

Abstract
AMP-activated protein kinase (AMPK), an important regulator of energy metabolism, is activated in response to cellular stress when intracellular levels of AMP increase. We investigated the neuroprotective effects of AMPK against scopolamine-induced memory impairment in vivo and glutamate-induced cytotoxicity in vitro. An adenovirus expressing AMPK wild type alpha subunit (WT) or a dominant negative form (DN) was injected into the hippocampus of rats using a stereotaxic apparatus. The AMPK WT-injected rats showed significant reversal of the scopolamine induced cognitive deficit as evaluated by escape latency in the Morris water maze. In addition, they showed enhanced acetylcholinesterase (AChE)-reactive neurons in the hippocampus, implying increased cholinergic activity in response to AMPK. We also studied the cellular mechanism by which AMPK protects against glutamate-induced cell death in primary cultured rat hippocampal neurons. We further demonstrated that AMPK WT-infected cells increased cell viability and reduced Annexin V positive hippocampal neurons. Western blot analysis indicated that AMPK WT-infected cells reduced the expression of Bax and had no effects on Bcl-2, which resulted in a decreased Bax/Bcl-2 ratio. These data suggest that AMPK is a useful cognitive impairment treatment target, and that its beneficial effects are mediated via the protective capacity of hippocampal neurons.
Keywords: Adenovirus, AMPK, Apoptosis, Learning and memory, Scopolamine
INTRODUCTION
Energy homeostasis is essential for the survival of any organism. Many reports have shown that the mechanisms of energy metabolism may play an important role in mediation of aspects of higher order cognitive function [1]. Cellular energy status is monitored and controlled by the adenosine monophosphate-activated protein kinase (AMPK) system. AMPK functions as an energy metabolic sensor when cells are subjected to energy-depleting stresses. Elevations in intracellular AMP concentration and corresponding decreases in ATP levels serve as critical factors in the stimulation of AMPK activity [2]. AMPK is allosterically activated by AMP, which accumulates following ATP hydrolysis. AMPK is a heterotrimeric complex consisting of a catalytic alpha subunit and regulatory beta and gamma subunits, each of which is encoded by two or three distinct genes (alpha 1, 2, beta 1, 2 and gamma 1, 2, 3) [3,4]. All combinations of isoforms appear to be able to form complexes, which when combined with splice variants and the use of alternative promoters leads to a diverse array of different AMPK complexes [4]. Aspartate 157 within the alpha subunit lies in the conserved DFG motif, which has been shown to be essential for MgATP binding in all protein kinases [5,6]. Mutation of this residue to alanine in either alpha1 or 2 yields an inactive kinase, but has no effect on binding of the beta and gamma subunits within the complex [5,7]. Since formation of the heterotrimeric complex is essential for AMPK activity, we reasoned that overexpression of the inactive alpha1 subunit would act as a dominant negative inhibitor by competing with the native alpha subunit for binding with beta and gamma [5].
AMPK was recently reported to be regulated by a calcium signal and to protect neuronal cells from glucose deprivation and exposure to glutamate [8,9]. Zhao et al. showed that AMPK acts as a molecular target for the treatment of neurodegenerative disorders including glutamate-induced epileptic seizure and ischemic injury, particularly through regulation of BDNF expression [10]. AMPK has also been shown to play a protective role in cerebral metabolic stress [11]. Through appropriate modulation of gene transcription, AMPK enhances the capacity of the cell to generate ATP [12,13]. Accordingly, the regulation of metabolism by AMPK ensures cell survival during energy depletion [4,13]. Moreover, AMPK inhibits cell growth and protects against suicidal cell death or apoptosis [4,12].
In the present study, intraperitoneal (i.p.) administration of scopolamine, a muscarinic cholinergic receptor antagonist, was exploited as a parmarcological model for AD in rats. Scopolamine interfere with central cholinergic functions and memory circuits, leading to decreased Ach release and subsequent memory, attention, and executive function-related deficits that are seen in aging and dementia [14]. Due to this, scopolamine-induced cognitive dysfunction is extensively used to probe potential therapeutic agents attenuating cognitive deficits [15]. In the Morris water maze, systemic administration of scopolamine has been reported to be more effective in disrupting learning and acquisition, and short-term retention of spatial memory [16]. Besides, scopolamine-induced amnesia is relevant to decreased glucose oxidation, increase in brain oxidative [17, 18] and decrease of ATP levels in the brain [19,20].
Neurotoxicity in the mammalian CNS has been relevant to glutamate. Glutamate-induced excitotoxicity has been linked to neuronal cell death, including periods of acute neuronal injury as anoxia and reperfusion that may account for the neuronal injury in neurodegenerative diseases such as AD [21-23]. Glutamate-induced neuronal damage is initiated by elevation of the intracellular cation level followed by activation of catabolic enzymes such as proteases, phospholipases and many kinases, as well as the formation of radical oxygen species, which induces cell death via a process known as necrosis or apoptosis [24,25]. Necrosis is characterized by cell swelling, organelle damage, depletion of ATP, loss of ion balance, membrane disorder, nuclear lysis, cessation of protein synthesis, and promotion of the death of adjacent cells, whereas apoptosis is characterized by cell loss, maintenance of the ATP level and ion balance, DNA fragmentation, and a lack of adverse effects on adjacent cells [24,26].
In this study, we investigated the effects of AMPK on scopolamine-induced memory deficit in rats and the protective effect of AMPK on glutamatergic excitatory neurotoxicity in primary cultured hippocampal neurons. To accomplish this, we used the two active and inactive AMPK subunits, c-myc-tagged AMPK wild type alpha subunit (Ad-AMPK-WT) and a dominant negative form (Ad-AMPK-DN), in which Asp157 was replaced with alanine [5]. The results suggest that AMPK plays important roles leading to the cognitive improvement of scopolamine-induced amnesia and that it reduces glutamate induced-cell death. These behavioral and neurochemical results indicate that AMPK has potent cognitive enhancing effects and neuroprotective activities against scopolamine-induced memory impairment. Based on the observations presented herein, we propose that AMPK is a crucial factor involved in cognition function and neuroprotection.
METHODS
Animals
Male Sprague-Dawley rats (250~280 g) purchased from Orient Animal Corp. (Korea) were housed in groups of four in a controlled environment (12:12-hour light:dark cycle, temperature 23±2℃, humidity 50±10%) with water and food available ad libitum for seven days before starting the experiments. All experiments in this study were approved by the Institutional Animal Care and Use Committee of Kyung Hee University. The rats were randomly assigned to three groups (21 rats in each group): group 1: AMPK dominant negative form+saline; group 2: AMPK dominant negative form+scopolamine 2 mgkg-1; group 3: AMPK wild type α subunit+scopolamine 2 mgkg-1.
Adenovirus-mediated gene transfer
c-myc-tagged AMPK wild type α subunit (Ad-AMPK-WT) and a dominant negative form (Ad-AMPK-DN), in which Asp157 was replaced with alanine, were kindly provided by Prof. Ha, Kyunghee University, Seoul, Korea. Small-volume aliquots were kept in liquid nitrogen, and fresh aliquots were used for each experiment. AMPK was infected with recombinant adenovirus at concentrations of 109~1010 pfu/ml.
Microinjection of adenovirus into the hippocampal CA3
Rats were positioned in a stereotaxic instrument (Stoelting, USA) under isoflurane anesthesia. After making a longitudinal incision of the scalp and cleaning the exposed dorsal cranium, a hole was drilled in the skull over the hippocampal CA3 [-5.3 mm anterior-posterior, ±5.0 mm mediolateral, -6.0 mm dorsoventral, according to the rat brain atlas]. Next, a 10 µl Hamilton syringe (30 gauge beveled needle) attached to a stepper motorized nano-injector (Stoleting) was filled with 3 µl of viral suspension, which was injected unilaterally into the hippocampus at a rate of 0.2 µl/min using a microinjection pump (Reno, NV, USA). After injection, the syringe was left in place for 10 min, and then withdrawn very slowly over 10 min. The skin was subsequently sutured with silk, and the rats were then allowed to recover from surgery. At seven days after surgery, the scopolamine (2 mgkg-1, ip) or saline was administered 30 min before the rats were subjected to the water maze task. The correct injection of adenovirus was verified by Cresyl violet staining of the hippocampus CA3 and subsequent microscopic analysis (Zeiss, Germany) at the end of the study (Fig. 1).
Fig. 1.

Verification of the correct injection of the adenovirus into the hippocampal CA3. The correct injection of adenovirus was verified by Cresyl violet staining of the hippocampus CA3 prepared following microinjection of the viral suspension, and subsequent microscopic analysis (100×magnification). Arrow heads and numbers indicate hippocampal layers. Hippocampal layers are: subregion 1, CA3 stratum oriens; subregion 2, CA3 pyramidal cell layer; subregion 3, CA3 stratum radiatum; subregion 4, hippocampal fissure. Arrow indicates the lesion created by needle injection within the hippocampal CA3 regions.
Primary hippocampal neuron culture and treatment
Pregnant Sprague-Dawley rats were purchased from Oriental bio Laboratory animal center. Rat hippocampal neuron cultures were prepared according to Lee et al. [27], with modifications. Briefly, rat brains were dissected from 17- to 18-day-old rat embryos, minced in L-15 medium, and incubated at 37℃ for 15 min with 0.125% trypsin. Dulbecco's modified Eagle's medium containing 10% fetal calf serum, 10% heat-inactivated horse serum, penicillin (100 U/ml) and streptomycin (100 µg/ml) was then added, after which the cells were centrifuged at 600 g for 1 min, resuspended in the above medium, triturated with the aid of a fire-polished Pasteur pipette, and plated onto poly(l-lysine)-coated (10 µg/ml) 35-mm culture dishes (1.5×106 cells/dish). After 1 day, the medium was replaced with fresh F-12 medium supplemented with 5 µg/ml insulin, 1 µg/ml transferrin, 20 nM hydrocortisone, 30 nM triiodothyronine, 2 µg/ml carnitine, and 15 nM selenium oxide (Medium A). The cells were maintained in Dulbecco's modified Eagle's medium for seven days before use. After seven days of culture in vitro, hippocampal neurons were transfected with Ad-AMPK-WT and Ad-AMPK-DN in each group, followed by exposure to 100 µM glutamate with 10 µM of glycine in supplemented neuronal culture medium for 15 min at 37℃ in a humidified incubator with an atmosphere composed of 5% CO2/95% air. After excitotoxicity was induced, the cells were further incubated with the neuronal culture medium at 37℃.
Morris water maze test
A modified version of the procedure described by Morris was used [28]. The water maze was a circular pool (2.0 m in diameter and 0.35 m in height) constructed of fiberglass. The pool contained water maintained at a temperature of 22±2℃ and 1 kg of powdered skim milk to make the water opaque. During testing in the water maze, a platform (15 cm in diameter) was fixed at 1.5 cm below the surface of the water in one of four locations, approximately 50 cm from the side walls. The pool was surrounded by different extra-maze cues. Each trial was initiated at one of four different starting positions and the out of the pool and swimming path of each rat was recorded by a video camera connected to a video recorder and a tracking device (S-MART, Pan-Lab, Spain). All rats were subjected to four trials per day with an interval of 15 min for four consecutive days, followed by one day of probe trials on the fifth day. The trials were completed when the rat found the hidden platform or the escape latency reached 180 s. For the probe trial, the platform was removed from the pool and the rat was allowed to swim freely for 60 s to search for the previous location of the platform. The proportion of time spent searching for the platform in the training quadrant, i.e. the previous location of the platform, was used as a measure of memory retention.
Western blot analysis
For phosphor-AMPK (p-AMPK), AMPK, and c-Myc protein analysis, hippocampal lysates (50 µg per lane) were separated by 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by electrophoretic transfer onto a nitrocellulose membrane (Millipore, MA, USA). The membranes were then probed with antibodies to AMPKα-Thr172 (2531, Cell Signaling Technology, MA, USA), AMPKα (07181, Upstate Biotechnology, Lake Placid, NY), c-Myc (sc764, Santa Cruz Biotechnology, CA) and α-actin (sc17829, Santa Cruz Biotechnology, CA). To assess cell apoptosis, 5~10 µg of cell extracts were analyzed by SDS-PAGE using the antibodies listed below. Antibodies for Bcl-2 (2876) and Bax (2772) were purchased from Cell Signaling Technology. The antibody-specific proteins were visualized using the enhanced chemiluminesence detection system according to the recommended procedure (Amersham Corp., Newark, NJ).
Flow cytometric analysis
Hippocampal neurons were incubated with FITC Annexin V in buffer and then analyzed by flow cytometry. Briefly, hippocampal neurons were washed twice with cold PBS and then resuspended in buffer (0.1 M Hepes/NaOH (pH 7.4), 1.4 M NaCl, 25 mM CaCl2) at a concentration of 1×106 cells/ml. Next, 100 µl of the solution were transferred to a 5 ml culture tube. FITC Annexin V was added (5 µl) to the culture tube and incubated for 15 min at RT (25℃) in the dark. After incubation, cells were resuspended in 400 µl of buffer and Annexin V positive cells were detected by flow cytometry (FACS Caliber, BD, San Diego, CA, USA) and then analyzed using the CellQuest software.
AChE histochemistry
For neurobiological analysis (n=20 per group), rats were perfused transcardially via the ascending aorta with normal saline (0.9%), followed by 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS). The brains were then removed, post-fixed overnight and immersed sequentially in 0.1 M PBS containing 20% sucrose at 4℃ until they sank. The brains were then cut into 40 µm coronal sections using a cryostat, after which the sections were washed in PBS and then incubated in a solution containing 25 mg acetylcholine iodine for 1 h. The solution was composed of 32.5 ml of 0.1 M sodium hydrogen phosphate buffer (NaH2PO4· H2O, pH 6.0), 2.5 ml of 0.1 M sodium citrate, 5 ml of 30 mM copper sulfate, 5 ml of 5 mM potassium ferricyanide and 5 ml of distilled water. The color of the mixing solution was green. The density of the stained nuclei of the hippocampal cells from the hippocampal CA1 and CA3 areas were measured using the Scion image program (Scion Corp., MD, USA).
Statistical analysis
The data were expressed as the means±SEM. Differences between groups were analyzed by repeated measures analysis of variance (ANOVA) using SPSS (Version 10.0; SPSS Inc., Chicago, USA). ANOVA followed by Tukey's post hoc test for multiple group comparison was used to analyze differences in data collected on successive training days, during the probe trials, and during the neurobiological analysis. Differences among groups were considered statistically significant if the associated probability (p-value) was <0.05.
RESULTS
AMPK reverses spatial learning deficits induced by scopolamine
To determine if adenoviral gene transfer of Ad-AMPK-DN and -WT into the hippocampus affects special memory tasks, we evaluated the effect of AMPK on scopolamine-induced impairment of memory by the Morris water maze test. In this study, scopolamine treatment had a significant effect on escape latency (the swimming time required to find a hidden platform in the acquisition test) and total swimming distance (the swim path length taken to find the hidden platform in the acquisition test) among Ad-AMPK-DN injected rats. Conversely, scopolamine-induced memory deficiency was significantly alleviated in Ad-AMPK-WT injected rats when compared with Ad-AMPK-DN injected rats. As shown in Fig. 2A, the results of the escape latency revealed a significant difference between groups [F (2, 60)=78.98, p<0.001] and time of day [F (3, 180)=56.32, p<0.001]. On days 1~4, the AMPK-WT+scopolamine group showed significantly reduced escape latency when compared with the AMPK-DN+scopolamine group (p<0.05 on day 1 and p<0.001 on days 2~4).
Fig. 2.

Alteration of escape latency and swimming distance during an acquisition test and time spent and distance around the platform during the retention test of the water maze test. Four trials per day for 4 consecutive days were conducted and the escape latency (A) and swimming distance (B) were measured. Four trials per day on the fifth day without the platform were conducted and the search latency (C) and distance (D) were measured. Data were analyzed using repeated measures ANOVA followed by Tukey's test. Each value represents the mean±SEM. *p<0.05, **p<0.01 and ***p<0.001 compared to AMPK-DN+scopolamine.
As shown in Fig. 2B, the total swimming distance differed significantly among groups [F (2, 60)=51.22, p<0.001] and time of day [F (3, 180)=8.24, p<0.001]. Post hoc analysis revealed that the AMPK-DN+scopolamine group had significantly increased swimming distance when compared with the AMPK-DN+saline group during all of the training days (p<0.001). On days 2~4, the AMPK-WT+scopolamine group showed a significantly decreased swimming distances when compared with the AMPK-DN+scopolamine group (p<0.05 on days 2~4). Conversely, there was no significant difference in the average swimming speed among groups on all training days (data not shown).
The performance on the probe trial for comparing the percentage of time spent swimming around the platform on day 5 is illustrated in Fig. 2C. Post hoc analysis of learning and memory retention performance revealed that the AMPK-DN+saline group and the AMPK-WT+scopolamine group spent a longer time around the platform than the AMPK-DN+scopolamine group (p<0.05 and p<0.05). Moreover, analysis of the performance during the probe trial for comparing the swimming distance around the platform is illustrated in Fig. 2D. Post hoc analysis of learning and memory retention performance also revealed that the AMPK-DN+saline group and the AMPK-WT+scopolamine group had a significantly longer swim distance around the platform than the AMPK-DN+scopolamine group (p<0.05 and p<0.05). These findings indicate that scopolamine severely impaired the spatial memory properties in the water maze test, but that adenoviral gene transfer of Ad-AMPK-WT ameliorated the scopolamine induced cognitive deficit.
AMPK causes an increase in the AChE-reactive neurons in the hippocampus
We measured the levels of AChE-reactive neurons at the hippocampal CA1 and CA3 using AChE histochemistry. Fig. 3A shows the distribution of AChE-reactive neurons in the hippocampus of the AMPK-DN+saline group, AMPKDN+ scopolamine group, and AMPK-WT+scopolamine group. The AMPK-DN+scopolamine group exhibited markedly diminished cholinergic activity that was characterized by reduced AChE-reactive neurons when compared with the AMPK-DN+saline group, but the AMPK-WT+scopolamine group was not influenced by scopolamine treatment.
Fig. 3.

Density of the AchE-reactive neurons in hippocampal CA1 and CA3 of the experimental groups. AchE positive nerve fibers in the hippocampal CA1 (A) and CA3 (B) molecular layer of experimental rats in AMPK-DN+saline group (a, d), AMPK-DN+scopolamine group (b, e), and AMPK-WT+scopolamine group (c, f). The brains were cut into 40 µm coronal sections and the scale bar represents 50 µm (100×magnification). The percentage of AMPK-DN+saline values of the density of acetylcholinesterase (AChE) stained nuclei in the hippocampal CA1 (C) and CA3 (D) areas after the water maze test. Data were analyzed using one-way ANOVA followed by the Tukey's test. Each value represents the mean±SEM. *p<0.05 compared to AMPK-DN+scopolamine.
As shown in Fig. 3B, the density of AChE-reactive neurons was lower in the AMPK-DN+scopolamine group than in the AMPK-DN+saline group. The density of the AChE neurons in the CA1 area was 63.71±4.10 (99.95±6.45%) in the AMPK-DN+saline group, 52.64±3.55 (82.64±5.57%) in the AMPK-DN+scopolamine group, and 63.65±2.45 (99.89± 3.85%) in the AMPK-WT+scopolamine group [F (2, 115)=3.850, p<0.05]. Post hoc analysis revealed that the density of the AChE reactive neurons in the hippocampus of the AMPK-WT+scopolamine group (p<0.05) showed higher expression than those of the AMPK DN+scopolamine group (in the CA1). The density of the AChE neurons at CA3 of the hippocampus was lower in the AMPK-DN+scopolamine group than the AMPK-DN+saline group (Fig. 3C). The density of AChE neurons in the CA3 area was 78.07±3.90 (99.91±5.01%) in the AMPK-DN+saline group, 64.91±2.67 (83.11±3.42%) in the AMPK DN+scopolamine group, and 69.64±2.35 (89.06±3.01%) in the AMPK-WT+scopolamine group [F (2, 121)=3.173, p<0.05]. Post hoc analysis revealed that the density of the AChE reactive neurons in the hippocampus of the AMPK-DN+saline group (p<0.05) was greater than that of the AMPK-DN+scopolamine group (in CA3). These results suggest that AMPK-WT may contribute to the amelioration of the decreased learning capacity through alleviation of the cholinergic neuronal damages in the hippocampal pathway.
Measurement of the phosphorylation levels of AMPK in the hippocampus
To determine if CA3 of the hippocampus was infected with Ad-AMPK-WT or Ad-AMPK-DN, expression of AMPKs in the adenoviral-infected brain region was confirmed by western blot analysis and AMPK activation was determined based on the phosphorylation levels of AMPK (p-AMPK) (Fig. 4). The results showed that the expression of the p-AMPK protein in the hippocampus of the AMPK-WT+scopolamine group was greater than that of the AMPK-DN+ scopolamine group. The adenoviral gene transfer of Ad-AMPK-WT resulted in elevation of the p-AMPK protein level, indicating an approximately 17-fold induction when compared with the normal group. However, adenoviral gene transfer of the Ad-AMPK-DN did not lead to a further increase in the p-AMPK protein level.
Fig. 4.

Phosphorylation of AMPK (p-AMPK) in CA3 of the hippocampus infected with Ad-AMPK-WT or Ad-AMPK-DN. Western blot analysis (A) of lysates obtained from normal hippocampus and those following Ad-AMPK-WT and -DN infection were performed using antibodies phosphorylated (P-AMPK) or total AMPK α (AMPK α). The intensities obtained from gel image of (A) were depicted as the ratio between total AMPK and P-AMPK (B).
AMPK protects against cytotoxic effects of glutamate
To determine if adenoviral gene transfer of AMPK affects cell apoptosis, we examined the effect of AMPK on glutamate-induced cytotoxicity in neuronal cells. After culture for seven days in vitro, hippocampal neurons were infected with Ad-AMPK-WT or Ad-AMPK-DN, after which they were exposed to glutamate and glycine. After excitotoxicity was induced, the cells were further incubated with the neuronal culture medium at 37℃ and then analyzed by western blot assays. Pro-apoptotic Bax and anti-apoptotic Bcl-2 are crucial in determining cell survival or death. As shown in Fig. 5, glutamate induced an increase in the expression of Bax, which was responsible for permeabilization of the mitochondrial membrane, leading to the release of cytochrome c from the mitochondria and initiation of the caspase activation pathway for apoptosis [29]. This activation of Bax was markedly reversed in cells infected with Ad-AMPK-WT. In addition, the expression of Bcl-2, which has anti-apoptotic and cell survival-promoting effects [30], was significantly upregulated in cells infected with Ad-AMPK-WT when compared to the control cells; however, there was no significant difference in the levels of Bcl-2 between glutamate-treated cells and Ad-AMPK-WT+glutamate treated cells (No significant change in the expression levels of Bcl-2 was observed). Hence, this anti-apoptotic effect of AMPK was identified based on a decreased Bax/Bcl-2 ratio.
Fig. 5.
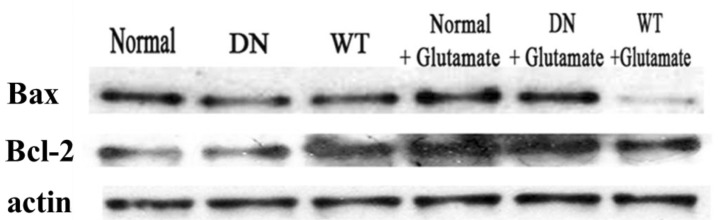
The effect of AMPK on glutamate-induced apopotosis of cells. Hippocampal neurons cultured in vitro for seven days were transfected with Ad-AMPK-WT and Ad-AMPK-DN in each group and then exposed to 100 µM glutamate with 10 µM of glycine in supplemented neuronal culture medium for 15 min at 37℃. Western blot assays were then conducted using anti-Bax and anti-Bcl-2. The experiment was repeated three times with similar results.
Our data show that infection with Ad-AMPK-WT generates a survival signal after glutamate treatment of the cells, suggesting that AMPK plays a protective role in glutamate-induced cytotoxicity.
We also analyzed the protective effect of AMPK against apoptosis by using flow cytometry. Annexin V (Mr 36-kDa), a member of the annexin family of calcium-dependent phospholipid binding proteins, has a high affinity for phosphatidylserine (PS) containing phospholipid bilayers. Fluorochrome conjugates of annexin V can be used to monitor changes in cell membrane phospholipid asymmetry, thereby providing a convenient tool for the detection of apoptotic cells. Therefore, we used FITC-conjugated annexin V to assess the effects of AMPK on the apoptosis of primary hippocampal neuron [31]. As shown in Fig. 6, Ad-AMPK-DN infected cells showed a distinct shift in annexin fluorescence intensity (AMPK-DN; M1 51.29%). Conversely, the shift in fluorescence intensity was reduced in Ad-AMPK-WT infected cells (AMPK-WT; M1 43.07%). These data indicate that apoptosis, as assessed by annexin V staining, was attenuated in the primary hippocampal neuron in response to AMPK expression.
Fig. 6.

The effect of AMPK on the apoptosis of primary cultured hippocampal neurons as determined by FITC-conjugated annexin V. Neonatal rat hippocampal neurons were cultured and infected with Ad-AMPK-WT or Ad-AMPK-DN, followed by exposure to glutamate and glycine. Anexin V positive cells were detected by flow cytometry (FACS Calibur, BD, San Diego, CA, USA) and analyzed using the CellQuest software.
DISCUSSION
The nervous system accounts for a high proportion of total body energy turnover, and neurons are particularly susceptible to energy deficits due to their inflexible metabolism and poor capacity to store nutrients [32]. Therefore, it is not surprising that adenosine monophosphate-activated protein kinase (AMPK), which is part of the signaling system that is important to the maintenance of energy balance at both the cellular and whole body levels, is highly expressed in the central nervous system [8,33]. AMPK is activated following ATP depletion, an increase in AMP levels, or a rise in the AMP-ATP ratio within a cell [8,10,33]. Once activated, AMPK increases cellular ATP supply by stimulating glycolysis. Conversely, it suppresses the key enzymes involved in ATP-consuming anabolic pathways to maintain ATP level homeostasis [2,3], through inhibition of the ATP gated Cl- channel [34] and phosphorylation of the GABAB receptor to induce increased GABA-dependent inhibition of presynaptic Ca2+ channels [32]. Modest AMPK activation induced neurogenesis and improved cognition in animals, but augmented AMPK activation reduced cognition and increased neural apoptosis and mortality [35].
As shown in this study, the AMPK wild type alpha subunit led to significant improvement in behavioral deficits in the Morris water maze and attenuated the decrease in acetylcholinergic neurons that occurred via scopolamine-induced memory impairment. Neurodegenerative disorders such as Alzheimer's disease are accompanied by a loss of cholinergic neurons such as a significant decrease in acetylcholine (Ach) level in the cerebral cortex and hippocampus, which correlates well with cognitive dysfunction. These findings indicate that the increase in Ach levels observed in this study may help alleviate behavioral and biochemical deficits and cholinesterase inhibitors, which can effectively increase cholinergic neurons are the prescribed pharmacological agents preventing against cognitive dysfunction. Scopolamine is a muscarinic cholinergic receptor antagonist that impairs learning and memory in rodents and humans, especially the process of learning, acquisition and short-term memory; accordingly, scopolamine-induced amnesia has been used as a model for screening anti-amnesic drugs. To confirm the protective effects of AMPK in neuronal cells, we evaluated AMPK to determine if it has an anti-apoptotic effect in glutamate-induced cytotoxicity in the primary cultured hippocampal neuron. Our results demonstrated that expression of AMPK reduced the expression of Bax and had no effect on Bcl-2, which resulted in a decreased Bax/Bcl-2 ratio, but that it increased cell viability and reduced the Annexin V positive hippocampal neurons.
Many recent studies have shown the protective effect of AMPK in the nervous system. Culmsee et al. demonstrated that fetal rat neurons cultured under glucose deprived conditions had improved viability after treatment with small to moderate concentrations of 5-aminoimidazole-4-carboxamide ribonucleoside, a potent activator of AMPK [8,36]. In addition, several reports have demonstrated that AMPK plays an important role in the survival of neurons under cytotoxic conditions. Kuramoto et al. showed that AMPK mediated protection against excitotoxicity through phosphorylation of the GABAB receptor [37] and Yoon et al. demonstrated that treatment of glioma cells with kainic acid, an excitotoxic agonist of ionotropic glutamate receptors, results in Ca2+/calmodulin-dependent protein kinase beta-beta-mediated activation of AMPK. AMPK activation, in turn, leads to an increase in brain-derived neurotrophic factor expression that likely occurs via the action of nuclear factor-kappaB [38]. In addition, AMPK activation decreased cytoplasmic HuR (a shuttling RNA-binding protein), which consequently decreased the binding of HuR to target transcripts and diminished the expression and half-lives of such HuR target mRNA [39,40]. Researchers have also found that sustained AMPK activation by either AMPK activator or active AMPK mutant induced apoptosis [41-43]. For example, the sustained activation of AMPK induced apoptosis in liver cells through activation of c-Jun N-terminal kinase [44]. Some studies have demonstrated that overactivation or sustained activation of AMPK was detrimental to neurons [10]. However, the wider role of AMPK in the nervous system is still not clear.
Taken together, the results of the present study demonstrate that AMPK significantly improved behavioral deficits during the Morris water maze test and attenuated the decrease in acetylcholinergic neurons that occurred in response to scopolamine-induced memory impairment. These behavioral and neurochemical results indicate that AMPK has potent cognitive enhancing effects and neuroprotective activities against scopolamine-induced memory impairment rats. The neuroprotective activities of AMPK, which are associated with elevated cell viability, might promote conservation of the remaining energy to support the survival and physiological functions of the cell in the central nervous system. The results of this study also show that AMPK has an anti-apoptotic effect in glutamate-induced cytotoxicity in primary cultured hippocampal neurons. Based on these findings, we propose that AMPK activation could be developed into a novel therapeutic approach for amelioration of cognitive dysfunction related neurodegenerative diseases.
ACKNOWLEDGEMENTS
This work was supported by the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korea government (MEST) (No. 2009-0063466) and a Post-Doctoral fellowship from Kyung Hee University in 2009 (KHU-20090503).
ABBREVIATIONS
- AMPK
AMP-activated protein kinase
- AChE
acetylcholinesterase
- Ad-AMPK-WT
AMPK wild type subunit
- Ad-AMPK-DN
AMPK dominant negative form
- FACS
flow cytometry
- BDNF
brain-derived neurotrophic factor
- CNS
central nervous system
- AD
Alzheimer's disease
References
- 1.Gomez-Pinilla F, Vaynman S, Ying Z. Brain-derived neurotrophic factor functions as a metabotrophin to mediate the effects of exercise on cognition. Eur J Neurosci. 2008;28:2278–2287. doi: 10.1111/j.1460-9568.2008.06524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu Rev Biochem. 1998;67:821–855. doi: 10.1146/annurev.biochem.67.1.821. [DOI] [PubMed] [Google Scholar]
- 3.Hardie DG. Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144:5179–5183. doi: 10.1210/en.2003-0982. [DOI] [PubMed] [Google Scholar]
- 4.Hardie DG. The AMP-activated protein kinase pathway-new players upstream and downstream. J Cell Sci. 2004;117:5479–5487. doi: 10.1242/jcs.01540. [DOI] [PubMed] [Google Scholar]
- 5.Woods A, Azzout-Marniche D, Foretz M, Stein SC, Lemarchand P, Ferré P, Foufelle F, Carling D. Characterization of the role of AMP-activated protein kinase in the regulation of glucose-activated gene expression using constitutively active and dominant negative forms of the kinase. Mol Cell Biol. 2000;20:6704–6711. doi: 10.1128/mcb.20.18.6704-6711.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson LN, Noble ME, Owen DJ. Active and inactive protein kinases: structural basis for regulation. Cell. 1996;85:149–158. doi: 10.1016/s0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- 7.Stein SC, Woods A, Jones NA, Davison MD, Carling D. The regulation of AMP-activated protein kinase by phosphorylation. Biochem J. 2000;345:437–443. [PMC free article] [PubMed] [Google Scholar]
- 8.Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci. 2001;17:45–58. doi: 10.1385/JMN:17:1:45. [DOI] [PubMed] [Google Scholar]
- 9.Witters LA, Kemp BE, Means AR. Chutes and ladders: the search for protein kinases that act on AMPK. Trends Biochem Sci. 2006;31:13–16. doi: 10.1016/j.tibs.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Y, Shen J, Su H, Li B, Xing D, Du L. Chronic corticosterone injections induce a decrease of ATP levels and sustained activation of AMP-activated protein kinase in hippocampal tissues of male mice. Brain Res. 2008;1191:148–156. doi: 10.1016/j.brainres.2007.11.027. [DOI] [PubMed] [Google Scholar]
- 11.Gadalla AE, Pearson T, Currie AJ, Dale N, Hawley SA, Sheehan M, Hirst W, Michel AD, Randall A, Hardie DG, Frenguelli BG. AICA riboside both activates AMP-activated protein kinase and competes with adenosine for the nucleoside transporter in the CA1 region of the rat hippocampus. J Neurochem. 2004;88:1272–1282. doi: 10.1046/j.1471-4159.2003.02253.x. [DOI] [PubMed] [Google Scholar]
- 12.Föller M, Sopjani M, Koka S, Gu S, Mahmud H, Wang K, Floride E, Schleicher E, Schulz E, Münzel T, Lang F. Regulation of erythrocyte survival by AMP-activated protein kinase. FASEB J. 2009;23:1072–1080. doi: 10.1096/fj.08-121772. [DOI] [PubMed] [Google Scholar]
- 13.McGee SL, Hargreaves M. AMPK and transcriptional regulation. Front Biosci. 2008;13:3022–3033. doi: 10.2741/2907. [DOI] [PubMed] [Google Scholar]
- 14.Lee B, Sur B, Shim I, Lee H, Hahm DH. Phellodendron amurense and its major alkaloid compound, berberine ameliorates scopolamine-induced neuronal impairment and memory dysfunction in rats. Korean J Physiol Pharmacol. 2012;16:79–89. doi: 10.4196/kjpp.2012.16.2.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mohapel P, Leanza G, Kokaia M, Lindvall O. Forebrain acetylcholine regulates adult hippocampal neurogenesis and learning. Neurobiol Aging. 2005;26:939–946. doi: 10.1016/j.neurobiolaging.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 16.Klinkenberg I, Blokland A. The validity of scopolamine as a pharmacological model for cognitive impairment: a review of animal behavioral studies. Neurosci Biobehav Rev. 2010;34:1307–1350. doi: 10.1016/j.neubiorev.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 17.Goverdhan P, Sravanthi A, Mamatha T. Neuroprotective effects of meloxicam and selegiline in scopolamine-induced cognitive impairment and oxidative stress. Int J Alzheimers Dis. 2012;2012:974013. doi: 10.1155/2012/974013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan Y, Hu J, Li J, Yang Z, Xin X, Wang J, Ding J, Geng M. Effect of acidic oligosaccharide sugar chain on scopolamine-induced memory impairment in rats and its related mechanisms. Neurosci Lett. 2005;374:222–226. doi: 10.1016/j.neulet.2004.10.063. [DOI] [PubMed] [Google Scholar]
- 19.Ray CA, Blin J, Chase TN, Piercey MF. Effects of cholinergic agonists on regional brain energy metabolism in the scopolamine-treated rat. Neuropharmacology. 1992;31:1193–1199. doi: 10.1016/0028-3908(92)90017-j. [DOI] [PubMed] [Google Scholar]
- 20.Piercey MF, Vogelsang GD, Franklin SR, Tang AH. Reversal of scopolamine-induced amnesia and alterations in energy metabolism by the nootropic piracetam: implications regarding identification of brain structures involved in consolidation of memory traces. Brain Res. 1987;424:1–9. doi: 10.1016/0006-8993(87)91186-3. [DOI] [PubMed] [Google Scholar]
- 21.Schubert D, Piasecki D. Oxidative glutamate toxicity can be a component of the excitotoxicity cascade. J Neurosci. 2001;21:7455–7462. doi: 10.1523/JNEUROSCI.21-19-07455.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Albright TD, Jessell TM, Kandel ER, Posner MI. Neural science: a century of progress and the mysteries that remain. Cell. 2000;100(Suppl):S1–S55. [PubMed] [Google Scholar]
- 23.Lee S, Kim Y, Li E, Park S. Ghrelin protects spinal cord motoneurons against chronic glutamate excitotoxicity by inhibiting microglial activation. Korean J Physiol Pharmacol. 2012;16:43–48. doi: 10.4196/kjpp.2012.16.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leon R, Wu H, Jin Y, Wei J, Buddhala C, Prentice H, Wu JY. Protective function of taurine in glutamate-induced apoptosis in cultured neurons. J Neurosci Res. 2009;87:1185–1194. doi: 10.1002/jnr.21926. [DOI] [PubMed] [Google Scholar]
- 25.Mark LP, Prost RW, Ulmer JL, Smith MM, Daniels DL, Strottmann JM, Brown WD, Hacein-Bey L. Pictorial review of glutamate excitotoxicity: fundamental concepts for neuroimaging. AJNR Am J Neuroradiol. 2001;22:1813–1824. [PMC free article] [PubMed] [Google Scholar]
- 26.McConkey DJ. Biochemical determinants of apoptosis and necrosis. Toxicol Lett. 1998;99:157–168. doi: 10.1016/s0378-4274(98)00155-6. [DOI] [PubMed] [Google Scholar]
- 27.Lee JH, Choi S, Kim JH, Kim JK, Kim JI, Nah SY. Effects of ginsenosides on carbachol-stimulated formation of inositol phosphates in rat cortical cell cultures. Neurochem Res. 2003;28:1307–1313. doi: 10.1023/a:1024979912161. [DOI] [PubMed] [Google Scholar]
- 28.Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 29.Beere HM. Death versus survival: functional interaction between the apoptotic and stress-inducible heat shock protein pathways. J Clin Invest. 2005;115:2633–2639. doi: 10.1172/JCI26471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 31.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 32.Hardie DG, Frenguelli BG. A neural protection racket: AMPK and the GABA(B) receptor. Neuron. 2007;53:159–162. doi: 10.1016/j.neuron.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 33.Turnley AM, Stapleton D, Mann RJ, Witters LA, Kemp BE, Bartlett PF. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J Neurochem. 1999;72:1707–1716. doi: 10.1046/j.1471-4159.1999.721707.x. [DOI] [PubMed] [Google Scholar]
- 34.Hallows KR, Raghuram V, Kemp BE, Witters LA, Foskett JK. Inhibition of cystic fibrosis transmembrane conductance regulator by novel interaction with the metabolic sensor AMP-activated protein kinase. J Clin Invest. 2000;105:1711–1721. doi: 10.1172/JCI9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dagon Y, Avraham Y, Magen I, Gertler A, Ben-Hur T, Berry EM. Nutritional status, cognition, and survival: a new role for leptin and AMP kinase. J Biol Chem. 2005;280:42142–42148. doi: 10.1074/jbc.M507607200. [DOI] [PubMed] [Google Scholar]
- 36.Spasić MR, Callaerts P, Norga KK. AMP-activated protein kinase (AMPK) molecular crossroad for metabolic control and survival of neurons. Neuroscientist. 2009;15:309–316. doi: 10.1177/1073858408327805. [DOI] [PubMed] [Google Scholar]
- 37.Kuramoto N, Wilkins ME, Fairfax BP, Revilla-Sanchez R, Terunuma M, Tamaki K, Iemata M, Warren N, Couve A, Calver A, Horvath Z, Freeman K, Carling D, Huang L, Gonzales C, Cooper E, Smart TG, Pangalos MN, Moss SJ. Phospho-dependent functional modulation of GABA (B) receptors by the metabolic sensor AMP-dependent protein kinase. Neuron. 2007;53:233–247. doi: 10.1016/j.neuron.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoon H, Oh YT, Lee JY, Choi JH, Lee JH, Baik HH, Kim SS, Choe W, Yoon KS, Ha J, Kang I. Activation of AMP-activated protein kinase by kainic acid mediates brain-derived neurotrophic factor expression through a NF-kappaB dependent mechanism in C6 glioma cells. Biochem Biophys Res Commun. 2008;371:495–500. doi: 10.1016/j.bbrc.2008.04.102. [DOI] [PubMed] [Google Scholar]
- 39.Wang W, Fan J, Yang X, Fürer-Galban S, Lopez de Silanes I, von Kobbe C, Guo J, Georas SN, Foufelle F, Hardie DG, Carling D, Gorospe M. AMP-activated kinase regulates cytoplasmic HuR. Mol Cell Biol. 2002;22:3425–3436. doi: 10.1128/MCB.22.10.3425-3436.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang W, Yang X, López de Silanes I, Carling D, Gorospe M. Increased AMP:ATP ratio and AMP-activated protein kinase activity during cellular senescence linked to reduced HuR function. J Biol Chem. 2003;278:27016–27023. doi: 10.1074/jbc.M300318200. [DOI] [PubMed] [Google Scholar]
- 41.Dagon Y, Avraham Y, Berry EM. AMPK activation regulates apoptosis, adipogenesis, and lipolysis by eIF2alpha in adipocytes. Biochem Biophys Res Commun. 2006;340:43–47. doi: 10.1016/j.bbrc.2005.11.159. [DOI] [PubMed] [Google Scholar]
- 42.Jung JE, Lee J, Ha J, Kim SS, Cho YH, Baik HH, Kang I. 5-Aminoimidazole-4-carboxamide-ribonucleoside enhances oxidative stress-induced apoptosis through activation of nuclear factor-kappaB in mouse Neuro 2a neuroblastoma cells. Neurosci Lett. 2004;354:197–200. doi: 10.1016/j.neulet.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 43.Kefas BA, Cai Y, Kerckhofs K, Ling Z, Martens G, Heimberg H, Pipeleers D, Van de Casteele M. Metformin-induced stimulation of AMP-activated protein kinase in beta-cells impairs their glucose responsiveness and can lead to apoptosis. Biochem Pharmacol. 2004;68:409–416. doi: 10.1016/j.bcp.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 44.Meisse D, Van de Casteele M, Beauloye C, Hainault I, Kefas BA, Rider MH, Foufelle F, Hue L. Sustained activation of AMP-activated protein kinase induces c-Jun N-terminal kinase activation and apoptosis in liver cells. FEBS Lett. 2002;526:38–42. doi: 10.1016/s0014-5793(02)03110-1. [DOI] [PubMed] [Google Scholar]