

Abstract
Abnormal hepatic gluconeogenesis contributes significantly to both fasting and non-fasting hyperglycemia of patients with type 2 diabetes. 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) regulates the key hepatic gluconeogenic enzymes including phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase) through the amplification of glucocorticoid receptor (GR) – mediated tissue glucocorticoid action, and is crucially dependent on hexose-6-phosphate dehydrogenase (H6PDH) – generating NADPH system. Here, we observed that compared with fasting state, H6PDH and 11β-HSD1 expression in livers were all increased under non-fasting state in both normal and diabetic rats, and the non-fasting diabetic group was the highest among the four experimental groups. Moreover, incubation of primary hepatocytes with increasing glucose caused dose-dependent increases in H6PDH, 11β-HSD1, GR, PEPCK and G6Pase expression. Also, glucose-6-phosphate (G6P) had a positive regulation on H6PDH and 11β-HSD1 in hepatocytes. In addition, primary hepatocytes treated with different doses of insulin in high glucose induced alteration of H6PDH and 11β-HSD1 while in low glucose there was no significant effect. These findings suggest that glucose instead of insulin directly regulates H6PDH and 11β-HSD1 and suppression of the two enzymes could be considered as an effective target for the treatment of type 2 diabetes.
Keywords: Hexose-6-phosphate dehydrogenase, 11β-Hydroxysteroid dehydrogenase type 1, Glucose, Type 2 diabetes
1. Introduction
It has long been demonstrated that glucocorticoids contribute to the pathophysiology of metabolic disorder syndrome, including hypertension, obesity, and type 2 diabetes mellitus. Glucocorticoids work antagonistically to the action performed by insulin characterized by increased hepatic gluconeogenesis and decreased ability of insulin to inhibit glucose production. Increased glucocorticoid production induces glucose intolerance and insulin resistance in genetically obese db/db and ob/ob mice and Zucker (fa/fa) rats (Freedman et al., 1986; Schwartz et al., 1992). Pharmacological blockade of glucocorticoid receptor reduces glucocorticoid-related hyperglycemia and insulin resistance in these animal models. However, glucocorticoid actions were regulated by 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1).
11β-HSD1 is a NADPH-dependent enzyme that resides within the ER lumen and expresses highly in liver, adipose tissue, and skeletal muscle. This isozyme is a bidirectional enzyme which acts predominantly as a reductase in vivo, catalyzing the interconversion of the active hormone cortisol (human) or corticosterone (rat) and inert cortisone (human) or 11-dehydrocorticosterone (rat). The physiological role of the enzyme is supposed to regulate local glucocorticoid levels in the target tissues, and then influence the key hepatic gluconeogenic enzymes including phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase) (Banhegyi et al., 2004). Activation of 11β-HSD1 results in the production of excess tissue glucocorticoids and induction of local glucocorticoid-mediated alterations of insulin action, adiposity, and glucose homeostasis, all of which are associated with visceral obesity and type 2 diabetes. In contrast, reduction of 11β-HSD1 expression prevents regeneration of active glucocorticoids, attenuates hepatic and adipose tissue glucocorticoid action (Nammi et al., 2007).
The role of 11β-HSD1 in the development of obesity and type 2 diabetes is due to its reductase activity, which requires NADPH as the cofactor. NADPH is regenerated by hexose-6-phosphate dehydrogenase (H6PDH) (Atanasov et al., 2004; Banhegyi et al., 2004), a microsomal enzyme located in the lumen of the endoplasmic reticulum (ER) and principally expressed in hepatocytes and adipocytes (Hewitt et al., 2005; McCormick et al., 2006; Liu et al., 2008). H6PDH catalyzes the initial step (conversion of glucose-6-phosphate to 6-phosphogluconolactone) of the pentose phosphate pathway within the lumen of the endoplasmic reticulum (Clarke and Mason, 2003), but this enzyme is distinguished biochemically from glucose-6-phosphate dehydrogenase (G6PDH), which catalyzes the same reactions in the cytosol by having much broader substrate specificity (White et al., 2007). Furthermore, hepatic 11β-HSD1 reductase activity was impaired in H6PDH knockout mice through inactivation of local NADPH regeneration and these mutant mice exhibited fasting hypoglycemia (Lavery et al., 2006). Subsequently, a series of in vitro studies have demonstrated close cooperativity between 11β-HSD1 and H6PDH in preparations of rat liver microsomes with manipulation of H6PDH expression directly altering the set point of 11β-HSD1 activity (Atanasov et al., 2004; Banhegyi et al., 2004).
Although some investigations about the effect of insulin or glucose on 11β-HSD1 have been reported (Voice et al., 1996; Whorwood et al., 2001), there is no report about the effect of insulin or glucose on H6PDH. Moreover, it was not known whether the effect of insulin or glucose on H6PDH and 11β-HSD1 was direct or indirect. In the present study, we first investigated the difference between fasting and non-fasting states by examining the expression and activity of H6PDH and 11β-HSD1 in livers of normal and diabetic rats. We also examined the effects of different concentrations of glucose on H6PDH and 11β-HSD1 in primary cultures of hepatocytes from normal rats. Finally, we tested whether insulin or glucose had direct effect on H6PDH and 11β-HSD1 in primary hepatocytes.
2. Materials and methods
2.1. Materials
Dulbecco’s modified Eagle’s medium (DMEM) and other culture reagents were obtained from Gibco Life Technologies (Gibco, Grand Island, NY, USA). Collagenase was purchased from Sigma (Sigma–Aldrich, St. Louis, MO, USA). Glucose oxidase kit was purchased from Beijing BHKT Clinical Reagent Co., Ltd (Beijing, China). The insulin radioimmunoassay kit was purchased from Shanghai Institute of Biological Products (Shanghai, China). Bio-Rad protein assay kit was purchased from Bio-Rad (Hercules, CA). Corticosterone enzyme-linked immunoassay (EIA) strip plate kit was obtained from R&D Systems (Minneapolis, MN, USA). Polyclonal antibodies of H6PDH, 11β-HSD1, GR, PEPCK, G6Pase and β-actin were all purchased from Santa Cruz Biotechnology (CA, USA). ECL Western Blotting Substrate was purchased from Pierce (Thermo Fisher Scientific Inc., Rockford, USA). Chemical agents for western blot and RT-PCR were obtained from Sigma. All other chemical reagents were from a commercial source.
2.2. Animal experiments
The diabetic rat model was developed using a high-fat diet plus multiple low doses of streptozotocin which was similar to that employed in previous studies from our laboratory (Zhang et al., 2008). Forty male Wistar rats were randomly divided into two groups: control group (CON), and high fat diet group plus streptozotocin injection group (DM). The control group was fed regular chow, and the DM group was given a high fat diet for 4 weeks. The high-fat diet consisted of 22% fat, 48% carbohydrate, and 20% protein with total caloric value of 44.3 kJ/kg (The Artificial Diet Center of the Experimental Animal Holding Facility) and control rats were given regular chow consisting of 5% fat, 53% carbohydrate, and 23% protein with total caloric value of 25 kJ/kg. Following 4 weeks of dietary intervention, diabetic group was injected intraperitoneally (i.p.) with low doses of streptozotocin (30 mg/kg, dissolved in 0.1 M sodium citrate buffer, pH 4.4). After 1 week, fasting blood glucose was measured in this group by glucose oxidase peroxidase. Rats with fasting blood glucose of <7.8 mmol/L were injected with streptozotocin again (30 mg/kg). Control rats were given vehicle citrate buffer (pH 4.4) in a matched volume (0.25 ml/kg) via intraperitoneal injection. The fasting blood glucose was measured every week. Four weeks after streptozotocin second injection, the rats with a two-time fasting blood glucose of ≥7.8 mmol/L or with non-fasting blood glucose of ≥11.1 mmol/L were considered to be diabetic. The rats were allowed to continue to feed on their respective diets until the end of the study. 8 weeks after second streptozotocin injection, the rats were sacrificed and the liver was immediately stored at −80 °C for later analysis.
2.3. Measurement of plasma glucose and insulin level
In overnight fasting or non-fasting condition, both control rats and diabetic rats were anesthetized with 20% urethane (100 mg/kg). Blood samples were obtained from abdominal aorta, allowed to clot for 30 min at 4 °C, centrifuged (3500 × g, 10 min, 4 °C), and the supernatant was used for measurement of glucose and insulin. Blood glucose was estimated by a commercially available glucose kit based on the glucose oxidase method. Insulin was measured by radioimmunoassay method.
2.4. Intraperitoneal glucose tolerance test (IPGTT)
After an overnight fast (12–16 h), the rats were intraperitoneally injected with 40% glucose (2 g/kg body weight). Blood samples were collected from the tail vein at 0, 30, 60, and 120 min for measurement of glucose.
2.5. Insulin tolerance test (ITT)
Insulin (0.75 IU/kg) was administered by intraperitoneal injection and blood samples were collected at 0, 30, 60, and 120 min for the measurement of plasma glucose.
2.6. Primary cultures of hepatocytes
Hepatocytes were isolated from normal male Wistar rats by a two-step collagenase perfusion method (0.5 mg/ml in Hanks’ balanced salt solution) as described previously (Liu et al., 2005). Primary hepatocytes were plated at 1 × 106 cells/dish in 3 ml DMEM with 10% fetal bovine serum and incubated at 37 °C for 4 h. Cells were then washed with PBS, and the medium was changed to DMEM without fetal bovine serum incubated in glucose-free media with indicated concentrations of glucose (5, 10, 20, and 30 mM) for 72 h. Also, hepatocytes were treated with varied concentrations of G6P (1, 2.5, and 5 mM) for 72 h. Moreover, hepatocytes were treated with insulin (10−9–10−7 M) in low (containing 5.5 mM glucose) or high (containing 25 mM glucose) glucose DMEM for 72 h, respectively.
2.7. Western blot analysis
Protein samples were prepared from liver tissue (50 mg) by homogenization with 1 ml ice-cold buffer containing: 10 mM Tris–HCl, 0.25 M sucrose, 10 mM NaCl, 1 mM EDTA, 1% SDS, and protease inhibitor cocktail (Roche), pH 7.5. Protein concentrations were measured using Bradford assay (Bio-Rad protein assay kit). The protein samples (80 μg) with 4× SDS-PAGE loading buffer (250 mM Tris–HCl, pH 6.8, 8%, w/v, SDS, 40% glycerol, 200 mM β-mercaptoethanol, and 0.4%, w/v, bromophenol blue) were denatured by boiling 5 min and separated by 12% SDS polyacrylamide gel. The proteins separated by SDS-PAGE were then electroblotted at 4 °C to polyvinylidene difluoride (PVDF) membranes (Bio-Rad) by employing a transfer buffer containing 25 mM Tris–HCl, 192 mM glycine, and 20% methanol (v/v). Membranes were blocked in 5% (w/v) non-fat milk for 2 h at room temperature and then incubated with rabbit polyclonal antibodies (H6PDH, 1:1000; 11β-HSD1, 1:1000; GR, 1:1000; PEPCK, 1:1000; G6Pase, 1:1000) with gentle agitation overnight at 4 °C. The membranes were washed 3 times for 10 min each with 15 ml of TBST [10 mM Tris–HCl, 150 mM NaCl and 0.1% (v/v) Tween-20] and then incubated with second antibody (1:2000 goat anti-rabbit IgG horseradish peroxidase conjugate) at room temperature for 2 h. The membranes were again washed with TBST as described above. Protein was then visualized with enhanced chemiluminescence ECL and X-ray film. To correct for differences in protein loading, the membranes were washed and reprobed with 1:5000 dilution goat polyclonal antibody to β-actin. An Imaging Densitometer was used to scan the protein bands and the densities were quantified using the Image Analysis Software.
2.8. RNA extraction and semiquantitative RT-PCR
Total RNA was extracted from individual rat liver sample using TRIzol reagent (Invitrogen). Approximately 50 mg of tissue was homogenized in 1 ml of TRIzol reagent, and total RNA was extracted according to the manufacturer’s instructions. The first-strand cDNAs were synthesized from 5 μg total RNA, using SuperScript reverse transcriptase and oligo deoxythymidine primers. The reverse transcription products were amplified by PCR, using Taq DNA polymerase and specific primers for rat H6PDH (forward: 5′ -ATCATTACCTGGGCAAGC-3′ , reverse: 5′-GCCATAC TCCTCGTAGAAACT-3′ ), 11β-HSD1 (forward: 5′-GAAGAAGCATGGAGGTCAAC-3′, reverse: 5′-GCAATCAGAGGTTGGGTCAT-3′), and GAPDH (forward: 5′-CCATGGAGAAGGCTGGG-3′, reverse: 5′-CAAAGTTGTCATGGATGACC-3′). The amplification conditions were optimized in preliminary studies to result in amplification within the linear range. PCR products were visualized on 1.5% agarose gels by ethidium bromide staining and gels were photographed under U.V. light. Relative gene expression was quantified and densitometrically analyzed using the Image Analysis Software. GAPDH transcript abundance was considered as an internal control.
2.9. Measurement of enzyme activity in vivo animals
H6PDH enzyme activity was carried out by spectrophotometric measurement of NADPH production in the presence of 1–2 mM glucosamine-6-phosphate and NADP using absorbance at 340 nm by a spectrophotometer (Synergy 2 SL Luminescence Microplate Reader, BioTek) (Clarke and Mason, 2003). The microsomal pellet was obtained by centrifugation of the 10,000 × g supernatant for 1 h at 100,000 × g. Homogenized tissue protein (200 mg) or protein from microsomes (20 mg) was incubated in 100 mM glycine buffer solution [containing 1% BSA (pH 10.0) and 0.5–5 mM glucosamine-6-phosphate (Ropson and Powers, 1988; Nammi et al., 2007), 1–5 mM NADP+] at room temperature for 0–5 min as described. Specific activities were calculated and expressed as nanomoles of NADPH production per minute per milligram of protein.
The reductase activity assay of 11β-HSD1 was performed by immunoassay of the corticosterone produced from 11-dehydrocorticosterone using a sensitive EIA (Nammi et al., 2007; Liu et al., 2005). Briefly, the liver was homogenized in Krebs–Ringer buffer solution, and the protein concentration of each supernatant was measured by Bradford assay. The reaction was started by adding 20 mg protein from microsomes in 50 mM sodium phosphate buffer (pH 7.4) [containing 1 mM EDTA, 1 M NaCl, 40% glycerol (w/v) and 0.4% Triton X-100 (w/v)], and then incubated for 1 h at 37 °C with 2 μM 11-dehydrocorticosterone and 200 μM NADPH. The produce of corticosterone was determined using EIA kit following manufacturer’s instructions. Specific activities were expressed as picomoles of corticosterone formed per minute per milligram of protein.
2.10. Measurement of enzyme activity in vitro hepatocytes
For the H6PDH activity in vitro, 20 mg protein extracts from primary hepatocytes were incubated with 2 mM G6P as substrates in 100 ml total volume of glycine buffer (pH 10.0) with 0.5 mM NADP+ as a cofactor (Stegeman and Klotz, 1979; Nammi et al., 2007). The changes in absorbance at 340 nm were measured over 25 min at 5 min intervals, and the relative H6PDH activity was expressed as described above.
11β-HSD1 reductase activity was measured in primary culture hepatocytes by measuring the corticosterone produced from 11-dehydrocorticosterone using a sensitive EIA. The primary hepatocytes were preincubated with 20 nM 11-dehydrocorticosterone for 1 h at 37 °C. The supernatant of cells was centrifugated for 10 min at 1000 × g. The produce of corticosterone was determined using EIA corticosterone kit following manufacturer’s instructions, and the relative 11β-HSD1 activity was expressed as described above.
2.11. Glucose consumption and MTT method
The hepatocytes were plated into 24-well tissue culture plates with some wells left blank in low glucose DMEM (containing 5.5 mM glucose) containing 10% fetal bovine serum. After the cells reached confluence, the medium was replaced by DMEM supplemented with 2% FBS and glucose at various concentrations (5.5, 11, and 25 mM). After 72 h the medium was removed and its glucose concentrations were determined by the glucose oxidase method. The amount of glucose consumption was calculated by the glucose concentrations of blank wells subtracting the remaining glucose in cell plated wells (Yin et al., 2002).
MTT was dissolved at a concentration of 5 mg/ml in sterile phosphate-buffered saline (PBS). One volume 5 mg/ml stock solution of MTT was mixed with 9 vol DMEM. It was added to the 24-well culture plates when the test of glucose consumption was finished. After a 4 h incubation at 37 °C, the MTT medium was replaced with dimethyl sulfoxide (DMSO). After shaking, the optical densities (OD) at 570 nm were measured using a Microplate Reader (Sanyo, Japan).
2.12. Statistical analyses
All data were expressed as mean ± S.E.M. “n” denotes the sample size in each group. Statistical analyses were performed using one-way analysis of variance (ANOVA) with post hoc test for multiple comparisons. SPSS software (version 13.0 for Windows) was used for statistical analysis. P < 0.05 was considered to be statistically significant.
3. Results
3.1. Characterization of type 2 diabetic rats
Mean values of the body weight and biochemical parameters from control and diabetic rats are summarized in Table 1. Body weight was not significantly different among the four groups. DM group had higher fasting blood glucose level compared with CON group, glucose of non-fasting state was higher than fasting state, and the non-fasting DM group was the highest among the four experimental groups. Serum insulin was reduced significantly in DM group compared with CON group, while serum insulin was higher in non-fasting state compared with fasting state.
Table 1.
Body weight and biochemical parameters in various groups
| Condition | Group | Body weight (g) | Blood glucose (mmol/L) | Insulin (μU/ml) |
|---|---|---|---|---|
| Fasted | CON-FA | 363.1 ± 12.6 | 4.66 ± 1.13 | 13.50 ± 1.39 |
| DM-FA | 376.4 ± 14.3 | 22.57 ± 2.85* | 6.96 ± 0.75* | |
| Fed | CON-FE | 368.8 ± 11.5 | 10.94 ± 1.72* | 17.10 ± 2.33* |
| DM-FE | 372.5 ± 13.1 | 28.65 ± 3.51◆,▴ | 8.38 ± 1.01◆,▴ |
CON-FA: control rats fasting for 12 h before killed; CON-FE: control rats fed with regular chow before killed; DM-FA: diabetic rats fasting for 12 h before killed; DM-FE: diabetic rats fed with high-fat diets before killed. Values are means±SE, n = 4–5 rats/group.
P > 0.01 vs. CON-FA.
P > 0.01 vs. CON-FE.
P > 0.01 vs. DM-FA.
IPGTT showed that glucose levels at each time point were significantly elevated in DM group compared with CON group during 120 min after glucose injection (Fig. 1A). The areas under the glucose curves (mmol/L min) were significantly greater in DM group compared with CON group [3056 ± 60 mmol/L min vs. 1399 ± 86 mmol/L min, P < 0.01]. This was confirmed by the ITT, which showed that glucose concentrations declined rapidly after insulin administration, and the decrease became significant by 30 min in CON group in comparison to that observed in DM group (Fig. 1B). All the data indicated that high-fat diet associated with 30 mg/kg STZ twice injection developed a diabetic model which was an analogue to type 2 diabetes mellitus with insulin resistance and hyperglycemia.
Fig. 1.
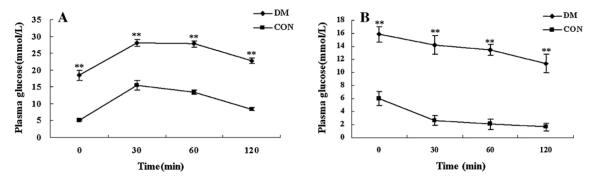
(A) Plasma glucose during intraperitoneal glucose tolerance test (IPGTT) in CON group and DM group after 8 weeks of STZ injection. (B) Plasma glucose during insulin tolerance test (ITT) in CON group and DM group after 8 weeks of injection. Values are means ± SE, n = 20 rats/group per time point. **P < 0.01, DM vs. CON.
3.2. Expression of H6PDH and 11β-HSD1 in liver of type 2 diabetic rats
To investigate the alteration of H6PDH and 11β-HSD1 under fasting and non-fasting states in normal and type 2 diabetic rats, their mRNA levels, protein expressions, and enzyme activity were all detected. It can be seen that mRNA and protein levels of H6PDH (Fig. 2A and B) and 11β-HSD1 (Fig. 2D and E) were significantly elevated in DM group, compared with CON group. Their levels were also elevated in non-fasting state compared with fasting state both in DM and CON groups, and strongest in non-fasting state of diabetic rats in four groups. Parallel to the increase in mRNA and protein expression, hepatic H6PDH (Fig. 2C) and 11β-HSD1 (Fig. 2F) activity was also highest in DM group under non-fasting state among the four groups.
Fig. 2.

Expression of H6PDH (A) and 11β-HSD1 (D) mRNA in the liver of control and diabetic rats under fasting and non-fasting states. mRNA levels are expressed relative to GAPDH. Expression of H6PDH (B) and 11β-HSD1 (E) protein in the liver of control and diabetic rats under fasting and non-fasting states. Protein levels are expressed relative to β-actin. Enzyme activity of H6PDH (C) and 11β-HSD1 (F) of control and diabetic rats under fasting and non-fasting states. Values are means ± SE, n = 4–5 rats/group. *P < 0.05 vs. CON-FA; **P < 0.01 vs. CON-FA; #P < 0.05 vs. DM-FA; and ##P < 0.01 vs. DM-FA.
3.3. Correlation analysis
We used SPSS software to analyze the correlation between H6PDH and blood glucose, H6PDH and circulating insulin, 11β-HSD1 and blood glucose, 11β-HSD1 and circulating insulin, respectively. The gray values of H6PDH and 11β-HSD1 protein by western blot were used in this analysis corrected by β-actin. Fig. 3A shows the positive correlation between blood glucose and H6PDH (R = 0.769, P = 0.003). Fig. 3B shows the positive correlation also between blood glucose and 11β-HSD1 (R = 0.769, P = 0.003). However, Fig. 3C shows that there was no correlation between circulating insulin and H6PDH (R = 0.379, P = 0.225). Fig. 3D shows that there was also no correlation between circulating insulin and 11β-HSD1 (R = 0.385, P = 0.217). These results showed both H6PDH and 11β-HSD1 are correlated with blood glucose but no correlation with circulating insulin.
Fig. 3.

Correlation analysis by SPSS software. (A) A scatter plot shows the positive correlation between blood glucose and H6PDH (R = 0.769, P = 0.003). (B) A scatter plot shows the positive correlation between blood glucose and 11β-HSD1 (R = 0.769, P = 0.003). (C) A scatter plot shows no correlation between circulating insulin and H6PDH (R = 0.379, P = 0.225). (D) A scatter plot shows no correlation between circulating insulin and 11β-HSD1 (R = 0.385, P = 0.217).
3.4. Effects of glucose on target gene expression in primary cultures of hepatocytes
The results of western blot and enzyme activity analysis for H6PDH and 11β-HSD1 in primary hepatocytes treated with different concentrations of glucose (5–30 mM) are shown in Fig. 4. Protein expression and enzyme activity of H6PDH (Fig. 4A and C) and 11β-HSD1 (Fig. 4B and D) in primary hepatocyte cultures were significantly increased in dose-dependent glucose. Similarly, the protein levels of GR, PEPCK, and G6Pase in primary cultures of hepatocytes after treatment with glucose were also increased in dose-dependent glucose (Fig. 5A–C).
Fig. 4.

Effects of glucose on H6PDH (A) and 11β-HSD1 (B) protein expression in primary cultures of hepatocytes from normal rats. Enzyme activity of H6PDH (C) and 11β-HSD1 (D) in primary hepatocytes incubated in different concentrations of glucose. Hepatocytes were incubated in glucose-free media with indicated concentrations of glucose (5, 10, 20, and 30 mM) for 72 h. Values are means ± SE from three separate culture preparations. *P < 0.05 vs. glucose-free group and **P < 0.01 vs. glucose-free group.
Fig. 5.
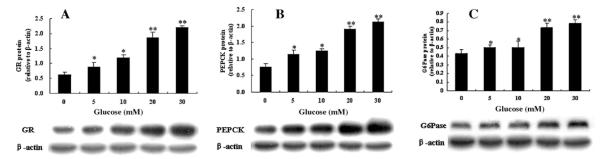
Effects of glucose on GR (A), PEPCK (B) and G6Pase (C) protein expression in primary cultures of hepatocytes from normal rats. Hepatocytes were incubated in glucose-free media with indicated concentrations of glucose (5, 10, 20, and 30 mM) for 72 h. Values are means ± SE from three separate culture preparations. *P < 0.05 vs. glucose-free group and **P < 0.01 vs. glucose-free group.
3.5. Effects of G6P treatment on H6PDH and 11β-HSD1 expression in primary cultures of hepatocytes
Some evidence suggests that the native substrates for H6PDH are G6P and NADP+ under physiological conditions in the lumen of the ER (Mandula et al., 1970; Kulkarni and Hodgson, 1982; Piccirella et al., 2006; Dzyakanchuk et al., 2009). As G6P was generated mainly from glucose phosphorylation in glucose metabolism, we determined the influence of extracellular G6P availability on H6PDH and 11β-HSD1 by treating the hepatocytes with varied concentrations of G6P (1, 2.5, and 5 mM). After 72 h, 2.5 mM and 5 mM G6P had a significant effect on H6PDH (Fig. 6A) and 11β-HSD1 (Fig. 6B) protein expression compared with control group in primary hepatocytes.
Fig. 6.
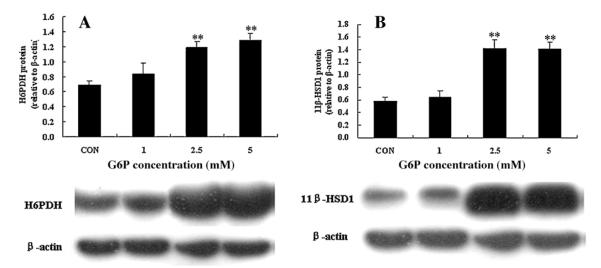
Effects of G6P on H6PDH and 11β-HSD1 in primary cultures of hepatocytes from normal rats. Hepatocytes were incubated in media with indicated concentrations of G6P (1, 2.5, and 5 mM) for 72 h. Values are means ± SE from three separate culture preparations. **P < 0.01 vs. control group.
3.6. Effects of insulin on H6PDH and 11β-HSD1 in hepatocytes cultured in low or high glucose media
In order to determine whether insulin directly affects H6PDH and 11β-HSD1 expression or through indirect action on glucose, first the glucose consumption of hepatocytes treated with varied concentrations of insulin (10−9–10−7 M) was measured. The results showed that insulin (10−9–10−7 M) did not affect glucose consumption of hepatocytes treated with 5.5 mM glucose. However, insulin (10−9–10−7 M) significantly increased glucose consumption in hepatocytes treated with 25 mM glucose (Fig. 7). Then effects of insulin on H6PDH and 11β-HSD1 in hepatocytes cultured in 5.5 or 25 mM glucose media were measured. As shown in Fig. 8, western blot analysis indicated that insulin (10−9–10−7 M) had no effect on H6PDH and 11β-HSD1 protein expression in incubation of cultured hepatocytes with 5.5 mM glucose, however, insulin (10−9–10−7 M) induced a marked reduction in H6PDH and 11β-HSD1 protein expression in incubation of cultured hepatocytes with 25 mM glucose. These results indicated that insulin may have an indirect effect on H6PDH and 11β-HSD1 by affecting the concentration of glucose in vitro.
Fig. 7.
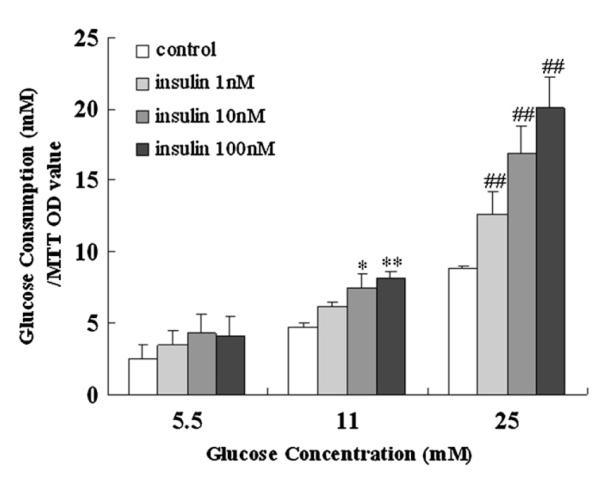
Effects of insulin on glucose consumption in primary cultures of hepatocytes from normal rats. The tests were performed at 5.5, 11, and 25 mM glucose in the absence or presence of 1, 10, and 100 nM insulin. Values are means ± SE from three separate culture preparations. *P < 0.05 vs. control group in the presence of 11 mM glucose; **P < 0.01 vs. control group in the presence of 11 mM glucose; and ##P < 0.01 vs. control group in the presence of 25 mM glucose.
Fig. 8.

Effects of insulin on H6PDH (A) and 11β-HSD1 (B) in primary cultures of hepatocytes from normal rats in low glucose (5.5 mM) DMEM. Effects of insulin on H6PDH (C) and 11β-HSD1 (D) in primary cultures of hepatocytes from normal rats in high glucose (25 mM) DMEM. Hepatocytes were incubated in medium with indicated concentrations of insulin (1, 10, and 100 nM) for 72 h. Values are means ± SE from three separate culture preparations. *P < 0.05 vs. control group and **P < 0.01 vs. control group.
4. Discussion
Accumulating evidence suggests that glucocorticoids play fundamental roles in the development of type 2 diabetes and obesity, acting mainly through glucocorticoid receptor, which confers tissue-specific responsiveness to circulating corticosteroids and thus mediates glucocorticoid-related obesity and insulin resistance (Brindley, 1995; Andrews and Walker, 1999). Indeed, glucocorticoids stimulate hepatic gluconeogenesis and reduce the ability of insulin to inhibit glucose production, all of which are thought to be the major source of increased glucose production in type 2 diabetes. However, the actions of glucocorticoids on target tissues, such as liver and adipose tissue are dependent on their pre-receptor metabolism, which is regulated by 11β-HSD1 (Kotelevtsev et al., 1997; Liu et al., 2005). This enzyme regenerates active corticosterone from inactive 11-keto forms and plays an important role in type 2 diabetes. Several studies have produced evidence in support of the hypothesis that the regulation of 11β-HSD1 may be mediated through the actions of a number of hormones and growth factors. However, much of this research is contradictory and fails to characterize the species and tissue specific mechanisms that underlie the regulation of 11β-HSD1. Nevertheless, this data have served to highlight key elements underlying the hormonal regulation of 11β-HSD1, including glucocorticoids (Bujalska et al., 1997, 1999), insulin (Hammami and Siiteri, 1991; Voice et al., 1996), growth hormone (Voice et al., 1996) and support the hypothesis that deregulation of enzyme activity may underlie the etiology of a spectrum of diseases, including essential insulin resistance and glucose intolerance (Walker et al., 1995; Andrews and Walker, 1999), and central obesity (Bujalska et al., 1997).
In this study, we found that diabetic rats had higher fasting blood glucose compared with control, blood glucose of non-fasting state was higher than fasting state, and the non-fasting diabetic rats were highest among the four experimental groups. Moreover, compared with fasting state, mRNA levels, protein expression and enzyme activity of both H6PDH and 11β-HSD1 were all increased in livers of not only normal but also diabetic rats under non-fasting state, and non-fasting diabetic rats were also the highest. Therefore, we presumed glucose might had the direct effect on both 11β-HSD1 and H6PDH, and the latter catalyzes G6P and NADP+ to regenerate NADPH, thereby playing an important role in determining 11β-HSD1 reductase activity in liver tissues. The correlation analysis showed both H6PDH and 11β-HSD1 had a correlation with blood glucose, while neither had a correlation with circulating insulin. Consistent with these observations, our cell work demonstrated that glucose has dose-dependent increases in the expression of H6PDH, 11β-HSD1 and GR. The results indicated that glucose is an important metabolic signal that increases intrahepatic corticosterone production, thereby increasing circulating glucocorticoid levels, and that hyperglycemia in diabetic rats may be involved in the activation of H6PDH, 11β-HSD1 and therefore induced GR – mediated local glucocorticoid action in liver. Our results also showed the availability of glucose plays a role in the regulation of PEPCK and G6Pase. After treated with increasing glucose for 72 h, the expression of PEPCK and G6Pase in primary hepatocytes were obviously dose-dependently increased. That is to say, there was a significant increase in the rate of gluconeogenesis. This elevation is similar with hyperglucagonemia of the diabetic condition, leading to increased expression of gluconeogenic enzymes (Yu et al., 1994).
A known substrate for H6PDH is G6P, transported from the cytoplasm into the ER-lumen via the membrane-bound G6P transporter (G6PT) (Chou et al., 2002). Thus, both glucose catabolism and anabolism seem to be tightly linked to intracellular glucocorticoid activation through the availability of G6P supplied by glucose. In our study, treatment of primary hepatocytes with increasing doses of G6P led to increase in H6PDH and 11β-HSD1 expression. It further confirmed our hypothesis that glucose directly altered H6PDH and 11β-HSD1. Also, our results are in agreement with previous studies reporting that the importance of H6PDH utilized G6P and NADP+ to produce NADPH, thus, H6PDH is likely to be the crucial enzyme supplying NADPH for 11β-HSD1 induced amplification of tissue GR ligand cortisol/corticosterone production linked to the development of type 2 diabetes and obesity (Kimura et al., 1979; Stegeman and Klotz, 1979).
As glucose concentrations were increased from 5.5 mM to 25 mM, glucose consumption was elevated under the glucose-lowering effect of insulin. Therefore, incubation of cultured hepatocytes in the presence of high glucose medium (25 mM) with increasing concentrations of insulin (10−9–10−7 M) induced a marked reduction in H6PDH and 11β-HSD1 protein expression, while incubating with low glucose media (5.5 mM) had no significant effect. This is in marked contrast with contemporary studies that insulin increases 11β-HSD1 in human skeletal muscle cells (Whorwood et al., 2001), decreases 11β-HSD1 in rat hepatocytes (Voice et al., 1996), and has no effect on 11β-HSD1 in human adipose stromal cells (Bujalska et al., 1999). This may be interpreted that insulin exerts species and tissue specific differential regulation of 11β-HSD1, while our results showed that insulin positively changed the concentration of glucose and then regulated the expression of 11β-HSD1.
In summary, our work demonstrated that under non-fasting state the expression and activity of H6PDH and 11β-HSD1 were increased in livers compared with fasting state, and the non-fasting diabetic rats was the highest. We also found the activation of H6PDH and 11β-HSD1 expression and activity in the primary hepatocytes were mediated through levels of glucose, not related to insulin levels. Moreover, these studies raise the possibility that insulin positively changed the concentration of glucose and then regulated the expression and activity of H6PDH and 11β-HSD1. These findings suggest that suppression of H6PDH and 11β-HSD1 could be considered as an effective target for the treatment of type 2 diabetes.
Acknowledgments
The research in this study was supported by a grant from the National Natural Science Foundation of China (30772604). Y.J. Liu is supported by National Institute of Diabetes and Digestive and Kidney Disease SC1DK087655.
References
- Andrews RC, Walker BR. Glucocorticoids and insulin resistance: old hormones, new targets. Clin. Sci. (Lond.) 1999;96:513–523. doi: 10.1042/cs0960513. [DOI] [PubMed] [Google Scholar]
- Atanasov AG, Nashev LG, Schweizer RA, Frick C, Odermatt A. Hexose-6-phosphate dehydrogenase determines the reaction direction of 11beta-hydroxysteroid dehydrogenase type 1 as an oxoreductase. FEBS Lett. 2004;571:129–133. doi: 10.1016/j.febslet.2004.06.065. [DOI] [PubMed] [Google Scholar]
- Banhegyi G, Benedetti A, Fulceri R, Senesi S. Cooperativity between 11beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase in the lumen of the endoplasmic reticulum. J. Biol. Chem. 2004;279:27017–27021. doi: 10.1074/jbc.M404159200. [DOI] [PubMed] [Google Scholar]
- Brindley DN. Role of glucocorticoids and fatty acids in the impairment of lipid metabolism observed in the metabolic syndrome. Int. J. Obes. Relat. Metab. Disord. 1995;19(Suppl. 1):S69–S75. [PubMed] [Google Scholar]
- Bujalska IJ, Kumar S, Hewison M, Stewart PM. Differentiation of adipose stromal cells: the roles of glucocorticoids and 11beta-hydroxysteroid dehydrogenase. Endocrinology. 1999;140:3188–3196. doi: 10.1210/endo.140.7.6868. [DOI] [PubMed] [Google Scholar]
- Bujalska IJ, Kumar S, Stewart PM. Does central obesity reflect “Cushing’s disease of the omentum”? Lancet. 1997;349:1210–1213. doi: 10.1016/S0140-6736(96)11222-8. [DOI] [PubMed] [Google Scholar]
- Chou JY, Matern D, Mansfield BC, Chen YT. Type I glycogen storage diseases: disorders of the glucose-6-phosphatase complex. Curr. Mol. Med. 2002;2:121–143. doi: 10.2174/1566524024605798. [DOI] [PubMed] [Google Scholar]
- Clarke JL, Mason PJ. Murine hexose-6-phosphate dehydrogenase: a bifunctional enzyme with broad substrate specificity and 6-phosphogluconolactonase activity. Arch. Biochem. Biophys. 2003;415:229–234. doi: 10.1016/s0003-9861(03)00229-7. [DOI] [PubMed] [Google Scholar]
- Dzyakanchuk AA, Balazs Z, Nashev LG, Amrein KE, Odermatt A. 11beta-Hydroxysteroid dehydrogenase 1 reductase activity is dependent on a high ratio of NADPH/NADP+ and is stimulated by extracellular glucose. Mol. Cell. Endocrinol. 2009;301:137–141. doi: 10.1016/j.mce.2008.08.009. [DOI] [PubMed] [Google Scholar]
- Freedman MR, Horwitz BA, Stern JS. Effect of adrenalectomy and glucocorticoid replacement on development of obesity. Am. J. Physiol. 1986;250:R595–R607. doi: 10.1152/ajpregu.1986.250.4.R595. [DOI] [PubMed] [Google Scholar]
- Hammami MM, Siiteri PK. Regulation of 11beta-hydroxysteroid dehydrogenase activity in human skin fibroblasts: enzymatic modulation of glucocorticoid action. J. Clin. Endocrinol. Metab. 1991;73:326–334. doi: 10.1210/jcem-73-2-326. [DOI] [PubMed] [Google Scholar]
- Hewitt KN, Walker EA, Stewart PM. Minireview: hexose-6-phosphate dehydrogenase and redox control of 11beta-hydroxysteroid dehydrogenase type 1 activity. Endocrinology. 2005;146:2539–2543. doi: 10.1210/en.2005-0117. [DOI] [PubMed] [Google Scholar]
- Kimura K, Endou H, Sudo J, Sakai F. Glucose dehydrogenase (hexose 6-phosphate dehydrogenase) and the microsomal electron transport system. Evidence supporting their possible functional relationship. J. Biochem. 1979;85:319–326. doi: 10.1093/oxfordjournals.jbchem.a132336. [DOI] [PubMed] [Google Scholar]
- Kotelevtsev Y, Holmes MC, Burchell A, Houston PM, Schmoll D, Jamieson P, Best R, Brown R, Edwards CR, Seckl JR, Mullins JJ. 11beta-Hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc. Natl. Acad. Sci. U.S.A. 1997;94:14924–14929. doi: 10.1073/pnas.94.26.14924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni AP, Hodgson E. Mouse liver microsomal hexose-6-phosphate dehydrogenase. NADPH generation and utilization in monooxygenation reactions. Biochem. Pharmacol. 1982;31:1131–1137. doi: 10.1016/0006-2952(82)90353-7. [DOI] [PubMed] [Google Scholar]
- Lavery GG, Walker EA, Draper N, Jeyasuria P, Marcos J, Shackleton CH, Parker KL, White PC, Stewart PM. Hexose-6-phosphate dehydrogenase knock-out mice lack 11beta-hydroxysteroid dehydrogenase type 1-mediated glucocorticoid generation. J. Biol. Chem. 2006;281:6546–6551. doi: 10.1074/jbc.M512635200. [DOI] [PubMed] [Google Scholar]
- Liu Y, Nakagawa Y, Wang Y, Liu L, Du H, Wang W, Ren X, Lutfy K, Friedman TC. Reduction of hepatic glucocorticoid receptor and hexose-6-phosphate dehydrogenase expression ameliorates diet-induced obesity and insulin resistance in mice. J. Mol. Endocrinol. 2008;41:53–64. doi: 10.1677/JME-08-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Nakagawa Y, Wang Y, Sakurai R, Tripathi PV, Lutfy K, Friedman TC. Increased glucocorticoid receptor and 11beta-hydroxysteroid dehydrogenase type 1 expression in hepatocytes may contribute to the phenotype of type 2 diabetes in db/db mice. Diabetes. 2005;54:32–40. doi: 10.2337/diabetes.54.1.32. [DOI] [PubMed] [Google Scholar]
- Mandula B, Srivastava SK, Beutler E. Hexose-6-phosphate dehydrogenase: distribution in rat tissues and effect of diet, age and steroids. Arch. Biochem. Biophys. 1970;141:155–161. doi: 10.1016/0003-9861(70)90118-9. [DOI] [PubMed] [Google Scholar]
- McCormick KL, Wang X, Mick GJ. Evidence that the 11beta-hydroxysteroid dehydrogenase (11beta-HSD1) is regulated by pentose pathway flux. Studies in rat adipocytes and microsomes. J. Biol. Chem. 2006;281:341–347. doi: 10.1074/jbc.M506026200. [DOI] [PubMed] [Google Scholar]
- Nammi S, Dembele K, Nyomba BL. Increased 11beta-hydroxysteroid dehydrogenase type-1 and hexose-6-phosphate dehydrogenase in liver and adipose tissue of rat offspring exposed to alcohol in utero. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007;292:R1101–R1109. doi: 10.1152/ajpregu.00255.2006. [DOI] [PubMed] [Google Scholar]
- Piccirella S, Czegle I, Lizak B, Margittai E, Senesi S, Papp E, Csala M, Fulceri R, Csermely P, Mandl J, Benedetti A, Banhegyi G. Uncoupled redox systems in the lumen of the endoplasmic reticulum. Pyridine nucleotides stay reduced in an oxidative environment. J. Biol. Chem. 2006;281:4671–4677. doi: 10.1074/jbc.M509406200. [DOI] [PubMed] [Google Scholar]
- Ropson IJ, Powers DA. A novel dehydrogenase reaction mechanism for hexose-6-phosphate dehydrogenase isolated from the teleost Fundulus heteroclitus. J. Biol. Chem. 1988;263:11697–11703. [PubMed] [Google Scholar]
- Schwartz MW, Figlewicz DP, Baskin DG, Woods SC, Porte D., Jr. Insulin in the brain: a hormonal regulator of energy balance. Endocr. Rev. 1992;13:387–414. doi: 10.1210/edrv-13-3-387. [DOI] [PubMed] [Google Scholar]
- Stegeman JJ, Klotz AV. A possible role for microsomal hexose-6-phosphate dehydrogenase in microsomal electron transport and mixed-function oxygenase activity. Biochem. Biophys. Res. Commun. 1979;87:410–415. doi: 10.1016/0006-291x(79)91811-4. [DOI] [PubMed] [Google Scholar]
- Voice MW, Seckl JR, Edwards CR, Chapman KE. 11beta-hydroxysteroid dehydrogenase type 1 expression in 2S FAZA hepatoma cells is hormonally regulated: a model system for the study of hepatic glucocorticoid metabolism. Biochem. J. 1996;317:621–625. doi: 10.1042/bj3170621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BR, Connacher AA, Lindsay RM, Webb DJ, Edwards CR. Carbenoxolone increases hepatic insulin sensitivity in man: a novel role for 11-oxosteroid reductase in enhancing glucocorticoid receptor activation. J. Clin. Endocrinol. Metab. 1995;80:3155–3159. doi: 10.1210/jcem.80.11.7593419. [DOI] [PubMed] [Google Scholar]
- White PC, Rogoff D, McMillan DR, Lavery GG. Hexose 6-phosphate dehydrogenase (H6PD) and corticosteroid metabolism. Mol. Cell. Endocrinol. 2007;265–266:89–92. doi: 10.1016/j.mce.2006.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorwood CB, Donovan SJ, Wood PJ, Phillips DI. Regulation of glucocorticoid receptor alpha and beta isoforms and type I 11beta-hydroxysteroid dehydrogenase expression in human skeletal muscle cells: a key role in the pathogenesis of insulin resistance? J. Clin. Endocrinol. Metab. 2001;86:2296–2308. doi: 10.1210/jcem.86.5.7503. [DOI] [PubMed] [Google Scholar]
- Yin J, Hu R, Chen M, Tang J, Li F, Yang Y, Chen J. Effects of berberine on glucose metabolism in vitro. Metabolism. 2002;51:1439–1443. doi: 10.1053/meta.2002.34715. [DOI] [PubMed] [Google Scholar]
- Yu B, Pugazhenthi S, Khandelwal RL. Effects of metformin on glucose and glucagon regulated gluconeogenesis in cultured normal and diabetic hepatocytes. Biochem. Pharmacol. 1994;48:949–954. doi: 10.1016/0006-2952(94)90365-4. [DOI] [PubMed] [Google Scholar]
- Zhang M, Lv XY, Li J, Xu ZG, Chen L. The characterization of high-fat diet and multiple low-dose streptozotocin induced type 2 diabetes rat model. Exp. Diabetes Res. 2008:704045. doi: 10.1155/2008/704045. [DOI] [PMC free article] [PubMed] [Google Scholar]