

Abstract
Ever since its discovery 20 years ago, caspase-2 has been enigmatic and its function somewhat controversial. Although many in vitro studies suggested that caspase-2 was important for apoptosis, demonstrating an in vivo cell death role for this caspase has been more problematic, with caspase-2-deficient mice showing limited, tissue-specific cell death defects. Recent results from different laboratories suggest that at least one of its physiological roles in animals is to protect against cellular stress and transformation. As such, loss of caspase-2 augments tumorigenesis in some mouse models of cancer, assigning a tumour suppressor function to this enigmatic caspase. This review focuses on this seemingly non-apoptotic function of caspase-2 as a tumour suppressor and reconciles some of the recent findings in the field.
Keywords: caspase-2, cancer, genomic instability, tumour suppression, DNA damage response
Facts
Caspase-2 is rapidly processed (and activated) in response to many apoptotic stimuli but is redundant for most apoptosis.
Caspase-2 is the only caspase that constitutively localizes to the nucleus.
Loss of caspase-2 is associated with impaired DNA damage response, cell cycle regulation and genomic instability in MEFs and tumour cells.
Loss of caspase-2 in mice leads to a slight, but consistent early ageing phenotype.
Deficiency of caspase-2 augments tumorigenesis in some mouse models.
Open Questions
How broadly does the tumour suppressor function of caspase-2 extend?
What is the mechanistic basis of caspase-2 function in DNA damage response, cell cycle regulation and maintenance of genomic stability?
How are these functions linked to its role in tumour suppression?
What is the relevance of caspase-2 function in tumour suppression in human cancers?
Along with caspase-1, caspase-2 was one of the first discovered mammalian homologues of Caenorhabditis elegans CED-3.1, 2 Given the function of caspase-1 (IL-1β-converting enzyme) in IL-1β maturation, caspase-2 was hailed as the caspase that was more likely to functionally emulate CED-3 in mammalian cell death.3, 4 However, as we now know, there are about a dozen caspases in mammals, some with distinct functions in cell death and/or inflammation.5 Surprisingly, caspase-2 does not sit in either camp, having no general role in apoptosis or inflammation.5 The fact that caspase-2 is the most evolutionarily conserved of mammalian caspases but yet is dispensable for most cases of cell death (apoptotic and non-apoptotic) remains puzzling.
In general, caspase-2 appears to be a classical apoptosis initiator caspase. It contains a caspase activation and recruitment domain (CARD), such as CED-3 in C. elegans, Dronc in Drosophila and caspase-9 in mammals.1, 5, 6 Similar to other initiator caspases, caspase-2 activation initially occurs by dimerization, and full activation is then achieved by autoprocessing (Box 1). A unique feature of caspase-2 is its nuclear localization, a property not shared by other caspases.7, 8 Caspase-2 has been implicated in apoptosis induced by multiple intrinsic and extrinsic stimuli including DNA damage, reactive oxygen species (ROS) and cytoskeletal disruption (Figure 1).9 However, as discussed below, deletion of caspase-2 in mice does not lead to a phenotype that would support a broad function for this caspase in apoptosis.10
Box 1 Regulation of caspase-2 activation.
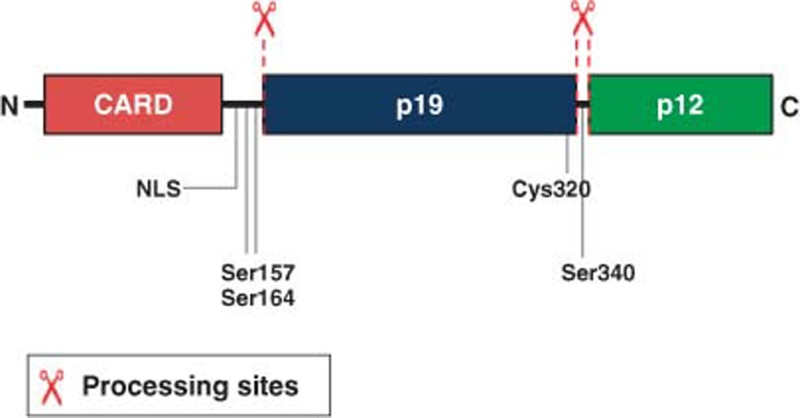 Caspase-2 is synthesized as a zymogen and is activated via a dimerization-dependent, autocatalytic cleavage mechanism.45 Caspase-2 is made up of a C-terminal catalytic domain containing the active site (Cys320) and an amino terminal CARD, which mediates protein–protein interactions and facilitates recruitment into activation platforms.9 Upon proximity-induced oligomerization via its CARD, caspase-2 becomes partially active.46 Autoprocessing then occurs between the small and large subunits of the catalytic domain yielding a fully active enzyme.45 Further processing results in removal of the N-terminal CARD, generating a fully mature tetramer.45 Caspase-2 is also actively imported into the nucleus via a nuclear localization sequence (NLS) located in the prodomain.7, 8 Caspase-2 activation has been shown to be regulated by phosphorylation mediated by various kinases, including calcium/calmodulin-dependent protein kinase II (Ser164),47 protein kinase CK2 (Ser157)48 and cyclin-dependent kinase 1 (Ser340).49
Caspase-2 is synthesized as a zymogen and is activated via a dimerization-dependent, autocatalytic cleavage mechanism.45 Caspase-2 is made up of a C-terminal catalytic domain containing the active site (Cys320) and an amino terminal CARD, which mediates protein–protein interactions and facilitates recruitment into activation platforms.9 Upon proximity-induced oligomerization via its CARD, caspase-2 becomes partially active.46 Autoprocessing then occurs between the small and large subunits of the catalytic domain yielding a fully active enzyme.45 Further processing results in removal of the N-terminal CARD, generating a fully mature tetramer.45 Caspase-2 is also actively imported into the nucleus via a nuclear localization sequence (NLS) located in the prodomain.7, 8 Caspase-2 activation has been shown to be regulated by phosphorylation mediated by various kinases, including calcium/calmodulin-dependent protein kinase II (Ser164),47 protein kinase CK2 (Ser157)48 and cyclin-dependent kinase 1 (Ser340).49
Figure 1.

The function of caspase-2 in cell death. Cellular stress induced by DNA damage, cytoskeletal disruption or ROS can lead to alternative pathways of caspase-2 activation. In response to cytoskeletal disruption or ROS, the activation of caspase-2 occurs upstream of mitochondrial outer membrane permeabilization (MOMP). Following DNA damage, caspase-2 is activated downstream of ATM/ATR to induce apoptosis by an unknown mechanism. ATM/ATR also activates p53 and MOMP via Puma and Bax/Bak activation, leading to cytochrome c (Cyt c) release and formation of the Apaf-1 apoptosome, which recruits and activates caspase-9 and caspase-3. Smac/Diablo release from mitochondria during MOMP blocks IAP (inhibitor of apoptosis) function to facilitate the caspase activation cascade. Once activated, the key effector caspase, caspase-3, also cleaves caspase-2, which potentially provides an amplification loop for the caspase activation cascade
A potential function of caspase-2 in tumour suppression was first suggested in 1995, and a number of subsequent correlative studies with clinical samples have followed.11, 12, 13, 14 However, more definitive experimental studies were first published in 2009, in which we presented data for the first time to show a potential tumour suppressor function for caspase-2.15 In this article, we review recent findings detailing the potential functions of caspase-2, with particular emphasis on how these processes may be linked to its role as a tumour suppressor.
Caspase-2 Knockout Mice
Studies in caspase-2 knockout (Casp2−/−) mice failed to provide any conclusive evidence that caspase-2 is essential for apoptosis.10, 16 Casp2−/− mice have excess numbers of germ cells in ovaries, and caspase-2-deficient oocytes have been reported to be somewhat resistant to apoptosis induced by chemotherapeutic drugs.10 However, most cell types in wild-type and Casp2−/− mice, including thymocytes and DRG neurons, show comparable levels of cell death in response to various cytotoxic stimuli.16
Further, Casp2−/−; Casp9−/− double knockout mice are indistinguishable from Casp9−/− mice in terms of their development and show embryonic brain malformation and perinatal lethality.17 In addition, Casp2−/−; Casp9−/− hematopoietic cells develop normally, and lymphocytes and fibroblasts lacking both caspases remain sensitive to many apoptotic stimuli, showing release of cytochrome c from mitochondria.17 On the other hand, Casp2−/− mouse embryonic fibroblasts (MEFs) show subtle resistance to killing by heat-shock and cytoskeletal-disrupting chemotherapeutic agents.18, 19 Interestingly, in Casp2−/− neurons, NGF-deprivation-induced cell death is dependent on the caspase-9 pathway.20 However, in wild-type neurons, a caspase-2-dependent pathway is required for death, whereas the caspase-9-dependent pathway appears to be suppressed, suggesting functional redundancy or compensation between caspase-2 and caspase-9.20 Also, Casp2−/− mice show signs of premature ageing and metabolic and oxidative stress and are noticeably leaner as they age.21, 22
Overall, Casp2−/− mice have provided only limited insight into the physiological functions of this enigmatic caspase. As stated above, compensatory mechanisms have been proposed to account for this lack of an overt phenotype. However, as discussed in this article, context-dependent, fine-tuning functions of caspase-2 may explain these observations in Casp2−/− mice.
Caspase-2 – the Anti-Cancer Connection
Evasion of apoptosis is a well-established hallmark of cancer.23 Apoptosis is a critical tumour-suppressive process that prevents survival and clonogenic expansion of potentially malignant cells carrying deleterious mutations.23, 24 Given their essential role as regulators of apoptosis initiation and execution, caspases have been proposed to possess tumour suppressor functions. However, only caspase-2 and caspase-8 have been experimentally demonstrated to have such a role.15, 25, 26 As alluded to above and discussed in a number of recent reviews,9, 27 previous studies focusing on the role of caspase-2 in apoptosis have yielded somewhat contradictory findings, questioning the significance of caspase-2 in cell death. More recently, attention has been focused on its potential non-apoptotic functions.
There are several lines of evidence that implicate a role for caspase-2 in human cancers. Much of the earlier work comes from studies investigating haematological malignancies. For example, the human gene encoding caspase-2 is located in a region of chromosome (7q34-35) that is frequently deleted in leukaemia.11 In addition to the identification of caspase-2 somatic mutations in gastric and colorectal cancers,28 reduced expression of caspase-2 has been correlated with chemotherapeutic drug resistance in childhood acute lymphoblastic leukaemia (ALL).29 In line with these findings, reduced caspase-2 protein levels have been associated with poor prognosis and outcome in patients with acute myelogenous leukaemia (AML) and ALL.12, 14 Further, analysis of caspase-2 expression from publicly available microarray data demonstrates that, in addition to various haematological malignancies, caspase-2 is downregulated in multiple solid tumours such as hepatocellular and invasive breast carcinomas, ovarian adenocarcinomas and glioblastomas.30
The loss of caspase-2 locus and expression in tumours is consistent with a putative tumour suppressor function. Paradoxically, somatic mutations of caspase-2 are rare in various human cancers.31 Therefore, direct mutational inactivation of caspase-2 (unless deleted, as in haematological malignancies carrying chromosome 7q deletions/aberrations) is unlikely to explain its reduced expression or loss of function in human tumours. However, indirect mechanisms such as miRNA-mediated or epigenetic silencing of caspase-2 cannot be ruled out.
Caspase-2 Function in Protecting Against Cell Transformation
Clinical studies implicating caspase-2 in human cancers have prompted further investigation into its potential function in tumour suppression. There is growing evidence to suggest that caspase-2 is an important barrier that protects against cell transformation. In line with this, loss of caspase-2 slightly increases the rate of proliferation in primary, spontaneously immortalized and E1A/Ras-transformed MEFs, suggesting defective cell cycle regulation in caspase-2-deficient cells.15, 32 Consistent with their increased proliferation rate, E1A/Ras-transformed Casp2−/− MEFs displayed an increased ability to form colonies in soft agar.15 Interestingly, this enhanced ability of anchorage-independent growth in E1A/Ras-transformed Casp2−/− MEFs coincides with an increased tumorigenic potential.15 These observations link the functions of caspase-2 in the regulation of growth and proliferation with its ability to safeguard against transformation. Importantly, the catalytic activity of caspase-2 has been shown to be required for its ability to regulate proliferation and suppress transformation.30
Caspase-2 as a Tumour Suppressor – Evidence from Mouse Models
On the basis of in vitro studies describing a role for caspase-2 in protecting against cell transformation, loss of caspase-2 function would be predicted to enhance tumour susceptibility in vivo. Contrary to this prediction, caspase-2 deletion is not sufficient to induce spontaneous tumorigenesis in mice.21 However, when crossed into an oncogenic background, the function of caspase-2 in tumorigenesis becomes apparent. Deletion of caspase-2 in Eμ-Myc transgenic mice accelerates lymphomagenesis, providing the first direct line of evidence for a tumour suppressor function for caspase-2.15 These results with Eμ-Myc/Casp2−/− mice have been confirmed by independent studies from another laboratory.33 Interestingly, loss of even a single allele of caspase-2 is sufficient to augment lymphomagenesis in this model, suggesting that caspase-2 may be a haploinsufficient tumour suppressor.15 These observations are consistent with the reduced expression of caspase-2 in human cancer, which may also be potentially associated with loss of heterozygosity.
The extent of caspase-2 in tumour suppression has been further explored using various models of carcinogen-induced tumorigenesis. However, caspase-2 failed to suppress tumorigenesis induced by ionizing radiation or 3-methylcholanthrene.33 These findings suggest that caspase-2 is not a general tumour suppressor and may indicate a context-specific function for this caspase.
More recently, it was shown that deletion of caspase-2 causes a modest increase in tumour onset in the mouse mammary tumour virus (MMTV) model of breast carcinoma.34 Interestingly, this was only observed in multiparous mice with no significant differences in tumour onset observed between MMTV/Casp2+/+ and MMTV/Casp2−/− nulliparous mice.34 Unlike female nulliparous mice, mammary glands from female multiparous mice undergo extensive proliferation and differentiation during pregnancy and lactation.35 Given that loss of caspase-2 augments tumorigenesis only in multiparous mammary glands, caspase-2 function in protecting against cell transformation appears to become more important in highly proliferative tissues that experience increased replicative and oncogenic stress compared with tissues with lower proliferative activity. These studies show that not only does the tumour suppressor function of caspase-2 extend beyond Myc-driven malignancies but it is also a suppressor of epithelial tumours.
Taken together, studies from mouse models have demonstrated that the function of caspase-2 in tumour suppression becomes important under conditions of oncogenic stress rather than suppressing tumorigenesis at the level of initiation. This is supported by the fact that Casp2−/− mice do not develop spontaneous tumours, but caspase-2 deficiency in tumour-prone mice can potentiate tumorigenesis.
Possible Mechanism(s) of Tumour Suppression by Caspase-2
As discussed above, in vivo mouse models have been instrumental in establishing caspase-2 as a tumour suppressor. Given its functions in protecting against cell transformation and suppressing tumour development, it is reasonable to speculate that these two functions are interdependent. Attention has now been focused on identifying and characterizing the mechanisms by which caspase-2 exerts these functions. Given that cancer cells frequently display aberrant proliferation, genomic instability and an impaired response to DNA damage,23 involvement of caspase-2 in the regulation of these processes likely contributes to its function in suppressing cell transformation and tumorigenesis. On the basis of current experimental evidence, caspase-2 appears to function by suppressing these interdependent hallmarks of cancer. A detailed understanding of these processes will be essential for deciphering the mechanisms by which caspase-2 exerts its tumour suppressor function.
Regulation of cell growth and proliferation
For cells to acquire a malignant phenotype, they must be able to suppress growth inhibitory signals and establish the ability of replicative immortality.23 In line with aberrant growth signalling and an increased susceptibility to transform in the absence of caspase-2, primary Casp2−/− MEFs were found to rapidly undergo spontaneous immortalization in culture.32 These findings suggest that loss of caspase-2 is associated with deregulation of cell proliferation. This tendency to rapidly immortalize in culture most likely stems from the ability of Casp2−/− MEFs to readily escape replicative senescence.32 Interestingly, escape from senescence coincided with reduced expression of the anti-proliferative cyclin-dependent kinase inhibitors p19ARF, p16INK4a and p21CIP1/WAF1 in late-passage Casp2−/− MEFs.32 These findings suggest that caspase-2 may protect against cell transformation by regulation of senescence, a known mechanism that prevents the propagation of cells harbouring potentially harmful mutations or experiencing oncogenic stress.23 In line with these findings, caspase-2-deficient tumours from mice have been shown to display an increased proliferation rate (unpublished data).33, 34 Therefore, disruptions in pathways controlling growth and proliferation may account, at least in part, for the increased tumorigenic potential of Casp2−/− MEFs (Figure 2).
Figure 2.

Caspase-2 in growth signalling and DNA damage response pathways. Inappropriate activation of oncogenic signalling pathways and DNA damage lead to cellular stress that promotes cell transformation. In response to oncogenic stress and DNA damage, tumour suppressor pathways are activated, which mediate anti-proliferative responses such as apoptosis, senescence and cell cycle arrest. These cellular responses form critical barriers that protect against tumorigenesis. Caspase-2 has been shown to regulate multiple components of these pathways controlling proliferation and the response to DNA damage
DNA damage response (DDR)
There is growing evidence implicating caspase-2 in the regulation of the DDR. However, contradictory findings have made it difficult to elucidate the physiological relevance and mechanistic basis of caspase-2 function in the DDR. On the basis of current experimental evidence, it is clear that the specificity of caspase-2 function in the response to genotoxic stress is highly context dependent. Cell type, in addition to the nature and extent of damage sustained by a cell, seem to be important determinants of the extent of caspase-2 involvement in the response to cell stress. Nevertheless, some in vitro studies have provided robust experimental evidence that caspase-2 regulates the p53-dependent DDR.32, 34 Following treatment with ionizing radiation, Casp2−/− MEFs show impaired transactivation of p53 target genes involved in cell cycle arrest and apoptosis, including puma, noxa and p21CIP1/WAF1.32 Impaired p53 activity in the absence of caspase-2 may partly explain cell proliferation defects and attenuated DDR in Casp2−/−MEFs.
The ubiquitin ligase Mdm2, a negative regulator of p53, was recently identified as a caspase-2 substrate.36 Caspase-2-mediated cleavage of Mdm2 in response to DNA damage generated an Mdm2 fragment that stabilized p53 via a positive feedback loop.36 This mechanism has been proposed to explain the reduced p53 activity and aberrant DDR observed in Casp2−/− MEFs.32 However, we have demonstrated Mdm-2-independent modulation of p53 activity by caspase-2.32 Therefore, the way in which caspase-2 controls p53 activity may be more complex than originally anticipated.
Caspase-2 has also been implicated in p53-independent pathways, highlighting the increasing complexity of caspase-2 in the modulation of the DDR. Checkpoint kinase 1 (Chk1) was shown to suppress a caspase-2-dependent apoptosis pathway in p53-deficient cells.37 Chk1 inhibition restored sensitivity to gamma-radiation-induced apoptosis independent of mitochondrial involvement and caspase-3.37 This pathway is dependent on the ataxia telangiectasia-mutated (ATM) kinase, which upon activation by DNA damage phosphorylates the p53-induced death domain protein (PIDD), triggering the RIP-associated protein with a death domain (RAIDD) recruitment and assembly of the PIDDosome complex, leading to caspase-2 activation.38 This Chk1-suppressed pathway has been proposed to be a mechanism of apoptosis that ensures deletion of cells that sustain DNA damage in the presence of compromised checkpoint surveillance.37, 38 Importantly, these findings highlight that some potentially unidentified functions of caspase-2 may become apparent only under very specific experimental or physiological conditions.
Caspase-2 has been shown to affect the function of multiple proteins that are known to regulate cell proliferation and apoptosis (Figure 2). On the basis of available in vitro data, we envisage that caspase-2 most likely fine-tunes multiple pathways that converge on the regulation of the DDR and on cell proliferation (Figure 2). Therefore, one of the primary functions of caspase-2 may be to ensure that a robust response is triggered and maintained under specific stress conditions.
Given the nuclear functions of various cell cycle and DDR regulators, involvement of caspase-2 in these processes may explain its constitutive nuclear localization, a unique feature among members of the caspase family.9 Therefore, changes in the sub-cellular localization of caspase-2 in response to specific stimuli may permit differential access to the cytoplasmic apoptotic machinery and nuclear DDR components, reflecting its bimodal functions in apoptotic and non-apoptotic pathways.
Maintenance of genomic stability
Strict regulation of proliferation and cell cycle checkpoints is essential for maintaining genomic stability.39, 40 Given the inextricable link between deregulation of proliferation, genomic instability and tumorigenesis,41 a role for caspase-2 in maintaining genomic stability may be one of the main mechanisms by which caspase-2 exerts its tumour suppressor function. Studies using MEFs have demonstrated that, with serial passaging in culture, loss of caspase-2 promotes aneuploidy.32 These in vitro observations have been extended and validated in vivo with B-cell lymphomas derived from Eμ-Myc/Casp2−/− mice, which display an increased frequency of aneuploidy as well as reduced telomere length (Figure 3).32 Consistent with these findings, increased genomic instability and aberrant mitoses were recently reported in breast epithelial tumours derived from MMTV/Casp2−/− mice.34 Further, we have found that deletion of caspase-2 in other mouse models is strongly associated with loss of genomic integrity and increased incidence of tumorigenesis (unpublished data). These important findings provide direct evidence that caspase-2-deficient tumours frequently display aneuploidy, implicating a role for caspase-2 in the maintenance of genomic stability in vitro and in vivo. Given that aneuploidy is a well-established hallmark of cancer, the ability of caspase-2 to prevent genomic instability is an attractive candidate mechanism that could potentially explain its tumour suppressor function.
Figure 3.

Caspase-2 fine-tunes cellular responses to protect against oxidative and oncogenic stress. We propose that, in the absence of caspase-2, cells become more susceptible to stress conditions. Therefore, when challenged, caspase-2 knockout animals have a reduced ability to counteract these adverse conditions, leading to enhanced oncogenic and oxidative stress. At the phenotypic level, this manifests as premature ageing and increased tumour susceptibility (lower panel). This is exemplified in tumours derived from caspase-2-deficient Eμ-Myc transgenic mice, which exhibit increased aneuploidy (an example is shown in the upper panel)
In addition to its role in DNA damage-induced apoptosis, caspase-2 is also known to be required for cell death induced by cytoskeletal disruption.19 In line with these findings, caspase-2 has also been implicated in a mode of cell death that is induced following aberrant mitoses called mitotic catastrophe.42, 43 Despite the lack of a clear molecular definition for mitotic catastrophe, it has been proposed to be important for the deletion of aneuploid cells generated from abnormal mitotic events.44 Therefore, mitotic catastrophe has been viewed as a tumour-suppressive process that is important for maintaining genomic stability.44 Thus, it is plausible that caspase-2 is required to execute cell death in cells that have become aneuploid through defective mitotic processes such as improper chromosome segregation. This hypothesis fits with current experimental data demonstrating increased genomic instability in caspase-2-deficient tumours. In the absence of caspase-2, aneuploid cells may persist and acquire further oncogenic lesions that promote tumorigenesis.
Caspase-2 has also been implicated in the regulation of the G2/M checkpoint,32, 33, 34 which promotes G2 arrest and subsequent inhibition of mitotic entry in the presence of unrepaired DNA damage.40 Inappropriate mitotic progression in cells harbouring DNA breaks has been shown to cause aberrant mitoses leading to aneuploidization via improper chromosome attachment and segregation.40 Consistent with in vitro studies demonstrating a defective G2/M checkpoint in caspase-2-deficient cells, an increased mitotic index has been observed in caspase-2-deficient tumours derived from Eμ-Myc and MMTV transgenic mice.33, 34 These findings provide evidence for increased proliferation and impaired cell cycle progression in caspase-2-deficient tumours, suggesting that caspase-2 may regulate these processes in vivo.
In support of a role for caspase-2 in cell death induced by aberrant mitotic events, epithelial tumours derived from MMTV/Casp2−/− mice displayed a striking increase in the rate of karyomegaly and multinucleation.34 Further, MMTV/Casp2−/− tumours showed a high frequency of cells with bizarre mitoses displaying abnormal spindle asters, a defect not observed in MMTV/Casp2+/+ tumours.34 From these studies, we can infer that the functions of caspase-2 in the maintenance of genomic stability and cell cycle checkpoint regulation are potentially linked and that these processes are likely to contribute to its role in tumour suppression.
Perspectives
Although earlier observations in knockout mice ruled out an essential physiological function for caspase-2 in developmental cell death and inflammation, as discussed here experimental evidence has begun to emerge, opening new and previously unexplored avenues, rekindling interest in this enigmatic caspase. In particular, the focus of recent studies has shifted towards exploring the role of caspase-2 in suppressing tumorigenesis. Clinical observations together with extensive in vitro and in vivo experimental evidence have strengthened our understanding of the physiological functions of caspase-2 in disease, with clear roles in genomic stability and tumour suppression. However, as discussed above and listed in Table 1, there are a number of caveats that caution against a major, essential and widespread function of caspase-2 in tumour suppression. Clearly, more studies with different in vivo models and further mechanistic studies directly linking caspase-2 to its apparently diverse modes of functions are required.
Table 1. Evidence implicating caspase-2 in tumour suppression.
| Evidence | For | Against |
|---|---|---|
| Correlative data from human tumours | Reduced expression of caspase-2 in AML and ALL correlates with poor prognosis12, 14 Reduced levels associated with drug resistance in childhood ALL29 Chromosomal locus containing caspase-2 gene frequently deleted in cancer11 Somatic mutations in some gastric tumours28 | Somatic mutations rare in human tumours31 Not all tumour types show reduced expression of caspase-2 |
| In vitro studies (cell biology) | Casp2−/− MEFs display: Increased proliferation rate15, 32, 34 Aberrant cell cycle checkpoint regulation15, 32 Increased genomic instability32, 34 Impaired DNA damage response32, 34 Increased potential to immortalize and transform by oncogenes15, 32 | Caspase-2 is not essential for apoptosis induced by many stimuli |
| In vivo studies (mouse models) | Caspase-2 is a suppressor of tumorigenesis in: Eμ-Myc transgenic mice15 MMTV transgenic mice34 Other mouse models of tumorigenesis | Aged Casp2−/− mice do not have an increased incidence of tumours21 Not all tumour models are affected by loss of caspase-2 |
On the basis of our current understanding of caspase-2, one of the predominant functions of this caspase appears to be its role in protection against cellular stress (Figure 3). Therefore, in the absence of caspase-2, cells are unable to efficiently counteract challenges such as oxidative and oncogenic stresses, making caspase-2-deficient mice more susceptible to these stress conditions. Our proposed model in Figure 3 may explain the physiological basis upon which caspase-2 mitigates stress conditions, such as in the context of ageing and tumorigenesis.
A lack of known caspase-2 substrates has been a major hindrance to dissecting out the mechanisms that underlie the physiological functions of caspase-2. Moreover, substrate identification has been hampered by the highly context-dependent functions of caspase-2 and the extensive promiscuity in substrate specificity among members of the caspase family. However, with the advent of increasingly sophisticated proteomics technologies, identification of specific substrates linking caspase-2 to its physiological functions is keenly awaited.
Acknowledgments
The caspase-2 work in our laboratory is supported by the National Health and Medical Research Council (NHMRC) Project Grants 1021456 and 1043057. JP is supported by an Australian Postgraduate Award, LD by a Cancer Council Senior Fellowship and SK by NHMRC Senior Principal Research Fellowship (1002863).
Glossary
- Casp2−/−
caspase-2 knockout
- MEFs
mouse embryonic fibroblasts
- DDR
DNA damage response
- ATM
ataxia telangiectasia mutated
- ROS
reactive oxygen species
The authors declare no conflict of interest.
Footnotes
Edited by G Melino
References
- Kumar S, Kinoshita M, Noda M, Copeland NG, Jenkins NA. Induction of apoptosis by the mouse Nedd2 gene, which encodes a protein similar to the product of the Caenorhabditis elegans cell death gene ced-3 and the mammalian IL-1 beta-converting enzyme. Genes Dev. 1994;8:1613–1626. doi: 10.1101/gad.8.14.1613. [DOI] [PubMed] [Google Scholar]
- Kumar S, Tomooka Y, Noda M. Identification of a set of genes with developmentally down-regulated expression in the mouse brain. Biochem Biophys Res Commun. 1992;185:1155–1161. doi: 10.1016/0006-291x(92)91747-e. [DOI] [PubMed] [Google Scholar]
- Miura M, Zhu H, Rotello R, Hartwieg EA, Yuan J. Induction of apoptosis in fibroblasts by IL-1 beta-converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3. Cell. 1993;75:653–660. doi: 10.1016/0092-8674(93)90486-a. [DOI] [PubMed] [Google Scholar]
- Kumar S. ICE-like proteases in apoptosis. Trends Biochem Sci. 1995;20:198–202. doi: 10.1016/s0968-0004(00)89007-6. [DOI] [PubMed] [Google Scholar]
- Kumar S. Caspase function in programmed cell death. Cell Death Differ. 2007;14:32–43. doi: 10.1038/sj.cdd.4402060. [DOI] [PubMed] [Google Scholar]
- Dorstyn L, Colussi PA, Quinn LM, Richardson H, Kumar S. DRONC, an ecdysone-inducible Drosophila caspase. Proc Natl Acad Sci USA. 1999;96:4307–4312. doi: 10.1073/pnas.96.8.4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colussi PA, Harvey NL, Kumar S. Prodomain-dependent nuclear localization of the caspase-2 (Nedd2) precursor. A novel function for a caspase prodomain. J Biol Chem. 1998;273:24535–24542. doi: 10.1074/jbc.273.38.24535. [DOI] [PubMed] [Google Scholar]
- Baliga BC, Colussi PA, Read SH, Dias MM, Jans DA, Kumar S. Role of prodomain in importin-mediated nuclear localization and activation of caspase-2. J Biol Chem. 2003;278:4899–4905. doi: 10.1074/jbc.M211512200. [DOI] [PubMed] [Google Scholar]
- Kumar S. Caspase 2 in apoptosis, the DNA damage response and tumour suppression: enigma no more. Nat Rev Cancer. 2009;9:897–903. doi: 10.1038/nrc2745. [DOI] [PubMed] [Google Scholar]
- Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A, et al. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev. 1998;12:1304–1314. doi: 10.1101/gad.12.9.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, White DL, Takai S, Turczynowicz S, Juttner CA, Hughes TP. Apoptosis regulatory gene NEDD2 maps to human chromosome segment 7q34-35, a region frequently affected in haematological neoplasms. Hum Genet. 1995;95:641–644. doi: 10.1007/BF00209480. [DOI] [PubMed] [Google Scholar]
- Estrov Z, Thall PF, Talpaz M, Estey EH, Kantarjian HM, Andreeff M, et al. Caspase 2 and caspase 3 protein levels as predictors of survival in acute myelogenous leukemia. Blood. 1998;92:3090–3097. [PubMed] [Google Scholar]
- Hofmann WK, de Vos S, Tsukasaki K, Wachsman W, Pinkus GS, Said JW, et al. Altered apoptosis pathways in mantle cell lymphoma detected by oligonucleotide microarray. Blood. 2001;98:787–794. doi: 10.1182/blood.v98.3.787. [DOI] [PubMed] [Google Scholar]
- Faderl S, Thall PF, Kantarjian HM, Talpaz M, Harris D, Van Q, et al. Caspase 2 and caspase 3 as predictors of complete remission and survival in adults with acute lymphoblastic leukemia. Clin Can Res. 1999;5:4041–4047. [PubMed] [Google Scholar]
- Ho LH, Taylor R, Dorstyn L, Cakouros D, Bouillet P, Kumar S. A tumor suppressor function for caspase-2. Proc Natl Acad Sci USA. 2009;106:5336–5341. doi: 10.1073/pnas.0811928106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly LA, Ekert P, Harvey N, Marsden V, Cullen L, Vaux DL, et al. Caspase-2 is not required for thymocyte or neuronal apoptosis even though cleavage of caspase-2 is dependent on both Apaf-1 and caspase-9. Cell Death Differ. 2002;9:832–841. doi: 10.1038/sj.cdd.4401033. [DOI] [PubMed] [Google Scholar]
- Marsden VS, Ekert PG, Van Delft M, Vaux DL, Adams JM, Strasser A. Bcl-2-regulated apoptosis and cytochrome c release can occur independently of both caspase-2 and caspase-9. J Cell Biol. 2004;165:775–780. doi: 10.1083/jcb.200312030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchier-Hayes L, Oberst A, McStay GP, Connell S, Tait SW, Dillon CP, et al. Characterization of cytoplasmic caspase-2 activation by induced proximity. Mol Cell. 2009;35:830–840. doi: 10.1016/j.molcel.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho LH, Read SH, Dorstyn L, Lambrusco L, Kumar S. Caspase-2 is required for cell death induced by cytoskeletal disruption. Oncogene. 2008;27:3393–3404. doi: 10.1038/sj.onc.1211005. [DOI] [PubMed] [Google Scholar]
- Troy CM, Rabacchi SA, Hohl JB, Angelastro JM, Greene LA, Shelanski ML. Death in the balance: alternative participation of the caspase-2 and -9 pathways in neuronal death induced by nerve growth factor deprivation. J Neurosci. 2001;21:5007–5016. doi: 10.1523/JNEUROSCI.21-14-05007.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalini S, Dorstyn L, Wilson C, Puccini J, Ho L, Kumar S. Impaired antioxidant defence and accumulation of oxidative stress in caspase-2-deficient mice. Cell Death Differ. 2012;19:1370–1380. doi: 10.1038/cdd.2012.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Padalecki SS, Chaudhuri AR, De Waal E, Goins BA, Grubbs B, et al. Caspase-2 deficiency enhances aging-related traits in mice. Mech Ageing Dev. 2007;128:213–221. doi: 10.1016/j.mad.2006.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Olsson M, Zhivotovsky B. Caspases and cancer. Cell Death Differ. 2011;18:1441–1449. doi: 10.1038/cdd.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teitz T, Wei T, Valentine MB, Vanin EF, Grenet J, Valentine VA, et al. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat Med. 2000;6:529–535. doi: 10.1038/75007. [DOI] [PubMed] [Google Scholar]
- Krelin Y, Zhang L, Kang TB, Appel E, Kovalenko A, Wallach D. Caspase-8 deficiency facilitates cellular transformation in vitro. Cell Death Differ. 2008;15:1350–1355. doi: 10.1038/cdd.2008.88. [DOI] [PubMed] [Google Scholar]
- Fava LL, Bock FJ, Geley S, Villunger A. Caspase-2 at a glance. J Cell Sci. 2012;125:5911–5915. doi: 10.1242/jcs.115105. [DOI] [PubMed] [Google Scholar]
- Kim MS, Kim HS, Jeong EG, Soung YH, Yoo NJ, Lee SH. Somatic mutations of caspase-2 gene in gastric and colorectal cancers. Pathol Res Pract. 2011;207:640–644. doi: 10.1016/j.prp.2011.08.004. [DOI] [PubMed] [Google Scholar]
- Holleman A, den Boer ML, Kazemier KM, Beverloo HB, von Bergh AR, Janka-Schaub GE, et al. Decreased PARP and procaspase-2 protein levels are associated with cellular drug resistance in childhood acute lymphoblastic leukemia. Blood. 2005;106:1817–1823. doi: 10.1182/blood-2004-11-4296. [DOI] [PubMed] [Google Scholar]
- Ren K, Lu J, Porollo A, Du C. Tumor-suppressing function of caspase-2 requires catalytic site Cys-320 and site Ser-139 in mice. J Biol Chem. 2012;287:14792–14802. doi: 10.1074/jbc.M112.347625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Chung NG, Yoo NJ, Lee SH. Somatic mutation of proapoptotic caspase-2 gene is rare in acute leukemias and common solid cancers. Eur J Haematol. 2011;86:449–450. doi: 10.1111/j.1600-0609.2011.01591.x. [DOI] [PubMed] [Google Scholar]
- Dorstyn L, Puccini J, Wilson CH, Shalini S, Nicola M, Moore S, et al. Caspase-2 deficiency promotes aberrant DNA-damage response and genetic instability. Cell Death Differ. 2012;19:1288–1298. doi: 10.1038/cdd.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzl C, Peintner L, Krumschnabel G, Bock F, Labi V, Drach M, et al. PIDDosome-independent tumor suppression by Caspase-2. Cell Death Differ. 2012;19:1722–1732. doi: 10.1038/cdd.2012.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons MJ, McCormick L, Janke L, Howard A, Bouchier-Hayes L, Green DR. Genetic deletion of caspase-2 accelerates MMTV/c-neu-driven mammary carcinogenesis in mice. Cell Death Differ. 2013;20:1174–1182. doi: 10.1038/cdd.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strange R, Li F, Saurer S, Burkhardt A, Friis RR. Apoptotic cell death and tissue remodelling during mouse mammary gland involution. Development. 1992;115:49–58. doi: 10.1242/dev.115.1.49. [DOI] [PubMed] [Google Scholar]
- Oliver TG, Meylan E, Chang GP, Xue W, Burke JR, Humpton TJ, et al. Caspase-2-mediated cleavage of Mdm2 creates a p53-induced positive feedback loop. Mol Cell. 2011;43:57–71. doi: 10.1016/j.molcel.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidi S, Sanda T, Kennedy RD, Hagen AT, Jette CA, Hoffmans R, et al. Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell. 2008;133:864–877. doi: 10.1016/j.cell.2008.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando K, Kernan JL, Liu PH, Sanda T, Logette E, Tschopp J, et al. PIDD death-domain phosphorylation by ATM controls prodeath versus prosurvival PIDDosome signaling. Mol Cell. 2012;47:681–893. doi: 10.1016/j.molcel.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- Lobrich M, Jeggo PA. The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat Rev Cancer. 2007;7:861–869. doi: 10.1038/nrc2248. [DOI] [PubMed] [Google Scholar]
- Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet. 2012;13:189–203. doi: 10.1038/nrg3123. [DOI] [PubMed] [Google Scholar]
- Castedo M, Perfettini JL, Roumier T, Valent A, Raslova H, Yakushijin K, et al. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene. 2004;23:4362–4370. doi: 10.1038/sj.onc.1207572. [DOI] [PubMed] [Google Scholar]
- Mansilla S, Priebe W, Portugal J. Mitotic catastrophe results in cell death by caspase-dependent and caspase-independent mechanisms. Cell Cycle. 2006;5:53–60. doi: 10.4161/cc.5.1.2267. [DOI] [PubMed] [Google Scholar]
- Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011;12:385–392. doi: 10.1038/nrm3115. [DOI] [PubMed] [Google Scholar]
- Baliga BC, Read SH, Kumar S. The biochemical mechanism of caspase-2 activation. Cell Death Differ. 2004;11:1234–1241. doi: 10.1038/sj.cdd.4401492. [DOI] [PubMed] [Google Scholar]
- Butt AJ, Harvey NL, Parasivam G, Kumar S. Dimerization and autoprocessing of the Nedd2 (caspase-2) precursor requires both the prodomain and the carboxyl-terminal regions. J Biol Chem. 1998;273:6763–6768. doi: 10.1074/jbc.273.12.6763. [DOI] [PubMed] [Google Scholar]
- Nutt LK, Margolis SS, Jensen M, Herman CE, Dunphy WG, Rathmell JC, et al. Metabolic regulation of oocyte cell death through the CaMKII-mediated phosphorylation of caspase-2. Cell. 2005;123:89–103. doi: 10.1016/j.cell.2005.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S, Lee Y, Kim W, Ko H, Choi H, Kim K. Caspase-2 primes cancer cells for TRAIL-mediated apoptosis by processing procaspase-8. EMBO J. 2005;24:3532–3542. doi: 10.1038/sj.emboj.7600827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen JL, Johnson CE, Freel CD, Parrish AB, Day JL, Buchakjian MR, et al. Restraint of apoptosis during mitosis through interdomain phosphorylation of caspase-2. EMBO J. 2009;28:3216–3227. doi: 10.1038/emboj.2009.253. [DOI] [PMC free article] [PubMed] [Google Scholar]