

Abstract
Among the three types of super oxide dismutases (SODs) known, SOD2 deficiency is lethal in neonatal mice owing to cardiomyopathy caused by severe oxidative damage. SOD2 is found in red blood cell (RBC) precursors, but not in mature RBCs. To investigate the potential damage to mature RBCs resulting from SOD2 deficiency in precursor cells, we studied RBCs from mice in which fetal liver stem cells deficient in SOD2 were capable of efficiently rescuing lethally irradiated host animals. These transplanted animals lack SOD2 only in hematopoietically generated cells and live longer than SOD2 knockouts. In these mice, approximately 2.8% of their total RBCs in circulation are iron-laden reticulocytes, with numerous siderocytic granules and increased protein oxidation similar to that seen in sideroblastic anemia. We have studied the RBC deformability and oxidative stress in these animals and the control group by measuring them with a microfluidic ektacytometer and assaying fluorescent heme degradation products with a fluorimeter, respectively. In addition, the rate of hemoglobin oxidation in RBCs from these mice and the control group were measured spectrophotometrically. The results show that RBCs from these SOD2-deficient mice have reduced deformability, increased heme degradation products, and an increased rate of hemoglobin oxidation compared with control animals, indicative of increased RBC oxidative stress.
Red blood cell (RBC) oxidative stress has been linked to different types of anemia, such as sickle cell anemia [1], thalassemia [2], hemolytic anemia [3,4], and sideroblastic anemia (SA) [5]. SAs are a heterogeneous group of inherited (rare) and acquired (relatively common) disorders of erythroid development characterized by iron accumulation within the mitochondria of developing erythroid cells (i.e., ringed sideroblasts) [6,7].
Superoxide dismutases (SODs) are a class of antioxidant enzymes that disproportionate superoxide (O2•−) ions to hydrogen peroxide (H2O2) and oxygen [8] and are found in almost all organisms [9]. There are three types of SODs in mammals: SOD1, containing copper and zinc at their active site, also known as CuZnSOD, present in the cytosol of virtually all eukaryotic cells; SOD2, containing manganese in the active site (MnSOD), present only in mitochondria; and SOD3, also containing Cu and Zn at the active site found in extracellular milieu [10].
Srinivasan et al. [11] reported that yeast cells lacking the SOD2 gene are oxygen-sensitive and, when required to respire, grow poorly. Significantly elevated levels of free iron were found in these yeast cells compared with wild type strains. In addition, in wild type yeast cells, an increase in the EPR-detectable iron pool could be induced by treatment with paraquat, a redox-cycling drug that generates superoxide, suggesting that oxidative stress can generate free iron in cells. To study the importance of SODs in mammals, all three genes SOD1, SOD2, and SOD3 have been inactivated genetically in mice through homologous recombination. However, inactivation of the mitochondrial gene SOD2 has resulted in the most severe phenotype: homozygous mutant mice generated by inactivation of SOD2 gene in the CD1 background die within the first 10 days of life with a dilated cardiomyopathy [12]. A line of SOD2 knockout mice in a hybrid background was able to survive for up to 3 weeks of age, but exhibited several pathologic phenotypes, including severe anemia and degeneration of neurons in the basal ganglia and brainstem [13]. In addition to early lethality, SOD2 knockout animals had increased oxidative DNA damage and respiratory chain defects in the mitochondria [14]. On the other hand, heterozygous Sod2−/+ mice appear normal, although they show oxidative damage to mitochondrial proteins and DNA, decreased levels of reduced glutathione, and altered mitochondrial function [15,16].
To study the effects of SOD2 deficiency in cells over a longer period of time in vivo, Friedman et al. [17] generated a fetal liver transplant model in mice in which fetal liver stem cells deficient in SOD2 were capable of efficiently rescuing lethally irradiated host animals. In this system, there is a selective defect in erythroid reconstitution of SOD2-knockout fetal liver recipients, which is similar to that seen in both hereditary and acquired SAs. In fact, it was possible to demonstrate that 2.8% of the total RBCs from these mice were iron-laden siderocytes, which are essentially SOD2 deficient (SOD2_KO) reticulocytes, showing numerous siderocytic granules, enhanced levels of reactive oxygen species, and increased protein oxidative damage [18]. The association between excess iron and concomitant protein oxidation in these cells raises the possibility that redox active iron is generating protein-damaging hydroxyl radicals via Fenton chemistry in the developing RBCs [7]. Because in vitro and in vivo oxidative stress has been reported to decrease RBC deformability [19–22], we were interested to examine the deformability of RBCs in these transplanted SOD2_KO mice. In addition, because increased oxidative stress in RBCs has been reported to generate increased heme degradation products [23], we were interested to examine the level of heme degradation products in RBCs from these mice. Moreover, because peroxiredoxin 2 (PRDX2) is the third most abundant protein in the RBC [24], its absence in knockout mice gives rise to hemolytic anemia and is extremely sensitive to oxidation by H2O2. We have also examined both deformability and heme degradation in RBCs from mice with a similar transplant model in which fetal liver stem cells deficient in PRDX2 (PRDX2_KO) as well as both SOD2 and PRDX2 (Double_KO) were reconstituted. PRDX2 deficiency in mice is also known to result in the formation of Heinz bodies in their peripheral blood, and morphologically abnormal dense cells that contain markedly higher levels of reactive oxygen species (ROS) [25].
Methods
Transgenic mice and collection of blood
Transgenic mice using hematopoietic cell transplant system for SOD2, PRDX2 or double deficiencies were generated by the Friedman group at the Scripps Research Institute as described earlier [5,17]. Mice in these experiments were conditioned with 10.5 cGy on this model as described in the original report [17]. As reported, lymphoid and myeloid engraftment (using Ly5 congenic marker) exceeds 95% for lymphoid and 90% for myeloid lineages. Engraftment is durable for over 12 months following transplant. As control measure for the effects of transplantation, animals transplanted with wild type fetal liver cells were used for comparison. All mouse manipulations were approved by the Animal Use Committee of Scripps Clinic. Blood from these mice were collected from the ocular venous sinus with ethylenedi-amine tetraacetic acid as an anticoagulant and sent on ice by overnight mail to the Molecular Dynamics Section at the National Institute on Aging at Baltimore for measurements of RBC deformability and heme degradation.
Measurement of RBC deformability
In each whole blood sample, RBC deformability was measured using a microfluidic RheoScan-D slit-flow ektacytometer (Rheo Meditech, Seoul, South Korea). The methodologic details of this instrument have been published previously [26]. Blood (~6 μL) was suspended (~1/100 dilution) by mixing it slowly in 600 μL of the highly viscous PVP360 solution (viscosity ~ 30 cP), and then 500 μL of this solution was loaded into the sample reservoir of the microfluidic chip. During operation of the ektacytometer, the vacuum-generating mechanism allowed RBCs to flow through the micro-channel at a range of shear stresses, whereas the elliptical diffraction patterns of the flowing cells were generated by a laser beam (wavelength, 635 nm from a 1.5-mW laser diode). The elliptical diffraction patterns of the flowing RBCs at different shear stress levels are projected on a screen and captured by a charge-coupled device-video camera. The image data were then analyzed with an ellipse-fitting software to determine L and W, the major and minor axes of the ellipse, respectively, at various shear stress values (0–20 Pa) to give a value of the elongation index (EI = [L – W] / [L + W]), which measures the deformability at each shear stress. For comparative analysis of the deformability values of multiple blood samples, EI values (mean and standard deviation of three measurements) at 3 Pa were used, which is approximately the halfway point on the EI-shear stress curve [27].
Measurement of heme degradation and hemoglobin autoxidation
The levels of heme degradation in RBCs were measured using a procedure similar to that described earlier [28]. Briefly, 20 μL of washed RBCs were lysed in 3 ml of ice-cold high quality water (prepared by Siemens ELGA Purelab Prima reverse osmosis and Purelab Ultra deionization). The hemoglobin (Hb) spectrum of the hemolysate was recorded from 490–640 nm using a Perkin Elmer Lamda 35 spectrophotometer. The concentrations of oxyHb and metHb were determined by a least square–fitting program using spectra at known concentrations. The Hb concentration of the hemolysate was then adjusted to 50 μmol/L, and the fluorescent emission spectrum was recorded from 400–600 nm at an excitation wavelength of 321 nm using a Perkin Elmer LS55B spectrofluorometer with the slit width set at 10 nm. The fluorescence intensity of emission at 480 nm was used as a measure of the heme degradation products.
Measurements of the rate of hemoglobin oxidation
Because the RBC sample obtained from each individual mouse was not sufficient to follow the time course of hemoglobin oxidation, samples from three or four mice of the same category were pooled and the rate of hemoglobin oxidation was studied both in RBCs and in purified hemoglobin [29] (devoid of cellular antioxidants) prepared from these pooled cells. In addition, the limited number of available mice made it impossible to perform multiple measurements on pooled samples. Although repeated experiments are always advantageous, with limited low-volume samples, it is considered valid [30,31] to report results of one pooled sample, which reflect the average effect for this class of mice thereby ruling out an artifactual effect of a single outlier. For these kinetic studies, we determined the significance of the time-dependent changes for different groups of animals using the “Linear regression–compare slopes” feature of GraphPad Prism 5.04 software (http://www.graphpad.com). For measuring the rate of hemoglobin oxidation, a sample was incubated at 30°C, and an aliquot was taken at different times and diluted with water. The metHb concentration was measured as a percentage of total hemoglobin in the sample by fitting the spectra with known standards. For the RBC sample, the aliquot was lysed by dilution with water and metHb concentration measured as described earlier.
Results
In this study, RBC deformability and RBC heme degradation (a cellular measure of oxidative stress) were measured in blood from chimeric mice reconstituted with hematopoietic stem cells deficient in genes for the SOD2, PRDX2, or both. As shown in Figure 1, deformability of RBCs from SOD2-deficient (Trans_SOD2_KO) and SOD2- and PRDX2-deficient (Trans_Double_KO) mice is significantly (p < 0.0001) reduced by ~15%, compared with transplanted control (Trans_Control) mice. Although there is a slight decrease in RBC deformability in transplanted PRDX2-deficient mice (Trans_PRDX2_KO) compared to that of Trans_Control mice, the difference is not statistically significant. In addition, the RBC deformability in Trans_Double_KO mice appears to be slightly higher than that of Trans_SOD2_KO mice, but the difference is not statistically significant.
Figure 1.
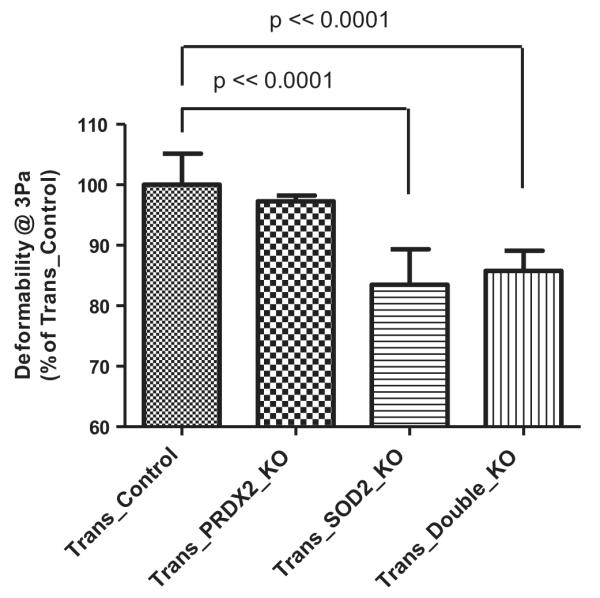
Comparison of RBC deformability (% of Trans_Control) in Trans_PRDX2_KO (not significant difference), Trans_SOD2_KO (p < 0.0001) and Trans_Double_KO (p < 0.0001) mice with that in Trans_Control mice.
As shown in Figure 2, heme degradation in RBCs measured from Trans_SOD2_KO or Trans_Double_KO mice significantly (p < 0.001) increased by ~150% relative to Trans_Control mice. Heme degradation in Trans_PRDX2_KO mice appears to be slightly higher than the Trans_Control mice, but the difference is not statistically significant. Similarly, the difference in heme degradation when comparing Trans_SOD2_KO and Trans_Double_KO mice is not statistically significant. Further statistical analysis suggests that there is a negative correlation between the level of RBC-heme degradation and RBC-deformability comparing all the mouse blood samples in this study (Fig. 3), which is statistically significant (p < 0.001). This result implies that an increase in RBC oxidative stress, as indicated by increased heme degradation, results in less deformable RBCs.
Figure 2.
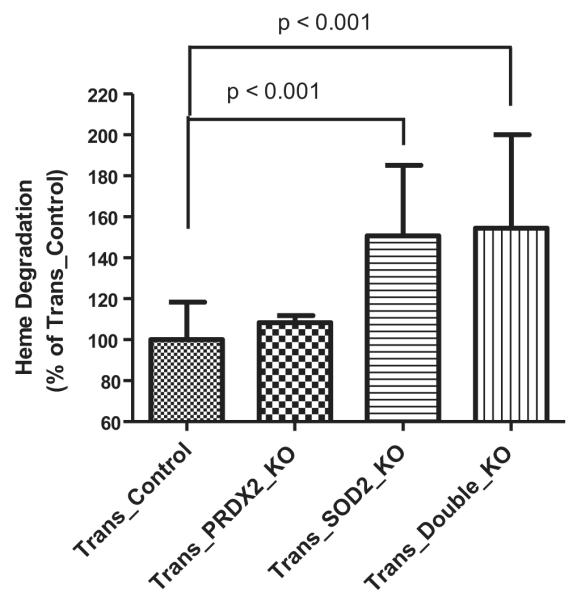
Comparison of heme degradation (% of Trans_Control) in RBCs of Trans_PRDX2_KO (not significant difference), Trans_SOD2_KO (p < 0.001) and Trans_Double_KO (p < 0.001) mice with that in Trans_Control mice.
Figure 3.
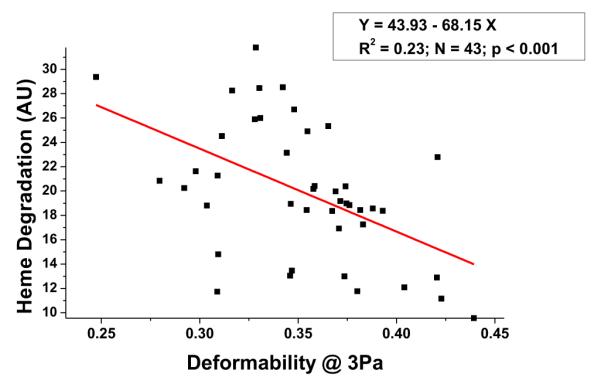
Correlation between heme degradation and RBC deformability at 3 Pa in all mouse blood samples (N = 43; p < 0.001).
We have also studied in vitro time-dependent increases in heme degradation in RBCs at 30°C from these animals (Fig. 4). Because of the scarcity of blood samples, blood from three to four mice of the same category was pooled, and this experiment and subsequent experiments (hemoglobin oxidation, see text as follows) were performed. Thus the data in Figures 4–6 can be considered as an average of three to four different samples in each category even though experiments were performed in one pooled sample in each category. As can be seen in Figure 4, there was a significant increase in the amount of heme degradation in RBCs from Trans_PRDX2_KO (p = 0.03), Trans_ SOD2_KO (p = 0.023), and Trans_Double_KO (p = 0.043) mice over the control mice (Trans_Control), although the difference between Trans_SOD2_KO and Trans_Double_KO was not significant. This finding suggests that the blood from these mice is exposed to an increase in oxidative stress relative to control samples. To rule out an effect of an increased rate of hemoglobin autoxidation, which would also result in increased heme degradation, we examined the rate of metHb formation in intact RBCs and in purified oxyHb from the pooled sample of Trans_SOD2_KO mice only compared with that of control mice. As can be seen in Figure 5, the initial level of metHb in Trans_SOD2_KO mice was higher than in control mice (Trans_Control), and the rate of metHb formation in RBCs at 37°C from Trans_SOD2_KO mice over control mice was also significantly higher (p = 0.0001). However, in purified hemoglobin samples (Fig. 6), although the level of metHb in Trans_SOD2_KO mice started at a higher level than the control mice, the rate of increase between the two (examined up to 5 hours) was not statistically significant. This finding suggests that increased hemoglobin autoxidation is not the reason of increased oxidative stress in RBCs from Trans_SOD2_KO mice.
Figure 4.
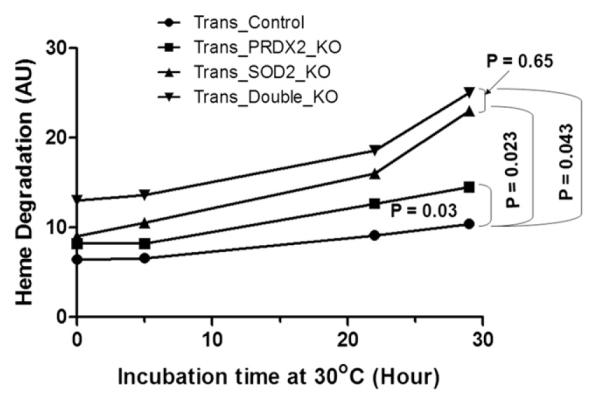
Comparison of time-dependent heme degradation (AU) changes at 30°C in intact RBCs with cellular pool from four mice in each category. Statistical significance in their differences in slopes is shown with their respective p values.
Figure 6.
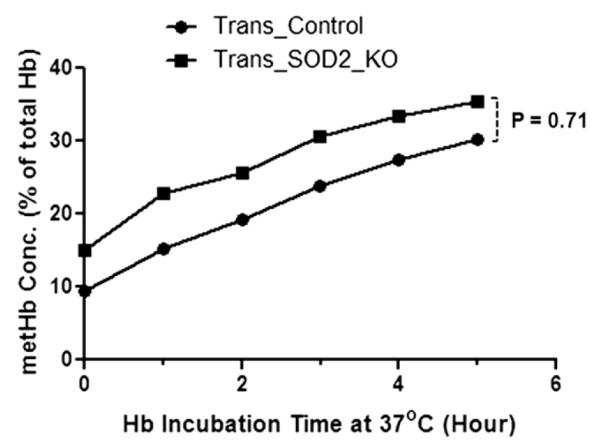
Time-dependent changes in metHb level at 37°C in hemoglobin samples purified from RBC pool of Trans_SOD2_KO mice are not statistically different (p = 0.71) from that of Trans_Control mice as in Figure 5.
Figure 5.
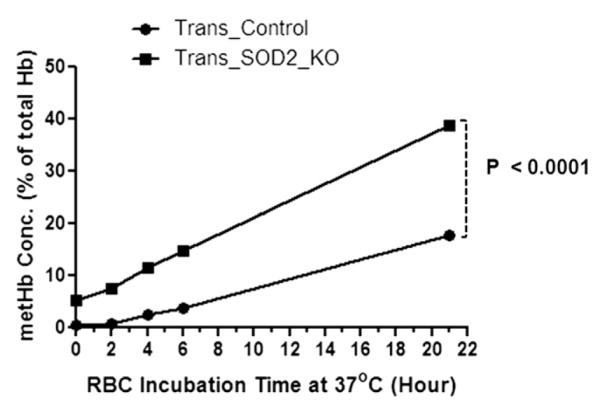
Time-dependent increase in metHb level at 37°C in intact RBCs from a cellular pool of Trans_SOD2_KO mice was significantly higher (p < 0.0001) than that of the Trans_control mice as in Figure 4.
Discussion
Our earlier publications [23,28,29,32,33] have established that the level of fluorescent heme degradation products in RBCs reflect the level of RBC-specific oxidative stress. Recently, Friedman et. al. [17] reported the creation of mouse hematopoietic chimeras in which all blood cells were derived from fetal liver stem cells of Sod2 knockout mice, efficiently rescuing hematopoiesis and allowing long-term survival of lethally irradiated host mice. However, these animals were persistently anemic, and the loss of SOD2 in erythroid progenitor cells resulted in enhanced protein oxidative damage, altered membrane deformation, and reduced survival of RBCs. In this study, in collaboration with Dr. Friedman, we have analyzed heme degradation in RBCs from these chimera mice with SOD2 knockout, PRDX2 knockout, and double knockout for SOD2 and PRDX2. In addition, we have investigated changes in RBC deformability measured by a recently available microfluidic ektacytometer and the rate of hemoglobin oxidation (measuring level of metHb formation with time) in intact RBCs and purified hemoglobin samples prepared from the same RBCs of these groups of chimera mice. The results presented in this article clearly show that RBC deformability in SOD2 as well as double knockout chimera mice was significantly reduced (Fig. 1), while heme degradation in these mice was significantly increased (Fig. 2) over control mice. Additional loss of PRDX2 did not significantly change the observed results, suggesting that the defect caused by loss of SOD2 is either more severe or somehow inactivates protection normally obtained through the PRDX2 system. Interestingly, when all the data from all groups of mice was considered, heme degradation was inversely proportional to RBC deformability (Fig. 3).
We have also studied the change in the formation of heme degradation products in these blood samples over a period of time. However, due to scarcity of blood samples, blood from three to four mice of the same category was pooled, and a single measurement was performed in this and subsequent experiments (measurement of hemoglobin oxidation). Therefore, data in Figures 4–6, are from single experiments of pooled samples in each category and should be considered with caution. As shown in the Results, incubation of RBCs at 30°C for longer periods of time resulted in a significant increase in heme degradation in RBCs from PRDX2-KO, SOD2_KO and double knockout chimera mice (Fig. 4) over control mice, although the difference between SOD2_KO and double knockout mice was not significant. Interestingly, the rate of hemoglobin oxidation (metHb formation) in intact RBCs was significantly higher in case of SOD2 knockout chimera mice in comparison to control mice (Fig. 5), whereas the rate of oxidation of purified hemoglobin from control and SOD2 knockout mice were essentially the same (Fig. 6). Because heme degradation requires oxidation of hemoglobin, this result indicated that the elevated in vitro formation of heme degradation for the SOD2_KO was not due to an intrinsic instability of the hemoglobin present, but instead reflected the presence of additional RBC oxidants that increased the oxidation and degradation of hemoglobin. Because mature RBCs are devoid of SOD2 and it has been reported that RBC from SOD2 knockout chimera mice have much higher content of iron laden reticulocytes [18], perhaps redox active iron has been carried over to mature RBCs, making them responsible for the generation of additional oxidants, such as hydroxyl radicals, via Fenton reaction in these cells, causing an increase in heme degradation.
At this point, we are unable to demonstrate the presence of redox-active iron particles in RBCs from Trans_SOD2_KO mice because of a limited amount of samples. However, it has been reported that in conditions such as sickle cell disease [34] and thalassemia sions are frequently used, exogenous iron can accumulate, circulate as non-transferrin bound iron (NTBI), enter tissues, and form ROS. In such a case, we can speculate that NTBI may also enter RBCs, causing increased heme degradation and reduced deformability with the formation of additional ROS. According to Tanno et al. [35], people with thalassemia also suffer from iron overload independent of blood transfusions.
In conclusion, although mature RBCs do not have SOD2, its deficiency or malfunction during erythroid development results in lasting changes to the mature RBC. These changes include increased RBC oxidative stress causing increased heme degradation [28,36], and reduced deformability that can reduce RBC lifetime in circulation [37]. This is perhaps the reason for anemia and increased RBC loss in SOD2 knockout mice chimera as reported earlier [17]. This general mechanism might also explain reduced RBC survival in clinical disorders characterized by excessive iron in erythroid cells [38–40].
Acknowledgment
This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Institute on Aging and The Scripps Research Institute, La Jolla, California.
Footnotes
Conflict of interest disclosure No financial interest/relationships with financial interest relating to the topic of this article have been declared.
References
- 1.Hebbel RP. The sickle erythrocyte in double jeopardy: autoxidation and iron decompartmentalization. Semin Hematol. 1990;27:51–69. [PubMed] [Google Scholar]
- 2.Shinar E, Rachmilewitz EA. Oxidative denaturation of red blood cells in thalassemia. Semin Hematol. 1990;27:70–82. [PubMed] [Google Scholar]
- 3.Fibach E, Rachmilewitz E. The role of oxidative stress in hemolytic anemia. Curr Mol Med. 2008;8:609–619. doi: 10.2174/156652408786241384. [DOI] [PubMed] [Google Scholar]
- 4.Winterbourn CC. Oxidative denaturation in congenital hemolytic anemias: the unstable hemoglobins. Semin Hematol. 1990;27:41–50. [PubMed] [Google Scholar]
- 5.Martin FM, Bydlon G, Friedman JS. SOD2-deficiency sideroblastic anemia and red blood cell oxidative stress. Antioxid Redox Signal. 2006;8:1217–1225. doi: 10.1089/ars.2006.8.1217. [DOI] [PubMed] [Google Scholar]
- 6.Cotter PD, May A, Li L, et al. Four new mutations in the erythroid-specific 5-aminolevulinate synthase (ALAS2) gene causing X-linked sideroblastic anemia: increased pyridoxine responsiveness after removal of iron overload by phlebotomy and coinheritance of hereditary hemochromatosis 2. Blood. 1999;93:1757–1769. [PubMed] [Google Scholar]
- 7.Martin FM, Prchal J, Nieva J, et al. Purification and characterization of sideroblasts from patients with acquired and hereditary sideroblastic anaemia. Br J Haematol. 2008;143:446–450. doi: 10.1111/j.1365-2141.2008.07358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valentine JS, Wertz DL, Lyons TJ, Liou LL, Goto JJ, Gralla EB. The dark side of dioxygen biochemistry. Curr Opin Chem Biol. 1998;2:253–262. doi: 10.1016/s1367-5931(98)80067-7. [DOI] [PubMed] [Google Scholar]
- 9.McCord JM, Keele BB, Jr, Fridovich I. An enzyme-based theory of obligate anaerobiosis: the physiological function of superoxide dismutase. Proc Natl Acad Sci U S A. 1971;68:1024–1027. doi: 10.1073/pnas.68.5.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fridovich I. Superoxide dismutases. An adaptation to a paramagnetic gas. J Biol Chem. 1989;264:7761–7764. [PubMed] [Google Scholar]
- 11.Srinivasan C, Liba A, Imlay JA, Valentine JS, Gralla EB. Yeast lacking superoxide dismutase(s) show elevated levels of “free iron” as measured by whole cell electron paramagnetic resonance. J Biol Chem. 2000;275:29187–29192. doi: 10.1074/jbc.M004239200. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Huang TT, Carlson EJ, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 13.Lebovitz RM, Zhang H, Vogel H, et al. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci U S A. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Melov S, Coskun P, Patel M, et al. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci U S A. 1999;96:846–851. doi: 10.1073/pnas.96.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams MD, Van RH, Conrad CC, Huang TT, Epstein CJ, Richardson A. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J Biol Chem. 1998;273:28510–28515. doi: 10.1074/jbc.273.43.28510. [DOI] [PubMed] [Google Scholar]
- 16.Van RH, Williams MD, Guo Z, et al. Knockout mice heterozygous for Sod2 show alterations in cardiac mitochondrial function and apoptosis. Am J Physiol Heart Circ Physiol. 2001;28:H1422–H1432. doi: 10.1152/ajpheart.2001.281.3.H1422. [DOI] [PubMed] [Google Scholar]
- 17.Friedman JS, Rebel VI, Derby R, et al. Absence of mitochondrial superoxide dismutase results in a murine hemolytic anemia responsive to therapy with a catalytic antioxidant. J Exp Med. 2001;193:925–934. doi: 10.1084/jem.193.8.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin FM, Bydlon G, Welsh ML, Friedman JS. A method for rapid mouse siderocyte enrichment. Exp Hematol. 2005;33:1493–1499. doi: 10.1016/j.exphem.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 19.Spolarics Z, Condon MR, Siddiqi M, Machiedo GW, Deitch EA. Red blood cell dysfunction in septic glucose-6-phosphate dehydrogenase-deficient mice. Am J Physiol Heart Circ Physiol. 2004;286:H2118–H2126. doi: 10.1152/ajpheart.01085.2003. [DOI] [PubMed] [Google Scholar]
- 20.Samukawa K, Suzuki Y, Ohkubo N, Aoto M, Sakanaka M, Mitsuda N. Protective effect of ginsenosides Rg(2) and Rh(1) on oxidation-induced impairment of erythrocyte membrane properties. Biorheology. 2008;45:689–700. [PubMed] [Google Scholar]
- 21.Chan JY, Kwong M, Lo M, Emerson R, Kuypers FA. Reduced oxidative-stress response in red blood cells from p45NFE2-deficient mice. Blood. 2001;97:2151–2158. doi: 10.1182/blood.v97.7.2151. [DOI] [PubMed] [Google Scholar]
- 22.Aydogan S, Yapislar H, Artis S, Aydogan B. Impaired erythrocytes deformability in H(2)O(2)-induced oxidative stress: protective effect of L-carnosine. Clin Hemorheol Microcirc. 2008;39:93–98. [PubMed] [Google Scholar]
- 23.Nagababu E, Mohanty JG, Bhamidipaty S, Ostera GR, Rifkind JM. Role of the membrane in the formation of heme degradation products in red blood cells. Life Sci. 2010;86:133–138. doi: 10.1016/j.lfs.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schroder E, Littlechild JA, Lebedev AA, Errington N, Vagin AA, Isupov MN. Crystal structure of decameric 2-Cys peroxiredoxin from human erythrocytes at 1.7 A resolution. Structure. 2000;8:605–615. doi: 10.1016/s0969-2126(00)00147-7. [DOI] [PubMed] [Google Scholar]
- 25.Yang HY, Kwon J, Choi HI, et al. In-depth analysis of cysteine oxidation by the RBC proteome: advantage of peroxiredoxin II knockout mice. Proteomics. 2011;12:101–112. doi: 10.1002/pmic.201100275. [DOI] [PubMed] [Google Scholar]
- 26.Shin S, Hou JX, Suh JS, Singh M. Validation and application of a microfluidic ektacytometer (RheoScan-D) in measuring erythrocyte deformability. Clin Hemorheol Microcirc. 2007;37:319–328. [PubMed] [Google Scholar]
- 27.Baskurt OK, Boynard M, Cokelet GC, et al. New guidelines for hemorheological laboratory techniques. Clin Hemorheol Microcirc. 2009;42:75–97. doi: 10.3233/CH-2009-1202. [DOI] [PubMed] [Google Scholar]
- 28.Nagababu E, Fabry ME, Nagel RL, Rifkind JM. Heme degradation and oxidative stress in murine models for hemoglobinopathies: thalassemia, sickle cell disease and hemoglobin C disease. Blood Cells Mol Dis. 2008;41:60–66. doi: 10.1016/j.bcmd.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagababu E, Rifkind JM. Formation of fluorescent heme degradation products during the oxidation of hemoglobin by hydrogen peroxide. Biochem Biophys Res Commun. 1998;247:592–596. doi: 10.1006/bbrc.1998.8846. [DOI] [PubMed] [Google Scholar]
- 30.Kendziorski C, Irizarry RA, Chen KS, Haag JD, Gould MN. On the utility of pooling biological samples in microarray experiments. Proc Natl Acad Sci U S A. 2005;102:4252–4257. doi: 10.1073/pnas.0500607102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng X, Wood CL, Blalock EM, Chen KC, Landfield PW, Stromberg AJ. Statistical implications of pooling RNA samples for microarray experiments. BMC Bioinformatics. 2003;4:26–34. doi: 10.1186/1471-2105-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagababu E, Rifkind JM. Heme degradation by reactive oxygen species. Antioxid Redox Signal. 2004;6:967–978. doi: 10.1089/ars.2004.6.967. [DOI] [PubMed] [Google Scholar]
- 33.Nagababu E, Gulyani S, Earley CJ, Cutler RG, Mattson MP, Rifkind JM. Iron-deficiency anaemia enhances red blood cell oxidative stress. Free Radic Res. 2008;42:824–829. doi: 10.1080/10715760802459879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walter PB, Harmatz P, Vichinsky E. Iron metabolism and iron chelation in sickle cell disease. Acta Haematol. 2009;122:174–183. doi: 10.1159/000243802. [DOI] [PubMed] [Google Scholar]
- 35.Tanno T, Bhanu NV, Oneal PA, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. 2007;13:1096–1101. doi: 10.1038/nm1629. [DOI] [PubMed] [Google Scholar]
- 36.Kiefmann R, Rifkind JM, Nagababu E, Bhattacharya J. Red blood cells induce hypoxic lung inflammation. Blood. 2008;111:5205–5214. doi: 10.1182/blood-2007-09-113902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Egberts J, Hardeman MR, Luykx LM. Decreased deformability of donor red blood cells after intrauterine transfusion in the human fetus: possible reason for their reduced life span? Transfusion. 2004;44:1231–1237. doi: 10.1111/j.1537-2995.2004.04014.x. [DOI] [PubMed] [Google Scholar]
- 38.Eschbach JW, Jr, Funk D, Adamson J, Kuhn I, Scribner BH, Finch CA. Erythropoiesis in patients with renal failure undergoing chronic dialysis. N Engl J Med. 1967;276:653–658. doi: 10.1056/NEJM196703232761202. [DOI] [PubMed] [Google Scholar]
- 39.Hsu CY, McCulloch CE, Curhan GC. Epidemiology of anemia associated with chronic renal insufficiency among adults in the United States: results from the Third National Health and Nutrition Examination Survey. J Am Soc Nephrol. 2002;13:504–510. doi: 10.1681/ASN.V132504. [DOI] [PubMed] [Google Scholar]
- 40.Prus E, Fibach E. Effect of iron chelators on labile iron and oxidative status of thalassaemic erythroid cells. Acta Haematol. 2010;123:14–20. doi: 10.1159/000258958. [DOI] [PubMed] [Google Scholar]