

Nucleophilic substitutions at phosphorus comprise one of the most important classes of reactions in biology. Phosphate diester substitution reactions are catalyzed by nucleases and polymerases and are critical in DNA replication and transcription. Phosphate monoester (phosphoryl transfer) reactions are catalyzed by GTPases, ATPases, protein, and small molecule kinases, protein, and small molecule phosphatases. These enzymes play diverse roles in energy regulation, cell signaling, ion and small molecule transport, and nucleotide synthesis. There have been intensive efforts to try to understand the details of phosphoryl transfer reactions extending from nonenzymatic (or enzyme model) systems to the mechanisms of the enzymatic reactions, as exemplified by the study by Cho et al. in the current issue of PNAS (1).
A full and convincing explanation at the quantum mechanical level has not been made as to why dissociative transition states should be preferred for nonenzymatic phosphoryl transfer reactions.
From the many decades of work on small molecules, consensus for a dissociative transition state, akin to an SN1 reaction in organic chemistry, has been reached by most investigators (2, 3). In such a transition state, the bond between the phosphorus and leaving group has largely broken before the formation of a bond between the incoming nucleophile and phosphorus. The role of the nucleophile is diminished, and the formation of a highly reactive metaphosphate-like species is central in a dissociative transition state. This contrasts with the transition state of an associative mechanism, which occurs in phosphate triester substitution reactions, in which a pentavalent-like species is generated. In associative transition states, there is a significant degree of bond formation between the incoming nucleophile and the attacked phosphorus before leaving group departure. Although a full and convincing explanation at the quantum mechanical level has not yet been made as to why dissociative transition states should be preferred for nonenzymatic phosphoryl transfer reactions, the simplest explanation is based on the concept that the negative charges on two of the phosphate oxygens repel incoming nucleophiles and stabilize a metaphosphosphate-like species.
More difficult to address is how enzymes catalyze phosphoryl transfer reactions. Enzymatic phosphoryl transfer reactions can occur either by direct transfer of the phosphoryl group to an incoming nucleophile or through the formation of a covalent intermediate (which does not rule out a dissociative transition state). Examples of the latter group of phosphotransferases include alkaline phosphatase, protein tyrosine phosphatases, nucleotide diphosphate kinase, and P-ATPases. In these two-stage reactions, an enzyme nucleophile forms a stable intermediate between the attacked phosphorus while the departing leaving group is expelled. This intermediate is then attacked by an incoming nucleophile, in the case of alkaline phosphatase a water molecule, which regenerates the free enzyme and final product. Phosphotransferases use a variety of enzyme nucleophiles, including: the carboxy group of aspartate (P-ATPases), the thiol of cysteine (protein tyrosine phosphatases), the hydroxy group of serine (alkaline phosphatase), and the imidizaole of histidine (nucleoside diphosphate kinase) (4–7). The linkages involved may or may not be stable in small-molecule systems, but in the context of the enzymes, they can be quite reactive to facilitate turnover. Consequently, studying their behavior in the structural context of the folded protein may be extremely difficult.
Phosphoaspartyl presents a prime example of the problem. The free energy for hydrolysis of phosphoaspartyl hydrolysis is estimated to be several kcal (1 kcal = 4.18 kJ)/mol more negative than hydrolytic cleavage of the γ phosphate of ATP, a very favorable process thermodynamically (8). As a consequence, suitable small-molecule analogs that mimic the properties of the phosphoaspartyl residues but have greater stability have been sought. BeF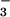 appears to be one such example (9, 10).
appears to be one such example (9, 10).
Cho et al. (1) have shown the general utility of this molecular probe by determining the crystal structure of phosphoserine phosphatase (PSP) bound to BeF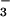 and comparing it to the structure of a related protein CheY, also bound to BeF
and comparing it to the structure of a related protein CheY, also bound to BeF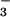 (11). Both PSP and CheY are members of the haloacid dehalogenase (HAD) superfamily of enzymes that includes a number of phosphotransferases among its members, such as the response regulators of two component signal transduction systems (e.g., CheY), phosphomannomutases, and P-ATPases (e.g., Ca-ATPase). A number of residues are highly conserved in the primary structures of HAD family phosphotransferases (12–14). These residues are also conserved at the tertiary level, as the large number of three-dimensional structures of HAD family members demonstrates (see refs. 1, 11, and 15–17 for examples). For the HAD family phosphotransferases, five conserved residues converge to help form the active sites of these enzymes. For PSP, these residues are Asp-11, Ser-99, Lys-144, Asp-167, and Asp-171. In other HAD enzymes, a Thr replaces this Ser (e.g., CheY). The general roles of most of these active-site residues and the analogous residues in other HAD enzymes can be surmised from both current and previously determined three-dimensional structures. For example, Asp-11, Asp-167, and Asp-171 are all Mg2+ ligands; however, the carboxylate of Asp-11 plays an additional role as the enzyme nucleophile. The PSP⋅BeF
(11). Both PSP and CheY are members of the haloacid dehalogenase (HAD) superfamily of enzymes that includes a number of phosphotransferases among its members, such as the response regulators of two component signal transduction systems (e.g., CheY), phosphomannomutases, and P-ATPases (e.g., Ca-ATPase). A number of residues are highly conserved in the primary structures of HAD family phosphotransferases (12–14). These residues are also conserved at the tertiary level, as the large number of three-dimensional structures of HAD family members demonstrates (see refs. 1, 11, and 15–17 for examples). For the HAD family phosphotransferases, five conserved residues converge to help form the active sites of these enzymes. For PSP, these residues are Asp-11, Ser-99, Lys-144, Asp-167, and Asp-171. In other HAD enzymes, a Thr replaces this Ser (e.g., CheY). The general roles of most of these active-site residues and the analogous residues in other HAD enzymes can be surmised from both current and previously determined three-dimensional structures. For example, Asp-11, Asp-167, and Asp-171 are all Mg2+ ligands; however, the carboxylate of Asp-11 plays an additional role as the enzyme nucleophile. The PSP⋅BeF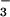 complex also identifies a hydrogen bond between Ser-99 and a nonbridge oxygen of the phosphoryl group and a series of salt bridges between Lys-144 and both the phosphoryl group and Asp-171 (Fig. 1). These interactions are likely important for ground-state stabilization.
complex also identifies a hydrogen bond between Ser-99 and a nonbridge oxygen of the phosphoryl group and a series of salt bridges between Lys-144 and both the phosphoryl group and Asp-171 (Fig. 1). These interactions are likely important for ground-state stabilization.
Figure 1.

Active-site organization of PSP in complex with BeF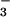 .
.
Cho et al. demonstrate the general utility of BeF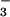 for the study of phosphoaspartyl-containing proteins by overlaying the positions of active-site residues in the PSP⋅BeF
for the study of phosphoaspartyl-containing proteins by overlaying the positions of active-site residues in the PSP⋅BeF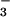 with the CheY⋅BeF
with the CheY⋅BeF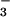 complex (1). This comparison shows that the functionally relevant portions of these residues can be almost perfectly overlapped, despite the fact that a circular permutation, which exists between CheY and PSP, causes Lys-144 and Asp-171 and their analogous residues in CheY to originate from different points in space. The observed structural similarity between these two enzymes when bound to BeF
complex (1). This comparison shows that the functionally relevant portions of these residues can be almost perfectly overlapped, despite the fact that a circular permutation, which exists between CheY and PSP, causes Lys-144 and Asp-171 and their analogous residues in CheY to originate from different points in space. The observed structural similarity between these two enzymes when bound to BeF also served to confirm conformational changes that are apparent between the CheY⋅BeF
also served to confirm conformational changes that are apparent between the CheY⋅BeF complex and CheY on its own. These changes include the movement of Thr-87 (the equivalent of Ser-99 in PSP) to form a hydrogen bond with one of the fluorine atoms in the CheY⋅BeF
complex and CheY on its own. These changes include the movement of Thr-87 (the equivalent of Ser-99 in PSP) to form a hydrogen bond with one of the fluorine atoms in the CheY⋅BeF complex, and the repositioning of Lys-109 such that it links the phosphoryl group to Asp-12 through a similar series of salt bridges. These observed active-site conformational changes in CheY are thought to cause conformational changes in the C terminus of this protein that are important in signal transduction. The observed similarities between the two structures led Cho et al. to suggest that this ground-state structural arrangement may be a common feature of other HAD phosphotransferases, and that movements by analogous active-site residues in the P-ATPases are likely coupled to ion transport by these enzymes (1).
complex, and the repositioning of Lys-109 such that it links the phosphoryl group to Asp-12 through a similar series of salt bridges. These observed active-site conformational changes in CheY are thought to cause conformational changes in the C terminus of this protein that are important in signal transduction. The observed similarities between the two structures led Cho et al. to suggest that this ground-state structural arrangement may be a common feature of other HAD phosphotransferases, and that movements by analogous active-site residues in the P-ATPases are likely coupled to ion transport by these enzymes (1).
The structural conservation observed between CheY and PSP is of interest not only because of its implications for the biological function of these and other enzymes, particularly the P-ATPases, but also mechanistically, because these two enzymes show very different rates of catalysis. Despite high structural conservation, the half-life of the phosphoaspartyl in PSP is less than 0.1 seconds, whereas in CheY the half-life is ≈10 seconds (14, 18). The slower rate of CheY cannot simply be explained by the circular permutation in the response regulator subfamily of HADs, because CheB, another response regulator, possesses a much shorter half-life (19). In fact, the half-lives of phosphoaspartyl groups in HAD phosphotransferases vary over 3 to 4 orders of magnitude (14, 18–21). In contrast, the phosphoaspartyl groups in these enzymes can take hours to hydrolyze in the absence of bound metal ion (21). These findings highlight the importance of the metal ion to catalysis and suggest that factors other than the simple arrangement of catalytic residues in space are required for rate acceleration. It will therefore be of future interest to identify the factors that serve to influence the rates of catalysis. Such factors could include effects on metal ion-dependent conformational changes or effects on the orientation of the hydrolytic nucleophile.
The paper by Cho et al. (1) underscores the utility of studying smaller, less complicated proteins as tools to gain insights into the molecular mechanisms of larger, structurally related proteins that may be multidomain, highly regulated, or difficult to obtain in sufficient quantities for biochemical studies (22–25). More complex enzymes such as these are often present in higher-order eukaryotic organisms, making a full understanding of the biochemical processes governing cellular growth, differentiation, and survival much more difficult. The use of “model enzymes” in this manner is akin to the use by enzymologists of model chemical reactions to gain insights into the molecular mechanisms of enzyme-catalyzed reactions.
Acknowledgments
This work was supported in part by the Canadian Institutes of Health Research (P.R.T.) and the National Institutes of Health (P.A.C.).
Abbreviations
- PSP
phosphoserine phosphatase
- HAD
haloacid dehalogenase
Footnotes
See companion article on page 8525.
References
- 1.Cho H, Wang W, Kim R, Yokota H, Damo S, Kim S H, Wemmer D, Kustu S, Yan D. Proc Natl Acad Sci USA. 2001;98:8525–8530. doi: 10.1073/pnas.131213698. . (First Published July 3, 2001; 10.1073/pnas.131213698) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Admiraal S J, Herschlag D. J Am Chem Soc. 2000;122:2145–2148. [Google Scholar]
- 3.Mildvan A S. Proteins. 1997;29:401–416. [PubMed] [Google Scholar]
- 4.Post R L, Kume S. J Biol Chem. 1973;248:6993–7000. [PubMed] [Google Scholar]
- 5.Stuckey J A, Schubert H L, Fauman E B, Zhang Z Y, Dixon J E, Saper M A. Nature (London) 1994;370:571–575. doi: 10.1038/370571a0. [DOI] [PubMed] [Google Scholar]
- 6.O'Brien P J, Herschlag D. Biochemistry. 2001;40:5691–5699. doi: 10.1021/bi0028892. [DOI] [PubMed] [Google Scholar]
- 7.Admiraal S J, Schneider B, Meyer P, Janin J, Veron M, Deville-Bonne D, Herschlag D. Biochemistry. 1999;38:4701–4711. doi: 10.1021/bi9827565. [DOI] [PubMed] [Google Scholar]
- 8.Walsh C T. In: Enzymatic Reaction Mechanisms. Bartlett A C, editor. New York: Freeman; 1979. p. 213. [Google Scholar]
- 9.Petsko G A. Proc Natl Acad Sci USA. 2000;97:538–540. doi: 10.1073/pnas.97.2.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chabre M. Trends Biochem Sci. 1990;15:6–10. doi: 10.1016/0968-0004(90)90117-t. [DOI] [PubMed] [Google Scholar]
- 11.Lee S Y, Cho H S, Pelton J G, Yan D, Berry E A, Wemmer D E. J Biol Chem. 2001;276:16425–16431. doi: 10.1074/jbc.M101002200. [DOI] [PubMed] [Google Scholar]
- 12.Aravind L, Galperin M Y, Koonin E V. Trends Biochem Sci. 1998;23:127–129. doi: 10.1016/s0968-0004(98)01189-x. [DOI] [PubMed] [Google Scholar]
- 13.Ridder I V, Dijkstra B W. Biochem J. 1999;339:223–226. [PMC free article] [PubMed] [Google Scholar]
- 14.Collet J F, Stroobant V, Pirard M, Delpierre G, Van Schaftingen E. J Biol Chem. 1998;273:14107–14112. doi: 10.1074/jbc.273.23.14107. [DOI] [PubMed] [Google Scholar]
- 15.Toyoshima C, Nakasako M, Nomura H, Ogawa H. Nature (London) 2000;405:647–655. doi: 10.1038/35015017. [DOI] [PubMed] [Google Scholar]
- 16.Hisano T, Hata Y, Fujii T, Liu J Q, Kurihara T, Esaki N, Soda K. J Biol Chem. 1996;271:20322–20330. doi: 10.1074/jbc.271.34.20322. [DOI] [PubMed] [Google Scholar]
- 17.Kern D, Volkman B F, Luginbuhl R, Nohaile M J, Kustu S, Wemmer D E. Nature (London) 1999;402:894–898. doi: 10.1038/47273. [DOI] [PubMed] [Google Scholar]
- 18.Lukat G S, Lee B H, Mottonen J M, Stock A M, Stock J B. J Biol Chem. 1991;266:8348–8354. [PubMed] [Google Scholar]
- 19.Stewart R C. J Biol Chem. 1993;268:1921–1930. [PubMed] [Google Scholar]
- 20.Bodley A L, Jencks W P. J Biol Chem. 1987;262:13997–14004. [PubMed] [Google Scholar]
- 21.Weiss V, Magasanik B. Proc Natl Acad Sci USA. 1988;85:8919–8923. doi: 10.1073/pnas.85.23.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hon W C, McKay G A, Thompson P R, Sweet R M, Yang D S C, Wright G D, Berghuis A M. Cell. 1997;89:887–895. doi: 10.1016/s0092-8674(00)80274-3. [DOI] [PubMed] [Google Scholar]
- 23.Boehr, D. D., Thompson, P. R. & Wright, G. D. (2001) J. Biol. Chem., in press. [DOI] [PubMed]
- 24.Wolf E, Vassilev A, Makino Y, Sali A, Nakatani Y, Burley S K. Cell. 1998;94:439–449. doi: 10.1016/s0092-8674(00)81585-8. [DOI] [PubMed] [Google Scholar]
- 25.Doyle D A, Morais Cabral J, Pfuetzner R A, Kuo A, Gulbis J M, Cohen S L, Chait B T, MacKinnon R. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]