. doi: 10.1016/j.jnutbio.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stanley JS, Griffin JB, Zempleni J. Biotinylation of histones in human cells. Effects of cell proliferation. Eur J Biochem. 2001;268:5424–9. doi: 10.1046/j.0014-2956.2001.02481.x. [DOI] [PubMed] [Google Scholar]
- 15.Bailey LM, Ivanov RA, Wallace JC, Polyak SW. Artifactual detection of biotin on histones by streptavidin. Anal Biochem. 2008;373:71–7. doi: 10.1016/j.ab.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 16.Kuroishi T, Rios-Avila L, Pestinger V, Wijeratne SS, Zempleni J. Biotinylation is a natural, albeit rare, modification of human histones. Mol Genet Metab. 2011;104:537–45. doi: 10.1016/j.ymgme.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Hassan YI, Moriyama H. Zempleni, J. Holocarboxylase synthetase interacts physically with euchromatic histone-lysine N-methyltransferase, linking biotinylation with histone methylation. J Nutr Biochem. 2013 doi: 10.1016/j.jnutbio.2012.12.003. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kouzarides T. Berger, SL. (2007) Chromatin modifications and their mechanism of action, In Epigenetics (Allis, CD, Jenuwein, T Reinberg, D, Eds.), pp 191-209, Cold Spring Harbor Press, Cold Spring Harbor, NY. [Google Scholar]
- 19.Gralla M, Camporeale G, Zempleni J. Holocarboxylase synthetase regulates expression of biotin transporters by chromatin remodeling events at the SMVT locus. J Nutr Biochem. 2008;19:400–8. doi: 10.1016/j.jnutbio.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schermelleh L, Haemmer A, Spada F, Rösing N, Meilinger D, Rothbauer U, et al. Dynamics of Dnmt1 interaction with the replication machinery and its role in postreplicative maintenance of DNA methylation. Nucleic Acids Res. 2007;35:4301–12. doi: 10.1093/nar/gkm432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li E. Bird, A. (2007) DNA methylation in mammals, In Epigenetics (Allis, CD, Jenuwein, T Reinberg, D, Eds.), pp 341-56, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 22.Bogdanović O, Veenstra GJ. DNA methylation and methyl-CpG binding proteins: developmental requirements and function. Chromosoma. 2009;118:549–65. doi: 10.1007/s00412-009-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kolker E, Higdon R, Haynes W, Welch D, Broomall W, Lancet D, et al. MOPED: Model Organism Protein Expression Database. Nucleic Acids Res. 2012;40(Database issue):D1093–9. doi: 10.1093/nar/gkr1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brenet F, Moh M, Funk P, Feierstein E, Viale AJ, Socci ND, et al. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS ONE. 2011;6:e14524. doi: 10.1371/journal.pone.0014524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martens JH, O’Sullivan RJ, Braunschweig U, Opravil S, Radolf M, Steinlein P, et al. The profile of repeat-associated histone lysine methylation states in the mouse epigenome. EMBO J. 2005;24:800–12. doi: 10.1038/sj.emboj.7600545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buzdin A, Kovalskaya-Alexandrova E, Gogvadze E, Sverdlov E. GREM, a technique for genome-wide isolation and quantitative analysis of promoter active repeats. Nucleic Acids Res. 2006;34:e67. doi: 10.1093/nar/gkl335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kazazian HH., Jr. Mobile elements: drivers of genome evolution. Science. 2004;303:1626–32. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- 28.Jaenisch R, Schnieke A, Harbers K. Treatment of mice with 5-azacytidine efficiently activates silent retroviral genomes in different tissues. Proc Natl Acad Sci USA. 1985;82:1451–5. doi: 10.1073/pnas.82.5.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jähner D, Stuhlmann H, Stewart CL, Harbers K, Löhler J, Simon I, et al. De novo methylation and expression of retroviral genomes during mouse embryogenesis. Nature. 1982;298:623–8. doi: 10.1038/298623a0. [DOI] [PubMed] [Google Scholar]
- 30.Stewart CL, Stuhlmann H, Jähner D, Jaenisch R. De novo methylation, expression, and infectivity of retroviral genomes introduced into embryonal carcinoma cells. Proc Natl Acad Sci USA. 1982;79:4098–102. doi: 10.1073/pnas.79.13.4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003;300:455. doi: 10.1126/science.1083557. [DOI] [PubMed] [Google Scholar]
- 32.Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, et al. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300:489–92. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- 33.Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, et al. Mouse Genome Sequencing Consortium Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–62. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 34.Chazin WJ. (2007) Chazin Lab Protocols, http://www.ihcworld.com/_protocols/lab_protocols/chazin-lab-protocols.htm (accessed 1/20/2009).
- 35.Wolff GL, Kodell RL, Moore SR, Cooney CA. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J. 1998;12:949–57. [PubMed] [Google Scholar]
- 36.Stover PJ. One-carbon metabolism-genome interactions in folate-associated pathologies. J Nutr. 2009;139:2402–5. doi: 10.3945/jn.109.113670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zeisel SH. Is maternal diet supplementation beneficial? Optimal development of infant depends on mother’s diet. Am J Clin Nutr. 2009;89:685S–7S. doi: 10.3945/ajcn.2008.26811F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wen YD, Perissi V, Staszewski LM, Yang WM, Krones A, Glass CK, et al. The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc Natl Acad Sci USA. 2000;97:7202–7. doi: 10.1073/pnas.97.13.7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–31. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–60. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–70. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ., 3rd SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002;16:919–32. doi: 10.1101/gad.973302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang L, Xia L, Wu DY, Wang H, Chansky HA, Schubach WH, et al. Molecular cloning of ESET, a novel histone H3-specific methyltransferase that interacts with ERG transcription factor. Oncogene. 2002;21:148–52. doi: 10.1038/sj.onc.1204998. [DOI] [PubMed] [Google Scholar]
- 44.Esaki S, Malkaram SA, Zempleni J. Effects of single-nucleotide polymorphisms in the human holocarboxylase synthetase gene on enzyme catalysis. Eur J Hum Genet. 2012;20:428–33. doi: 10.1038/ejhg.2011.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chew YC, Camporeale G, Kothapalli N, Sarath G. Zempleni, J. Lysine residues in N- and C-terminal regions of human histone H2A are targets for biotinylation by biotinidase. J Nutr Biochem. 2006;17:225–33. doi: 10.1016/j.jnutbio.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dahl JA, Collas P. MicroChIP--a rapid micro chromatin immunoprecipitation assay for small cell samples and biopsies. Nucleic Acids Res. 2007;36:e15. doi: 10.1093/nar/gkm1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.SAS Institute. (2008) SAS/STAT 9.2 User's Guide, SAS Institute, Cary, NC. [Google Scholar]
- 49.McDonald JH. (2009) Handbook of Biological Statistics, 2nd. ed., Sparky House Publishing, Baltimore, MD. [Google Scholar]
- 50.Kovalskaya E, Buzdin A, Gogvadze E, Vinogradova T, Sverdlov E. Functional human endogenous retroviral LTR transcription start sites are located between the R and U5 regions. Virology. 2006;346:373–8. doi: 10.1016/j.virol.2005.11.007. [DOI] [PubMed] [Google Scholar]