

Abstract
Over the past decades, the identification of several new cytokines, including interleukin (IL)-17 and IL-23, and of new T helper cell subsets, including Th17 cells, has changed the vision of immunological processes. The IL-17/Th17 pathway plays a critical role during the development of inflammation and autoimmunity, and targeting this pathway has become an attractive strategy for a number of diseases. This review aims to describe the effects of IL-17 in the joint and its roles in the development of autoimmune and inflammatory arthritis. Furthermore, biotherapies targeting directly or indirectly IL-17 in inflammatory rheumatisms will be developed.
1. Introduction
Cytokines play a key role in the coordination of the innate and adaptive immune responses to protect an organism against internal and external pathogenic assault. Over the past decades, the identification of several new cytokines, including interleukin (IL)-17 (also known as IL-17A) and IL-23, has changed the vision of immunological processes.
In response to antigen stimulation, naive CD4+ T cells differentiate into different T cell subsets with specialized effector functions, mainly on the basis of their cytokine expression profile. T helper type 1 (Th1) cells develop in response to IL-12 and produce high amounts of interferon (IFN)-γ, required to control infection with intracellular pathogens such as viruses. This cell subset is also important during inflammation and autoimmunity. IL-4 is the major inducer of Th2 cells that produce IL-4, IL-5, and IL-13, which are crucial for the clearance of parasitic worms and during development of allergic inflammation. The Th1/Th2 dichotomy paradigm has been revisited with the recent identification of additional effector CD4+ T cell subsets producing IL-17 (Th17), IL-22 (Th22), or IL-9 (Th9) [1–6]. The importance of Th17 cells during development of autoimmune and inflammatory diseases is now well documented. These cells play also a critical role during defense against extracellular pathogens. Besides Th17 cells, γδ T cells, innate lymphoid cells, natural killer cells, and CD8+ T cells represent other and important sources of IL-17.
This review aims to overview the role of IL-17 during host defense and autoimmunity, with a particular focus on IL-17 and articular inflammation. Biotherapies targeting directly or indirectly this cytokine in inflammatory rheumatisms will also be developed.
2. IL-17: Signaling, Cellular Sources, and Biological Activities
2.1. IL-17 and IL-17 Receptor Signaling
Originally called cytotoxic T-lymphocyte-associated antigen 8 (CTLA8), IL-17 was first identified in rodent T cell hybridoma clones and subsequently cloned from human CD4+ T cell library [7–9]. It is the founding member of the IL-17 cytokine family, which is composed of six members: IL-17 (IL-17A), IL-17B, IL-17C, IL-17D, IL-17E (IL-25), and IL-17F. IL-17 and IL-17F are highly homologous and bind the same receptor, implying shared biological activities (Figure 1). In addition, IL-17 exists as a homodimer or as a heterodimer with IL-17F [10, 11].
Figure 1.

IL-17 cytokines and receptors family.
The IL-17 receptor family contains five members, from IL-17RA to IL-17RE, and functional receptors for IL-17 cytokine family consist of homo- or heterodimers (Figure 1). Both IL-17 and IL-17 receptor family members have little homology to other known cytokines and cytokine receptors and are thus classified as a new cytokine and cytokine receptor families.
IL-17 acts through a heterotrimeric receptor composed of two IL-17RA chains and one IL-17RC subunit [11, 12]. Such receptor complex is shared with IL-17F and IL-17A/IL-17F heterodimer. IL-17RA is ubiquitously expressed, with elevated levels in hematopoietic cells; however, IL-17 main responsive cells are epithelial and endothelial cells, fibroblasts, and to a lesser extent macrophages, dendritic cells, and B cells. In contrast, IL-17RC is weakly expressed in hematopoietic cells, and higher expression is observed in nonhematopoietic tissues, such as liver, prostate, and joints. Thus, IL-17RA and IL-17RC differential expression may explain tissue-specific function of IL-17. Binding of IL-17 to IL-17RA induces recruitment of IL-17RC to form an active IL-17RA/IL-17RC complex, inducing mitogen-activated protein (MAP) kinases, nuclear factor κ B (NFκB), phosphoinositide 3 kinase (PI3K), and C/EBP signaling pathways [11]. In addition, NFκB activator 1 (Act 1), an adaptor protein for IL-17 receptor, is an essential component of IL-17-mediated signaling and downstream effects [13, 14].
As detailed below, IL-17 is mainly known for its roles in host defense, inflammation, and autoimmunity, and its expression is increased in inflammatory tissues [15].
2.2. IL-17: Adaptive and Innate Sources
Whereas IL-17 levels are low or undetected in normal homeostatic conditions, IL-17 production is highly increased following diverse stimuli, including infection and inflammation. Elevated IL-17 expression is also observed in a number of autoimmune and inflammatory diseases. IL-17-producing cells mainly belong to the hematopoietic lineage, comprising both innate and adaptive immune cells. Interestingly, both IL-1β and IL-23 are potent inducers of IL-17 production by these cell subsets.
2.2.1. Adaptive Sources of IL-17
IL-17 has been known to be produced by T cells for the past 18 years; however, the identification of IL-17-producing CD4+ T (Th17) cells as a T helper cell subset distinct from Th1 and Th2 cells [1–3] has had a tremendous impact on our understanding of the cytokines and T cell pathways that are involved during development and maintenance of chronic inflammation. Th17 cells were first recognized when assessing the role of IL-23 in various mouse models of chronic inflammation and autoimmunity, including inflammatory bowel diseases (IBDs), collagen-induced arthritis (CIA), or experimental autoimmune encephalomyelitis (EAE, a murine model of multiple sclerosis) [2, 16, 17]. In addition to IL-23, IL-1β, IL-21, prostaglandin E2 (PGE2), transforming growth factor (TGF)-β, and IL-6 regulate development of this cell subset [16–25]. Furthermore, retinoic acid receptor-related orphan receptor-γt (RORγt), RORα, signal transducer and activator of transcription 3 (STAT3), Interferon regulatory factor 4 (IRF4), and aryl hydrocarbon receptor (AHR) are key transcription factors in the differentiation program of Th17 cells [26–30]. Mammalian target of rapamycin (mTOR) and hypoxia-inducible factor 1α (HIF1α) were also recently identified as factors positively regulating Th17 development [31–34]. Although Th17 cells derived their name because of their ability to secrete IL-17, they also produce elevated levels of IL-17F, IL-22, IFN-γ, tumor necrosis factor (TNF)-α, IL-6, and CCL20, which have both overlapping and distinct roles during inflammation and host defense [16, 35]. Th17 cells have been largely described for their key role in the pathogenesis of inflammatory and autoimmune disorders, including arthritis, IBD, psoriasis, and multiple sclerosis, and targeting the Th17 pathway is showing promising results for treatment of chronic inflammation [36].
Although IL-17 is considered a CD4+ T cell product, activated CD8+ T cells are another adaptive source of this cytokine [37–39]. In line with distinct subsets of CD4+ T cells, naive CD8+ T cells can be polarized into different effector phenotypes, such as type 1 (Tc1), type 2 (Tc2) cells, and the recently described IL-17-producing CD8+ T cell subset, defined as Tc17 [40–43]. Tc17 cells display reduced cytotoxic activity and express molecules of the Th17 program. Data from an increasing number of reports suggest a possible role of Tc17 cells during inflammation and autoimmunity [44–47].
Lastly, B cells were very recently identified as an important source of IL-17 in response to Trypanosoma cruzi infection both in mice and human [48]. Such IL-17 production is independent of RORγt, RORα, and AHR and is unaffected after T. cruzi in IL-6 or IL-23 receptor deficient mice, showing that, in contrast to other cellular sources of IL-17, B cells do not use the canonical IL-17 program.
2.2.2. Innate Sources of IL-17
IL-17 production by adaptive immune cells could not explain the existence of early IL-17-mediated immune responses, and a wide range of studies have shown that IL-17 is also produced by a variety of innate cell subsets, including γδ T cells, innate lymphoid cells, and natural killer cells [49, 50]. Whether mast cells and neutrophils can produce IL-17 is still under investigation. IL-1β, IL-23, and downstream-activated transcription factors, RORγt, STAT3, and AHR, have been described as important factors to induce innate IL-17-producing cell development [49, 51]. These innate sources of IL-17 play a crucial role during stress responses and mucosal host defense. In addition, innate IL-17 producers have been involved in the development of autoimmune diseases, such as EAE, arthritis, and colitis [50–54].
2.3. IL-17 in Host Defense and Autoimmunity
Since its identification, biological activities of IL-17 have been extensively investigated. This cytokine has pleiotropic effects that bridge innate and adaptive immunity and plays critical roles during host defense against pathogens, as well as during development and maintenance of autoimmune and inflammatory diseases.
IL-17 promotes expression of antimicrobial peptides by keratinocytes, lung, and gut epithelial cells, such as defensins, S100A proteins, and lipocalin 2. It also induces secretion of proinflammatory cytokines (e.g., IL-1, IL-6, and TNF-α), chemokines (e.g., IL-8, CCL20, CCL2, and CXCL5), and matrix metalloproteinases (e.g., MMP1, MMP3, and MMP9) from multiple target cells, including epithelial and endothelial cells, fibroblasts, neutrophils, and osteoblasts [55, 56]. Such effects explain the diversity of IL-17 biological activities in the organism: promotion of inflammation, protection against infection, and chemotactic effects that induce recruitment of Th17 cells, as well as innate cells, such as neutrophils. Interestingly, IL-17 also cooperates with other cytokines to promote inflammation, such as TNF-α, IL-6, and IL-1β.
2.3.1. IL-17 in Host Defense
IL-17, as well as Th17-related cytokines IL-17F and IL-22, protects hosts against several microbial and fungal pathogens at epithelial and mucosal tissues, including skin, intestine, and lung. IL-17-signaling deficiency in mice causes a dramatic reduction in neutrophil chemotaxis and a subsequent increased susceptibility to bacterial infection. For example, mice deficient in IL-17 and/or IL-17RA show increased susceptibility to infections with Klebsiella pneumonia, Staphylococcus aureus, Citrobacter rodentium, and Candida Albicans [57–61]. In addition, emerging evidence points to an involvement of IL-17 and Th17 cells during immune protection against parasites and viruses [61–64]. Interestingly, Bermejo et al. just identified IL-17-producing B cells as critical to control trypanosome infection [48].
Several reports support an important role of Th17 cells during host defense in humans. The IL-23/Th17 pathway is important during defense against Mycoplasma hominis [65]; human memory T cells specific for C. albicans belong to the Th17 lineage [66], and patients with chronic mucocutaneous candidiasis, a heterogeneous group of disorders characterized by recurrent or persistent infections (predominantly with C. albicans and to a lesser extent with S. aureus), have reduced production of IL-17 and IL-22 [67].
Recent genetic studies revealed disease susceptibility association with IL-17RA autosomal recessive deficiency [68]. Lastly, patients suffering from hyper-IgE syndrome are highly susceptible to bacterial and fungal infections and have impaired Th17 cell differentiation [69–71]. However, the exact contribution of Th17 cells versus innate immune cells for protective immunity still needs to be fully determined.
2.3.2. IL-17 in Autoimmunity
In contrast to their protective role during host defense, IL-17 and other Th17-related cytokines (i.e., IL-22, IL-17F) can have adverse effects resulting in tissue damage. Th17 cells are linked to the pathogenesis of various human autoimmune and inflammatory diseases, and IL-17, IL-17F, IL-22, and IL-23 levels are increased in RA, psoriasis, multiple sclerosis, and IBD [4, 17, 72–75]. Together with Th17 cells, mast cells, neutrophils, Tc17, and γδ-T cells represent additional sources of IL-17 in inflammatory diseases [76–78]. IL-17-producing γδ-T cells are involved in the development of skin, brain, and articular inflammation in vivo [52, 79–81]. In addition, Tc17 cells cooperate with Th17 cells for the induction of EAE [47].
Consistent with these observations, studies in mice deficient in IL-17 or its receptor and blockade of IL-17 or IL-17 receptor revealed an important role of IL-17 in vivo during induction and propagation of autoimmunity in animal models, such as EAE and CIA [82–86]. Interestingly, IL-23 appears as an essential cytokine to drive the pathogenicity of both innate and adaptive IL-17-producing cells [87, 88].
2.4. IL-17 in the Joint
Together with IL-1β, TNF-α, and IL-23, IL-17 is an additional cytokine able to promote articular inflammation and damage (Figure 2). As detailed in the next part of this review, elevated levels of IL-17 are found in patients with autoimmune or inflammatory rheumatisms, such as RA, spondyloarthritis (SpA), systemic lupus erythematosus (SLE), or systemic sclerosis (SSc) [89–93], and in vivo studies demonstrated an important role of IL-17 in autoimmune arthritis by aggravating synovial inflammation and joint destruction [94, 95]. Conversely, IL-17 deficiency or inhibition protects from joint inflammation and damage in animal models of arthritis [82, 96, 97]. Besides Th17 cells, innate immune cells are also an important source of IL-17 in inflammatory joint diseases, and both Th17 cells and innate IL-17 producers have been shown to be important players of IL-17-induced effects in the joint [52, 98–104].
Figure 2.
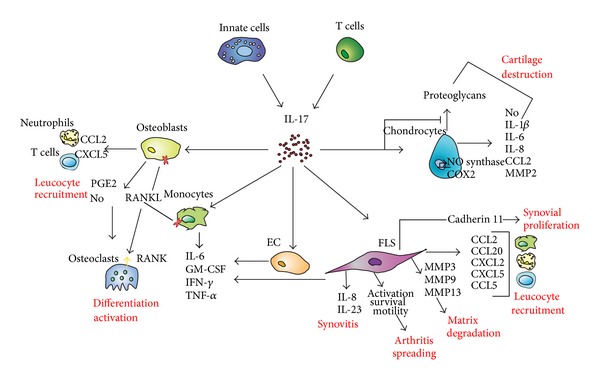
Effects of IL-17 in the joint. COX2: cyclooxygenase 2; EC: endothelial cells; FLS: fibroblast-like synoviocytes; GM-CSF: granulocyte-monocyte colony-stimulating factor; IFN-γ: interferon-γ; IL: interleukin; MMP: matrix metalloproteinases; NO: nitric oxide; PGE2: prostaglandin E2; RANK: receptor activator of NFκB; RANKL: RANK ligand; TNF-α: tumor necrosis factor-α.
2.4.1. IL-17 and Bone Metabolism
IL-17 affects bone remodeling through its effects on osteoblasts and osteoclasts; it induces production of PGE2, nitric oxide (NO), and receptor activator of NFκB ligand (RANKL) by osteoblasts, leading to osteoclast differentiation and activation, indirectly in favor of bone destruction. Interestingly, it was recently shown that IL-17 can also directly induce osteoclastogenesis from human monocytes in the absence of osteoblasts, and such effect is TNF-α dependent [105]. IL-17 also upregulates production of proinflammatory cytokines, such as IL-6, granulocyte-monocyte colony-stimulating factor (GM-CSF), IFN-γ, and TNF-α by fibroblasts, epithelial, endothelial cells, monocytes, and bone cells, also in favor of bone loss [89, 106–109]. In addition, IL-17 increases expression of chemotactic factors by osteoblasts, such as CCL2 and CXCL5, promoting recruitment of leucocytes, including neutrophils and T cells [108, 110], that are able to produce factors (e.g., IL-6, IL-1, TNF-α, and RANKL) that will further affect bone resorption.
2.4.2. IL-17 and Synovial Inflammation
IL-17 promotes joint degradation by acting on synoviocyte activation, survival, and migration. It increases expression of inflammatory cytokines and chemokines by synoviocytes, such as IL-6, IL-8, CCL20, TNF-α, and IL-23p19 subunit. IL-17 also contributes to the production of matrix metalloproteinases by the cells, including MMP3, MMP9, and MMP13, which drive degradation of the extracellular matrix within the joint [111–116]. Furthermore, IL-17 can synergize with other inflammatory cytokines, such as TNF-α, IL-1β, or IL-17F in synoviocytes [114, 117]. IL-17-induced chemokine production (e.g., IL-8, CXCL2, CCL20, CCL2, CXC5, and CCL5) by various cell types, including synoviocytes and synovial macrophages, contributes to recruitment of neutrophils, lymphocytes, and macrophages to the synovium, thereby enhancing inflammation [3, 108, 113, 118, 119]. Interestingly, IL-17 increases cadherin-11 expression in patients with RA as well as in mice with CIA, an adhesion molecule contributing to synovial inflammation and cartilage degradation [120]. IL-17 also increases IL-6 production by RA synovium explants while inhibiting type 1 collagen synthesis [121]. Recent data reported by Kato et al. suggest that IL-17 produced by Th17 cells is more important in the induction of proinflammatory cytokines rather than in the induction of cell-cell interaction molecules by synoviocytes, two relevant components of synovial inflammation [122]. In addition, IL-17 increases survival and motility of synoviocytes from RA patients [117, 123], and it was reported that migration of activated RA synoviocytes has the ability to spread arthritis to unaffected joints [124].
Formation of new vessels largely contributes to the formation and maintenance of the pannus in RA and, therefore, to cartilage and bone damage. IL-17 also contributes to angiogenesis by increasing production of proangiogenic factors by synoviocytes, such as vascular endothelial growth factor [125, 126].
2.4.3. IL-17 and Cartilage Remodeling
Another important target in joint inflammation is cartilage, and destruction of cartilage is a major consequence of chronic synovitis. Stimulation of normal and osteoarthritic human chondrocytes with IL-17 induces NO production as well as expression of genes and proteins associated with joint inflammation and cartilage degradation, such as inducible NO synthase, cyclooxygenase 2, IL-1β, IL-6, IL-8, CCL2, and MMP. These effects are mediated through activation of the MAP kinases, NFκB, and AP-1 signaling pathways [127–129]. In line with these observations, IL-17 inhibits proteoglycan synthesis by cartilage, and intra-articular administration of IL-17 in mice leads to cartilage destruction [121, 130]. Effects of IL-17 and IL-17 receptor signaling in promoting cartilage damage have been further confirmed in vivo in mouse models of CIA, and blocking IL-17 or IL-17 receptor deficiency reduces cartilage degradation [94, 97, 111]. Furthermore, IL-17 can synergize with other proinflammatory cytokines, such as TNF-α, to promote cartilage destruction [128, 131].
2.4.4. IL-17 and Enthesitis
The enthesis, located at the junction of tendon to bone, is the primary site of articular inflammation in SpA. Increased levels of IL-23 and IL-17 have been observed in sera from patients with SpA, like ankylosing spondylitis (AS) [90, 132–134]. Importantly, IL-23 was very recently identified as a major cytokine driving entheseal inflammation in vivo [135]. Notably, IL-23-sensitive cells in entheses are resident CD3+CD4−CD8−RORγt+ T cells. They allow the joint tissue to respond to IL-23 by secreting proinflammatory cytokines. The IL-23-mediated enthesitis is reduced in the presence of IL-17 and IL-22 neutralizing antibodies; however, in contrast to IL-22, IL-17 alone is not sufficient to induce enthesitis [135].
3. IL-17 and Inflammatory Rheumatisms
3.1. IL-17 in Autoimmune Diseases
3.1.1. Rheumatoid Arthritis
RA is the most frequent autoimmune arthritis in the world affecting around 1% of general population. It is a public health issue because of its chronicity and the progressive joint destruction experienced by some patients. The disease is characterized by inflammation of the synovium with a T cell, B cell, and proinflammatory cytokine infiltration. Etiology and pathophysiology of RA are not fully understood but an immunological conflict may precede the development of clinical stages of the disease [136]. Various environmental factors influence the development of the disease on specific genetic basis. These external and internal factors are not completely known, but are subjects of intense research. Complex immune modulator interactions resulting from this immunological conflict are at play in the joint, and therapeutic strategy largely uses this pathogenesis concept. A great range of immunomodulatory molecules is available in RA treatment from steroids to biologic and nonbiologic disease modifying antirheumatic drugs, of which the most used are, respectively, TNF-α blockers and methotrexate. Among proinflammatory cytokines, IL-17 axis seems to be of importance in RA pathophysiology, and both Th17 cells and mast cells have been described as IL-17 sources in inflamed joints of RA patients [102, 103]. Synoviocytes have been shown to produce CCL20 in autoimmune arthritis like RA, thus recruiting Th17 cells via CCR6 [103].
Autoimmune arthritis in animal models has been long considered as Th1 dependent. However, accumulating evidence is now in favor of a crucial role of Th17 cells. In CIA, development of joint destruction remains present in IFN-γ receptor-deficient mice [137, 138], whereas disease activity is markedly reduced in IL-17-deficient mice [82] as well as after blockade of IL-17 [97]. In a quite different model, RAG-deficient mice receiving naive CD4+ T cells from SKG mice, that are genetically prone to spontaneously develop chronic autoimmune arthritis, exhibit a Th17-dependent polyarthritis [139]. It has been shown that IL-1-receptor-antagonist- (IL-1Ra-) deficient mice develop spontaneous arthritis secondary to their increased sensitivity to IL-1 [140], and T cells play a critical role in this model since IL-1Ra deficient mice lacking T cells do not develop arthritis [141]. Interestingly, IL-1Ra-deficient mice present increased number of Th17 cells, and spontaneous development of arthritis is abrogated when associated with IL-17 deficiency (or IL-17 neutralization) [140, 142], demonstrating the great involvement of Th17 cells in this IL-1-driven arthritis model. However, after the onset of arthritis, neutralization of IL-17 prevents any further increase of the disease but does not reduce the arthritis score [140]. A critical role of IL-17 in development of arthritis has also been observed in F759 mice (characterized by increased STAT3 activation) [143] and specific-pathogen-free K/BxN mice treated with neutralizing anti-IL-17 antibody [144], two models of mice predisposed to develop T cell-dependent arthritis. All these results are in favor of a major role of Th17 cells in the development of T cell-dependent arthritis in mice.
Further experiments have explored the role of Th17 cells in autoimmune arthritis. On one hand, Th17 cells are proinflammatory; they are responsible for inducing the migration of innate immune cells with, as a result, an increase in the production of proinflammatory cytokines, chemokines, and matrix-degrading enzymes from these cells [145]. In addition, circulating Th17 cells from RA patients have the propensity to induce IL-6, IL-8, and MMP expression by RA synoviocytes [104], further pointing out the pathogenic role of Th17 cells in joint inflammation and degradation. On the other hand, Th17 cells promote autoimmunity; they generate the production of autoantibodies in several mouse models by enhancing germinal center formation, for example [146].
Beside Th17 cells, γδ T cells also contribute to IL-17 production in inflamed joints in the CIA mouse model [52, 147]. Although these IL-17 innate producers exacerbate CIA in mice [52], a recent study by Pöllinger et al. showed that in this mouse model of arthritis, Th17 cells, rather than IL-17+ γδ T cells, drive osteoclast-mediated joint degradation [147].
In human RA, there is some evidence of IL-17 involvement. Metawi et al. determined that IL-17 serum levels are higher in patients with inflammatory arthritis compared to healthy controls [148]. Th17 and Th22 cells have been found increased in peripheral blood of patients with RA, and levels are positively correlated with disease activity [149, 150]. IL-17 has also been found in joint tissue in higher quantity in RA than in osteoarthritis. It was linked in the same study to the production of matrix degradation molecules further proving the role of IL-17 in the pathophysiology of the disease [151]. Results on γδ T cells in mice are in line with human data showing that γδ T cells are not a prominent source of IL-17 in patients with RA [152].
Taken together, the IL-17/Th17 pathway seems greatly involved in the initiation process of autoimmune arthritis as well as in the inflammation stage of the disease, and IL-17-producing cells represent an attractive target in RA treatment.
3.1.2. Systemic Lupus Erythematous
SLE is an autoimmune disease characterized by its chronicity and the reciprocation of flare and remission periods. It can affect a lot of organs, among them joints, kidneys, skin, or nervous system [153]. Patients with SLE usually exhibit antinuclear antibodies, whose pathogenicity remains unclear. The prevalence rate displays large worldwide variations, due to genetic and environmental factors, and SLE affects around 0.2 to 2 in 1000 individuals [154]. It is predominantly a disease of women with a mean sex ratio of 1 to 9, male to female. Level of disease severity is extensive, and treatments range from preventive measure and hydroxychloroquine to heavy immunosuppressive drugs.
Pathophysiology is far from fully understood but great advances have been made in the past few years. It involves complex interaction between environmental and genetic factors, and the presence of circulating autoantibodies directed against intracellular antigens, such as DNA, appears to be one of the major events in disease initiation [153]. These autoantibodies are involved in the pathogenesis since they complex with antigens, thus activating effector responses. The resulting tissue destruction exposes more intracellular antigens and sustains the reaction. Throughout the importance of these autoantibodies, SLE has traditionally been considered as a B cell-dependent disease. However, there is increasing evidence that T cells have a major place in SLE mechanisms. In this context, the role of Th17 cells during SLE has recently become subject of increasing attention [155].
It has been shown that the phenomenon of organ injury following ischemia is greatly dependent on Th17 cells in MRL/lpr mice, a lupus-prone model of mice [92, 156]. This is partly reversed by CD4 depletion or IL-17 deficiency, especially regarding tissue damages. At baseline, MRL/lpr mice present a higher frequency of IL-17-producing cells than nonautoimmune mice like B6 strain (unpublished data, [155]). Higher production of IL-17 is observed from SNF1 (lupus-prone mice) splenocytes cultured with nucleosomes than from B6 splenocytes [157]. A decrease of both IL-17 production and of Th17 cell infiltration in the kidney is found altogether with clinical improvement observed after tolerance induction with a histone-derived peptide. Taken together, these results confirm that Th17 cells are increased in lupus models, but they also seem to be involved in pathogenicity.
Similar IL-17/Th17 involvement is demonstrated in BXD2 mice (a strain of mice genetically engineered to develop autoimmune manifestations), where the humoral response is strongly increased. This is not independent of the presence of IL-17-producing cells since they have been demonstrated to have a major impact on germinal center development in the spleen [158]. Again, besides their proinflammatory profile, Th17 cells play a direct role in autoimmunity generation. In this context, the recent description of T follicular helper (TFH) cells, T cells helping B cells in an extrafollicular location, is of great importance [159]. This cell population has been observed in lupus-prone strains of mice. Those cells produce IL-17 and IL-21, the latter playing the job of helping B cells. However, it has been proposed that high production of IL-17 favors IL-21 secretion, giving an indirect role to IL-17 in the generation of autoimmunity by TFH.
In human, IL-17 is able to increase immunoglobulin production and thus anti-DNA antibodies in cells from SLE patients [155]. Indirect evidence of the role of IL-17 in human SLE is the increased level of that cytokine along with IL-23 and IL-21 in patient sera [160–162]. The origin of IL-17 seems to be CD4+ T cells and CD3+ double negative T cells. Patients with SLE have increased number of circulating IL-17-producing Th17 cells and CD3+ CD4− CD8− T cells than healthy controls, and the frequency of Th17 cells correlates with disease activity [98, 162–164]. More interestingly in the pathogenesis point of view, IL-17 has been histologically found in lupus nephritis, and IL-17 expression positively correlates with disease activity [160, 164, 165]. Finally, genetic associations with SLE have been highlighted with Th17-associated molecule polymorphisms as IL-21, and genetic variants decreasing Th17 differentiation are associated with a higher risk of developing SLE [166]. The very recent identification of IL-17-producing B cells as important players to combat trypanosome infection [48] begs the question of whether IL-17 production by B cells in SLE is dysregulated.
3.1.3. Systemic Sclerosis
SSc is a rare connective tissue disease characterized by excessive extracellular matrix deposition in internal organs, like skin and lungs [167]. It affects approximately 1 per 10000 adult individuals and is highly dependent on the geographical location [168]. The ratio of women to men is about 4 : 1. Although survival in SSc has improved over the past several decades, SSc is still associated with a poor outcome. Despite the heterogeneity of the disease, disfigurement associated to cutaneous lesions, arthritis, fatigue, and dyspnea recapitulate the majority of patient complains. Besides, the major causes of invalidity and impairment of vital prognosis are digital ulcers, lung fibrosis, and pulmonary arterial hypertension. Therapeutic weapons used in SSc are largely nonspecific but lead to a slight decrease in mortality rates. Clinical progresses are mostly linked to vascular treatments and immunosuppressive strategy, although widely used in severe cases, such therapies have limited efficiency and are associated with significant side effects [169].
To date, SSc pathophysiology is still largely unknown, explaining the poor effectiveness of therapeutic strategy in SSc. Pathology includes vascular abnormalities, immune activation, and fibrosis [170], but the relationships between the three entities are still matter of debate. Accumulating evidence is in favor of a role of T cells in those mechanisms [167]. First, genetic studies indicate that most of the gene polymorphisms associated with SSc involve genes coding for molecules controlling T cell differentiation or activation, some shared with other autoimmune disorders like SLE [171, 172]. Second, histological examination of SSc skin during the early oedematous inflammatory phase of the disease demonstrates the presence of mononuclear cell infiltrates containing T cells, with perivascular distribution, preceding the development of fibrosis and overt vasculopathy [173]. These findings led to the hypothesis that T cells provide important stimuli that drive collagen synthesis in fibroblasts, propelling these cells to the forefront of SSc pathophysiology. Defining the T cell subsets at play has been the next challenge, and this issue is far from completely solved. On one hand, Th2 cells, mainly through their prototypic cytokines, are certainly involved in the disease fibrosis process [174]. On the other hand, some evidence points out the role of IFN-γ and Th1 cells [175]. However, accumulating reports over the last few years highlighted IL-17 and Th17 cells as important actors of the disease [176, 177].
IL-17 has been shown to be involved in the development of bleomycin-induced mouse lung fibrosis in an IL-1-dependent way [178]. In two different models of mouse SSc, importance of IL-17 was suggested. In bleomycin-induced skin fibrosis, the loss of IL-17 decreases the fibrotic process, and higher IL-17 mRNA levels are found than in wild-type skin [179]. IL-17 deficiency also attenuates skin thickness in tight skin 1 (TSK-1) mice, a strain of mice presenting spontaneous mutation in fibrillin-1 gene and used as a model of SSc. Furthermore, IL-17 stimulates directly collagen synthesis in rodent fibroblasts [180]. Animal models are poorly relevant for SSc human pathogenesis, but these are first clues of IL-17 involvement.
Increased levels of IL-17 are detected in the sera and bronchoalveolar lavage fluids of SSc individuals [181]. We and others observed an increase in Th17 and Th22 cells frequency in peripheral blood of SSc patients, further enhanced by some SSc treatment via monocyte production of IL-23 among others [176, 182]. In the skin of SSc patients, we recently showed an increase in IL-17-producing cells with an inverse correlation to the skin fibrosis score [177]. In vitro, IL-17 is able to partially inhibit the expression of α-smooth muscle actin induced by TGF-β and to induce the secretion of MMP1 in human dermal fibroblasts, and conversely to rodent, human fibroblasts do not produce collagen in response to IL-17. The difference in mouse and human responses to IL-17 may be explained by species-specific characteristics in the IL-17 biology, as it has been previously seen for Th17 differentiation, bringing caution towards murine models regarding the extrapolation of therapeutic strategies [183]. The hypothesis that, in humans, IL-17 and Th17 cells in SSc could be more related to inflammation, autoimmunity, and possibly to the generation of autoantibodies is seductive, and until now, no direct argument for the role of this pathway in the SSc arthritis pathophysiology has been reported in the literature.
3.2. IL-17 in Inflammatory Arthritis
3.2.1. Psoriatic Arthritis
Psoriatic arthritis (PsA) belongs to the spondylarthritis group of diseases and is characterized by a chronic inflammation of joints and skin. Peripheral and axial joints can be affected by the disease, with a potential breach of enthesis and synovial membranes in the meantime. PsA is a frequent inflammatory rheumatism, nearly as frequent as RA, as it concerns about 0.3 to 1% of general population [184]. Its presentation and course are highly variable. Mostly, skin involvement precedes joint inflammation, but the osteoarticular lesions may be present before the development of psoriasis in 10% of cases. Psoriasis is completed with arthritis in one-third of the patients during the development of the disease. The persistence of inflammation in joints can lead to destruction and severe disabilities. Until now, therapeutic strategies in PsA are often directly inspired by those used in RA.
Nevertheless, despite common features, PsA differs from RA in some aspects. The early events in PsA pathogenesis occur in genetically predisposed subjects and are mediated by T cells interaction with antigen-presenting cells. The location of the first immune conflict is not really defined but TNF-α seems to play an important role, and TNF-α blockers remain highly used and efficient treatments of the disease. The first antigen is still unknown; nevertheless, it induces a T cell-specific reaction followed by a proinflammatory cytokine secretion cascade. PsA, as with other rheumatic inflammatory disorders, was considered until recently as a Th1-dependent disease with IFN-γ playing an important role in the generation of that cascade. Psoriatic disease encompasses psoriasis and the involvement of musculoskeletal and gastrointestinal and ocular systems. Thus, PsA pathogenesis is closely connected to that of psoriasis. Much less information is available in PsA pathogenesis than in skin psoriasis, but both have susceptibility associated with alleles of the IL-12B and IL-23R genes [185, 186]. Moreover, IL-12/23 p40 subunit is elevated in sera of PsA patients [187]. Given the importance of IL-23 in Th17 biology, it suggests a role of this subset in PsA. Finally, with the importance of the IL-17 pathway in psoriasis and in autoimmune arthritis murine models, the role of IL-17/Th17 has been naturally evoked in PsA. IL-17 is increased in PsA synovial tissue and fluid. IL-17RA is overexpressed by PsA synoviocytes [188], is functionally active, and regulates the synoviocyte secretion of proinflammatory cytokines and matrix metalloproteinases tightly involved in the joint damages observed in PsA. Another indirect evidence of the role of IL-17 in PsA pathogenesis has been recently reported with the involvement of the adaptor protein Act1 in the disease through genome-wide association or functional studies [189, 190]. The correlation between disease activity and levels of IL-17 or Th17 cytokines in synovial fluid is variable [188, 191]. Differences in disease patterns or treatment regimen in studied population could be the reason of this discrepancy. It appears that the earlier and the more free of any treatment the patient is, the more correlated is the disease activity with Il-17 rates. These findings are in accordance with the bone erosive role of IL-17 demonstrated in vitro and highlight its role not only in the skin, but also in all major components of psoriatic disease. Whether or not innate immune cells contribute to IL-17 production in PsA is still unknown.
3.2.2. Ankylosing Spondylitis
AS is a systemic disease characterized by enthesopathy and ossification of the joints [192]. It is the most frequent member of the spondylarthritis group, and its prevalence ranges between 0.2 and 1.2% of general population. Affecting more often men than women (3 : 1), its peak is around the third decade of life. The clinical characteristic of AS is the axial joint damages, most notably the sacroiliac joints. Ossification and ankylosis are typical of the disease, but wide extraarticular manifestations can occur such as digestive and ocular but also and less frequently heart or lung damages. These extraarticular manifestations, expressing the systemic nature of the disease, are also involved in the therapeutic strategy. All spondyloarthropathies are associated with human leukocyte antigen (HLA)-B27 expression, the highest association is yet in the case of AS, as 90% of patients are HLA-B27 positive.
The pathogenic role of HLA-B27 in AS has been and is still debated. Hypotheses are as varied as arthritogenic peptide presentation, aberrant folding of surface heavy chains, or enhancing of intracellular microbial survival [193]. AS is an inflammatory disease, and to date, no specific antibodies have been identified. The immune system is still highly involved in the disease pathogenesis. The privileged association with HLA-B27 might be a clue for CD8+ T cells involvement. Even so, transgenic HLA-B27 rats, used as prototypic AS rodent model, present colitis and arthritis independently of CD8+ T cells, raising the question of other important players in the disease pathogenesis. Some proinflammatory cytokines are constantly increased in AS patients, such as TNF-α, IL-6, and IL-2 receptors [194].
Pathophysiology of AS has been recently enriched with genetic and more precisely genome-wide association studies, linking the disease to IL-23 receptor gene [195]. Other findings are in favor of the IL-23/IL-17 pathway involvement notably in murine models [36].
SKG mice are genetically prone to develop autoimmune arthritis, and curdlan injection can drive spondylarthritis symptoms [196]. The pathology is at least partly driven by IL-17-secreting γδ T cells and IL-17 deficiency ameliorates symptoms [197]. IL-23 was also very recently shown to be directly involved in AS and more precisely in the development of enthesitis in a collagen antibody-induced arthritis mouse model [135]. IL-23 induces AS in mice through the activation of IL-17/IL-22-producing RORγt+CD3+CD4−CD8− T cells directly located in the entheses. The mice phenotype is not only enthesitis, but it also recapitulates all features of AS [135].
In human, IL-17 is expressed in sacroiliac joints biopsies from AS patients. However, many arguments converge towards the involvement of innate immune cells. Notably, mast cells, neutrophils, and γδ T cells seem to be good candidates as IL-17-secreting cells in AS joints [101]. More indirectly, IL-23R expression has been found increased in active sites of the disease in AS patients, such as tendon-bone junction and aortic root [135]. Finally, Bowness et al. recently raised an interesting issue by establishing a link between HLA-B27 and IL-17 pathway [198]. They showed that APC expressing B27 β2 microglobulin-free heavy chain homodimers are prone to induce the proliferation of specific Th17 cells. These cells produce IL-17 and/or IFN-γ due to a high plasticity and are found in AS patients, suggesting their involvement in the disease pathogenesis [198].
4. Therapeutic Applications: Strategies and Molecules Targeting the IL-17/Th17 Pathway in Inflammatory Rheumatisms
Regarding all the findings involving the IL-17/Th17 pathway in inflammatory rheumatisms, the idea to target this pathway has become more and more attractive. The topic is discussed as a strategy more than a single treatment because of the complexity of the inflammatory process at play in IL-17 biology. There are several potential targets in the cascade leading to IL-17 effects, and we will describe those currently under consideration.
Standard treatment in RA is association of methotrexate together with a TNF-α blocker when an adequate response is not achieved.
4.1. Direct Targeting of IL-17
Directly targeting the cytokine is a classic strategy in biologic development based on monoclonal antibodies production. Owing to TNF-α blockers such as monoclonal antibodies (infliximab, Remicade and adalimumab, Humira) or receptor-targeting fusion protein (etanercept, Enbrel), a great experience and hindsight regarding this type of treatment exist in rheumatology.
Several molecules are in the pipeline of development with some advanced data of clinical trials (http://www.biocentury.com/targets/il-17).
Two monoclonal antibodies directed against IL-17 are currently tested in humans. Ixekizumab (LY2439821), a humanized hinge-modified IgG4 IL-17-specific antibody developed by Lilly is in a phase III trial for psoriasis and for PsA (clinicaltrial.gov, identifiers NCT01597245, NCT01624233, and NCT01646177). It has already completed a phase I and a phase II trial in RA ([199] and clinicaltrial.gov, identifiers NCT00966875). Secukinumab (AIN457), a fully human IL-17-specific IgG1k monoclonal antibody generated by Novartis is also in advanced clinical development. This molecule is in phase III for chronic plaque psoriasis, PsA, RA, and AS, and in phase II for chronic noninfectious uveitis [200, 201].
First results from trials with both antibodies are quite encouraging with an improvement in symptoms and a good safety profile. In a double-blind, placebo-controlled, parallel-group, phase IIA study (N = 36) in moderate to severe psoriasis, a single infusion of secukinumab (3 mg/kg) resulted in rapid and sustained improvement of psoriasis symptoms, with at week 4, 83% of secukinumab patients versus 11% of placebo patients achieving significant efficacy [201]. The effect was less impressive in a double-blind, placebo-controlled phase IIA study assessing safety and efficacy of subcutaneous secukinumab in PsA. The difference in rate of American College of Rheumatology 20 score (ACR20) response at week 6, with secukinumab or with placebo, was not statistically significant. Significant efficacy compared to placebo was finally reached at weeks 12 and 28.
Other IL-17-targeted antibodies are in early clinical development, such as SCH-900117 and RG4934. Brodalumab (AMG827), a human anti-IL-17RA antibody developed by Amgen/MedImmune has shown remarkable efficacy for the treatment of psoriasis in a phase II double-blind, placebo-controlled, dose-ranging study [202] and is currently in Phase II trial in RA and PsA [203].
In addition, new strategies under consideration aim to inhibit biological activities of more than one cytokine. For example, Roche is testing a blocking antibody targeting both IL-17A and IL-17F (RG7624, no clinical information available; Roche website and [36]). In addition, based on the synergistic activities of IL-17 and TNF-α, an anti-TNF-α/IL-17 bispecific neutralizing antibody (ABT-122, Abbott) is being tested in phase I in RA (Abbott website and [36]).
4.2. Indirect Targeting of the IL-17 Pathway
Current or in development tools for targeting the IL-17/Th17 pathway are also showing promising results to treat inflammatory rheumatisms.
4.2.1. IL-23 and/or IL-12 Targeting
Ustekinumab (Stelara) is a human monoclonal antibody engineered by Janssen Biotech that targets the p40 subunit of IL-12 and IL-23, and therefore inhibits both IL-23 and IL-12 signaling. It has been approved for psoriasis treatment since 2009 [204]. In a phase II double-blind, placebo-controlled, crossover study in patients with active PsA, subcutaneous injections of ustekinumab significantly reduced signs and symptoms of PsA and improved skin lesions and physical function in patients [205, 206]. Ongoing phase III trials will establish the benefice/risk profile of ustekinumab in this disease.
Given the major role of the IL-23/IL-17 pathway in inflammation and autoimmunity, new drugs in development specifically aim to neutralize only the IL-23 pathway and three anti-p19 neutralizing antibodies, MK-3222, CNTO 1959, and AMG 139, respectively, developed by MERCK, Janssen Biotech, and Amgen/MedImmune are currently in clinical trials for psoriasis, as well as Crohn's disease for AMG 139 [203, 207]. Only available clinical data reported that administration of MK-322 in psoriatic patients markedly decreases cutaneous inflammation. Such effects are associated with a significant reduction of T cells, dendritic cells, neutrophils, and macrophages in the inflammatory infiltrate [208]. These promising results suggest that neutralization of IL-23 but not IL-12 could be sufficient to inhibit downstream signaling cascades involved in disease development.
In this context, clinical trials assessing the effect of IL-23 blockade could also represent an interesting approach for the treatment of inflammatory rheumatisms.
4.2.2. IL-6 Targeting
IL-6 is another proinflammatory cytokine involved in the development of joint inflammation, and Tocilizumab (RoActemra) has proven its efficacy for few years in RA patients and is also approved since 2011 for treatment of systemic juvenile idiopathic arthritis [209]. It is a humanized anti-IL-6 receptor monoclonal antibody developed by Roche that binds both soluble and membrane bound IL-6 receptor and prevents IL-6 binding to its receptors [210]. The use of Tocilizumab is growing in RA patients, particularly in those with an inadequate response to methotrexate or TNF-α inhibitors therapies [211–213]. It is administered through intravenous infusion every four weeks and can be used as a monotherapy or in combination with methotrexate. Interestingly, a recent clinical trial compared the efficacy of tocilizumab versus adalimumab (anti-TNF-α antibody) as monotherapy in RA and revealed a significant greater disease improvement in patients treated with tolicizumab [214].
While IL-6 targeting is showing efficiency in RA, tolicizumab therapy has been disappointing in SpA patients [214, 215].
Other IL-6 targeting therapies in development include BMS945429, a humanized anti-IL-6 antibody engineered by Alder Biopharmaceuticals, and sarilumab, a human anti-IL6 receptor α (IL-6Rα) antibody codeveloped by Regeneron and Sanofi [214]. Phase II, double-blind, randomized, placebo-controlled studies showed that BMS945429 or sarilumab treatment associated with metothrexate induced significant improvement of disease activity in patients with active RA and inadequate response to methotrexate [214, 216]. If sarilumab is giving promising results for RA treatment, a phase II clinical trial assessing the effect of this anti-IL-6Rα antibody in AS did not show any significant efficacy [214].
4.2.3. IL-1 Targeting
IL-1 targeting started in 1993 with anakinra (Kineret), a recombinant IL-1 receptor antagonist developed by Amgen, which inhibits both IL-1α and IL-1β activities. It was approved for RA treatment in 2001 and is being tested in a number of diseases, including autoimmune and autoinflammatory diseases (e.g., severe atopic dermatitis, osteoarthritis of the knee), but also in diseases that are not inflammatory, like heart failure and type 2 diabetes [217]. Treatment with anakinra is fastidious for RA patients as it has to be daily injected because of its short half-life, and its efficacy is similar to other biologics; therefore, anakinra is not among first-line therapies.
Two other IL-1 targeting agents have been approved for the treatment of cryopyrin-associated periodic syndromes: rilonacept (Arcalyst, Regeneron), a soluble decoy receptor, and canakinumab (Ilaris, Novartis), a humanized monoclonal antibody against IL-1β. Several additional agents blocking IL-1 are in clinical trials in several diseases (e.g., stroke, diabetes, and chronic inflammatory diseases) and target the IL-1 receptor, IL-1α, IL-1β, or caspase 1 (crucial for IL-1β activity). IL-1β and IL-1 receptor neutralizing antibodies are currently tested in arthritis and joint diseases [217].
4.3. Targeting Signaling Pathway Molecules
Transcription factors modulators are very trendy among developing therapeutic strategies, and in USA, it represents 13% of current U.S. Food and Drug Administration-approved drugs.
4.3.1. RORγt and/or RORα Inhibition
As mentioned before, RORγt is both necessary and sufficient for induction of IL-17 production in human cells. This transcription factor represents potential therapeutic target and can be blocked by specific inhibitors. As ROR family is earlier in the IL-17 production cascade, its targeting can not only diminish the secretion of this cytokine but also favors the shift from Th17 towards regulatory T cells. This more complex effect could be of interest in the treatment of autoimmune diseases where an imbalance between effector and regulatory T cells has been observed. Nevertheless, as not being the only transcription factor involved in Th17 differentiation, RORγt inhibition might not have a complete IL-17 suppressive effect.
Small molecules have been described as capable of inhibiting the ROR family and thus target the IL-17 pathway. Digoxin, a cardiac glycoside, has been shown to be a specific inhibitor of RORγt independently of RORα [218]. SR1001 is a high-affinity synthetic ligand for both RORα and RORγt, and Solt et al. demonstrated its inhibitory effect on differentiation and function on Th17 cells. It induces conformational changes in the molecule, leading to a higher affinity for corepressors and lower for coactivators [219]. Ursolic acid is a third molecule involved in the inhibition of the IL-17 pathway through its action on the ROR family member RORγt [220]. Evidence for this molecule efficacy on IL-17 modulation in autoimmune diseases is still limited to in vitro or murine studies. To our knowledge, no molecules directly targeting ROR family are currently at clinical trial phase in human. Nevertheless, because it regulates RORγt expression, forkhead box P3 (FoxP3) upregulation can indirectly induce a decrease of IL-17 production. Molecules such as simvastatin (3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor), commonly administrated in atherosclerosis treatment, enhance FoxP3 expression and inhibit the production of IL-17 [221].
4.3.2. Janus Kinases (JAK)-STAT3 Inhibition
As stated before, IL-23, IL-6, and IL-21 are involved in Th17 differentiation, and they all act through the JAK-STAT signaling pathway. Thus, STAT3 has become an interesting potential target, at the convergence point of different upstream activators. It has been recently shown in vitro that STAT3 inhibition in synovial T cells of RA patients suppresses Th17 pathway [222]. Research in that field remains at the discovery stage; this demonstration is being done using siRNA, and no therapeutic molecules are currently tested in human clinical trials in the domain of autoimmune diseases. However, clinical trials are completed in the area of oncology, and this experience might be useful in a near future in the immunology field as it happened for rituximab [223].
On the other hand, the anti-JAK strategy is far much advanced with the development of tofacitinib, an oral JAK inhibitor, tested in a phase III clinical trial in the treatment of RA [224]. This molecule directly inhibits the production of IL-17 and IFN-γ, resulting in a decrease of proinflammatory cytokine production and synovitis [225]. Tofacitinib effect is also assessed for other inflammatory diseases involving IL-17 such as psoriasis or ankylosing spondylitis. STAT3 and JAK are closely linked as the second phosphorylates the first leading to its nuclear translocation and biological activity [226]. Inhibition of JAK molecules might have several effects, but it was demonstrated that tofacitinib inhibits IL-17 secretion in vitro [227]. This could be one of its mechanisms of action.
4.3.3. Phosphoinositide 3-Kinase δ-Subunit (PI3Kδ) Inhibition
The PI3K/Akt pathway is involved in both the pathogenesis of RA and IL-17 production [228, 229]. It is therefore a potential target in the scope of research in the autoimmunity therapeutic agent field. ZSTK474, a general PI3K inhibitor, has shown the ability to inhibit synovial inflammation, osteoclastic activity, and finally collagen-induced arthritis in vitro and in murine models [230]. Class 1 PI3K exhibits two isoforms of the catalytic subunits, p110γ and p110δ that are enriched in leucocytes. The selective inhibition of one of those subunits has attracted a major interest due to data of in vitro models notably in collagen- or in antigen-induced arthritis [231–233]. Finally, the rationale seems to be strong enough in RA and SLE literature to bridge the gap between the bench and bedside research. Promising results have been obtained with the p110δ inhibitor CAL-101 in the field of lymphomas giving reassuring data about the safety of this molecule, but clinical trials are still ongoing, and data remain unpublished [234].
5. Conclusion
It is now clearly demonstrated that IL-17 is deeply involved in autoimmune and inflammatory processes. Joint is a prime target of IL-17 action, and all compartments appear to be concerned by the action of this cytokine. Mechanisms are intensively studied because of potential therapeutic strategies that may arise. All of the pathogenesis is far from elucidated, but despite this fact, many molecules that target the IL-17/Th17 pathway are already under development or even tested in clinical trials for the treatment of autoimmune or inflammatory diseases.
Very recently, a new mechanism has been pointed out in the IL-17-autoimmunity interaction, bringing a new component in the system going awry: salt. Two different studies showed that in an isotonic culture medium, an elevated sodium chloride (NaCl) concentration promotes the differentiation of Th17 cells in vitro [235, 236]. One even brought in vivo evidence of the proautoimmune effects of a high-salted diet. These intriguing studies reassert the role of environmental factors in these diseases. It also emphasizes that there is not one Th17 cell but Th17 cells. These cells act differently to protect or to damage tissues, leading to an even more complicated story than it was initially imagined, and certainly quite far from what is observed in animal models.
Acknowledgment
The authors thank Cristina Tato for her careful reading of the paper.
References
- 1.Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nature Immunology. 2005;6(11):1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 2.Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. Journal of Experimental Medicine. 2005;201(2):233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park H, Li Z, Yang XO, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nature Immunology. 2005;6(11):1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Annunziato F, Cosmi L, Santarlasci V, et al. Phenotypic and functional features of human Th17 cells. Journal of Experimental Medicine. 2007;204(8):1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dardalhon V, Awasthi A, Kwon H, et al. IL-4 inhibits TGF-β-induced Foxp3+ T cells and, together with TGF-β, generates IL-9+ IL-10+ Foxp3-effector T cells. Nature Immunology. 2008;9(12):1347–1355. doi: 10.1038/ni.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nature Immunology. 2009;10(8):857–863. doi: 10.1038/ni.1767. [DOI] [PubMed] [Google Scholar]
- 7.Rouvier E, Luciani MF, Mattei MG, Denizot F, Golstein P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus Saimiri gene. Journal of Immunology. 1993;150(12):5445–5456. [PubMed] [Google Scholar]
- 8.Yao Z, Fanslow WC, Seldin MF, et al. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. 1995;3(6):811–821. doi: 10.1016/1074-7613(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 9.Yao Z, Painter SL, Fanslow WC, et al. Human IL-17: a novel cytokine derived from T cells. Journal of Immunology. 1995;155(12):5483–5486. [PubMed] [Google Scholar]
- 10.Aggarwal S, Gurney AL. IL-17: prototype member of an emerging cytokine family. Journal of Leukocyte Biology. 2002;71(1):1–8. [PubMed] [Google Scholar]
- 11.Gaffen SL. Structure and signalling in the IL-17 receptor family. Nature Reviews Immunology. 2009;9(8):556–567. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toy D, Kugler D, Wolfson M, et al. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. Journal of Immunology. 2006;177(1):36–39. doi: 10.4049/jimmunol.177.1.36. [DOI] [PubMed] [Google Scholar]
- 13.Seon HC, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. Journal of Biological Chemistry. 2006;281(47):35603–35607. doi: 10.1074/jbc.C600256200. [DOI] [PubMed] [Google Scholar]
- 14.Qian Y, Liu C, Hartupee J, et al. The adaptor Act1 is required for interleukin 17—dependent signaling associated with autoimmune and inflammatory disease. Nature Immunology. 2007;8(3):247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- 15.Kolls JK, Lindén A. Interleukin-17 family members and inflammation. Immunity. 2004;21(4):467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 16.Boniface K, Blom B, Liu YJ, de Waal Malefyt R. From interleukin-23 to T-helper 17 cells: human T-helper cell differentiation revisited. Immunological Reviews. 2008;226(1):132–146. doi: 10.1111/j.1600-065X.2008.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson NJ, Boniface K, Chan JR, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nature Immunology. 2007;8(9):950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 18.Boniface K, Bak-Jensen KS, Li Y, et al. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. Journal of Experimental Medicine. 2009;206(3):535–548. doi: 10.1084/jem.20082293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McGeachy MJ, Cua DJ. Th17 cell differentiation: the long and winding road. Immunity. 2008;28(4):445–453. doi: 10.1016/j.immuni.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 20.Zygmunt B, Veldhoen M. T helper cell differentiation. More than just cytokines. Advances in Immunology. 2011;109:159–196. doi: 10.1016/B978-0-12-387664-5.00005-4. [DOI] [PubMed] [Google Scholar]
- 21.Annunziato F, Romagnani S. Mouse T helper 17 phenotype: not so different than in man after all. Cytokine. 2011;56(1):112–115. doi: 10.1016/j.cyto.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 22.Chen Z, Tato CM, Muul L, Laurence A, O’Shea JJ. Distinct regulation of interleukin-17 in human T helper lymphocytes. Arthritis and Rheumatism. 2007;56(9):2936–2946. doi: 10.1002/art.22866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Volpe E, Servant N, Zollinger R, et al. A critical function for transforming growth factor-β, interleukin 23 and proinflammatory cytokines in driving and modulating human TH-17 responses. Nature Immunology. 2008;9(6):650–657. doi: 10.1038/ni.1613. [DOI] [PubMed] [Google Scholar]
- 24.Bettelli E, Oukka M, Kuchroo VK. TH-17 cells in the circle of immunity and autoimmunity. Nature Immunology. 2007;8(4):345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 25.Chizzolini C, Chicheportiche R, Alvarez M, et al. Prostaglandin E2 synergistically with interleukin-23 favors human Th17 expansion. Blood. 2008;112(9):3696–3703. doi: 10.1182/blood-2008-05-155408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126(6):1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 27.Yang XO, Pappu BP, Nurieva R, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors RORα and RORγ . Immunity. 2008;28(1):29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang XO, Panopoulos AD, Nurieva R, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. Journal of Biological Chemistry. 2007;282(13):9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 29.Veldhoen M, Hirota K, Westendorf AM, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453(7191):106–109. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- 30.Brüstle A, Heink S, Huber M, et al. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nature Immunology. 2007;8(9):958–966. doi: 10.1038/ni1500. [DOI] [PubMed] [Google Scholar]
- 31.Shi LZ, Wang R, Huang G, et al. HIF1α-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. Journal of Experimental Medicine. 2011;208(7):1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dang EV, Barbi J, Yang HY, et al. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell. 2011;146(5):772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delgoffe GM, Pollizzi KN, Waickman AT, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nature Immunology. 2011;12(4):295–304. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delgoffe GM, Kole TP, Zheng Y, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30(6):832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Graeber KE, Olsen NJ. Th17 cell cytokine secretion profile in host defense and autoimmunity. Inflammation Research. 2012;61(2):87–96. doi: 10.1007/s00011-011-0419-1. [DOI] [PubMed] [Google Scholar]
- 36.Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nature Reviews Drug Discovery. 2012;11:763–776. doi: 10.1038/nrd3794. [DOI] [PubMed] [Google Scholar]
- 37.Happel KI, Zheng M, Young E, et al. Cutting edge: roles of toll-like receptor 4 and IL-23 in IL-17 expression in response to Klebsiella pneumoniae infection. Journal of Immunology. 2003;170(9):4432–4436. doi: 10.4049/jimmunol.170.9.4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferretti S, Bonneau O, Dubois GR, Jones CE, Trifilieff A. IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. Journal of Immunology. 2003;170(4):2106–2112. doi: 10.4049/jimmunol.170.4.2106. [DOI] [PubMed] [Google Scholar]
- 39.Shin HCK, Benbernou N, Esnault S, Guenounou M. Expression of IL-17 in human memory CD45RO+ T lymphocytes and its regulation by protein kinase A pathway. Cytokine. 1999;11(4):257–266. doi: 10.1006/cyto.1998.0433. [DOI] [PubMed] [Google Scholar]
- 40.Liu SJ, Tsai JP, Shen CR, et al. Induction of a distinct CD8 Tnc17 subset by transforming growth factor-β and interleukin-6. Journal of Leukocyte Biology. 2007;82(2):354–360. doi: 10.1189/jlb.0207111. [DOI] [PubMed] [Google Scholar]
- 41.Kondo T, Takata H, Matsuki F, Takiguchi M. Cutting edge: phenotypic characterization and differentiation of human CD8+ T cells producing IL-171. Journal of Immunology. 2009;182(4):1794–1798. doi: 10.4049/jimmunol.0801347. [DOI] [PubMed] [Google Scholar]
- 42.Huber M, Heink S, Grothe H, et al. Th17-like developmental process leads to CD8+ Tc17 cells with reduced cytotoxic activity. European Journal of Immunology. 2009;39(7):1716–1725. doi: 10.1002/eji.200939412. [DOI] [PubMed] [Google Scholar]
- 43.Hamada H, Garcia-Hernandez MDLL, Reome JB, et al. Tc17, a unique subset of CD8 T cells that can protect against lethal influenza challenge. Journal of Immunology. 2009;182(6):3469–3481. doi: 10.4049/jimmunol.0801814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ciric B, El-behi M, Cabrera R, Zhang GX, Rostami A. IL-23 drives pathogenic IL-17-producing CD8+ T cells. Journal of Immunology. 2009;182(9):5296–5305. doi: 10.4049/jimmunol.0900036. [DOI] [PubMed] [Google Scholar]
- 45.Yen HR, Harris TJ, Wada S, et al. Tc17 CD8 T cells: functional plasticity and subset diversity. Journal of Immunology. 2009;183(11):7161–7168. doi: 10.4049/jimmunol.0900368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hijnen D, Knol EF, Gent YY, et al. CD8(+) T cells in the lesional skin of atopic dermatitis and psoriasis patients are an important source of IFN-γ, IL-13, IL-17, and IL-22. Journal of Investigative Dermatology. 2013;133(4):973–979. doi: 10.1038/jid.2012.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huber M, Heink S, Pagenstecher A, et al. IL-17A secretion by CD8+ T cells supports Th17-mediated autoimmune encephalomyelitis. Journal of Clinical Investigation. 2013;123(1):247–260. doi: 10.1172/JCI63681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bermejo DA, Jackson SW, Gorosito-Serran M, et al. Trypanosoma cruzi trans-sialidase initiates a program independent of the transcription factors RORγt and Ahr that leads to IL-17 production by activated B cells. Nature Immunology. 2013;14(5):514–522. doi: 10.1038/ni.2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nature Reviews Immunology. 2010;10(7):479–489. doi: 10.1038/nri2800. [DOI] [PubMed] [Google Scholar]
- 50.Sutton CE, Mielke LA. Mills KHG. IL-17-producing γδ T cells and innate lymphoid cells. European Journal of Immunology. 2012;42(9):2221–2231. doi: 10.1002/eji.201242569. [DOI] [PubMed] [Google Scholar]
- 51.Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KHG. Interleukin-1 and IL-23 induce innate IL-17 production from γδ T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31(2):331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 52.Roark CL, French JD, Taylor MA, Bendele AM, Born WK, O’Brien RL. Exacerbation of collagen-induced arthritis by oligoclonal, IL-17-producing γδ T cells. Journal of Immunology. 2007;179(8):5576–5583. doi: 10.4049/jimmunol.179.8.5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Geremia A, Arancibia-Cárcamo CV, Fleming MPP, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. Journal of Experimental Medicine. 2011;208(6):1127–1133. doi: 10.1084/jem.20101712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Korn T, Petermann F. Development and function of interleukin 17-producing γδ T cells. Annals of the New York Academy of Sciences. 2012;1247(1):34–45. doi: 10.1111/j.1749-6632.2011.06355.x. [DOI] [PubMed] [Google Scholar]
- 55.Iwakura Y, Nakae S, Saijo S, Ishigame H. The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunological Reviews. 2008;226(1):57–79. doi: 10.1111/j.1600-065X.2008.00699.x. [DOI] [PubMed] [Google Scholar]
- 56.Pappu R, Rutz S, Ouyang W. Regulation of epithelial immunity by IL-17 family cytokines. Trends in Immunology. 2012;33(7):343–349. doi: 10.1016/j.it.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 57.Ishigame H, Kakuta S, Nagai T, et al. Differential Roles of Interleukin-17A and -17F in Host Defense against Mucoepithelial Bacterial Infection and Allergic Responses. Immunity. 2009;30(1):108–119. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 58.Aujla SJ, Chan YR, Zheng M, et al. IL-22 mediates mucosal host defense against gram-negative bacterial pneumonia. Nature Medicine. 2008;14(3):275–281. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. Journal of Infectious Diseases. 2004;190(3):624–631. doi: 10.1086/422329. [DOI] [PubMed] [Google Scholar]
- 60.Kagami S, Rizzo HL, Kurtz SE, Miller LS, Blauvelt A. IL-23 and IL-17A, but not IL-12 and IL-22, are required for optimal skin host defense against Candida albicans. Journal of Immunology. 2010;185(9):5453–5462. doi: 10.4049/jimmunol.1001153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cypowyj S, Picard C, Maródi L, Casanova LJ, Puel A. Immunity to infection in IL-17-deficient mice and humans. European Journal of Immunology. 2012;42:2246–2254. doi: 10.1002/eji.201242605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guiton R, Vasseur V, Charron S, et al. Interleukin 17 receptor signaling is deleterious during toxoplasma gondii infection in susceptible BL6 mice. Journal of Infectious Diseases. 2010;202(3):427–435. doi: 10.1086/653738. [DOI] [PubMed] [Google Scholar]
- 63.Fan Y, Weifeng W, Yuluan Y, Qing K, Yu P, Yanlan H. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of coxsackievirus b3-induced viral myocarditis reduces myocardium inflammation. Virology Journal. 2011;8(article 17) doi: 10.1186/1743-422X-8-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Crowe CR, Chen K, Pociask DA, et al. Critical role of IL-17RA in immunopathology of influenza infection. Journal of Immunology. 2009;183(8):5301–5310. doi: 10.4049/jimmunol.0900995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Truchetet ME, Beven L, Renaudin H, et al. Potential role of mycoplasma hominis in interleukin (IL)-17-producing CD4+ t-cell generation via induction of IL-23 secretion by human dendritic cells. Journal of Infectious Diseases. 2011;204(11):1796–1805. doi: 10.1093/infdis/jir630. [DOI] [PubMed] [Google Scholar]
- 66.Acosta-Rodriguez EV, Rivino L, Geginat J, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nature Immunology. 2007;8(6):639–646. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 67.Eyerich K, Foerster S, Rombold S, et al. Patients with chronic mucocutaneous candidiasis exhibit reduced production of Th17-associated cytokines IL-17 and IL-22. Journal of Investigative Dermatology. 2008;128(11):2640–2645. doi: 10.1038/jid.2008.139. [DOI] [PubMed] [Google Scholar]
- 68.Puel A, Cypowyj S, Bustamante J, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 2011;332(6025):65–68. doi: 10.1126/science.1200439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Milner JD, Brenchley JM, Laurence A, et al. Impaired TH17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452(7188):773–776. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ma CS, Chew GYJ, Simpson N, et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. Journal of Experimental Medicine. 2008;205(7):1551–1557. doi: 10.1084/jem.20080218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Renner ED, Rylaarsdam S, Aňover-Sombke S, et al. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced TH17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. Journal of Allergy and Clinical Immunology. 2008;122(1):181–187. doi: 10.1016/j.jaci.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kebir H, Kreymborg K, Ifergan I, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nature Medicine. 2007;13(10):1173–1175. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lowes MA, Kikuchi T, Fuentes-Duculan J, et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. Journal of Investigative Dermatology. 2008;128(5):1207–1211. doi: 10.1038/sj.jid.5701213. [DOI] [PubMed] [Google Scholar]
- 74.Fujino S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52(1):65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chabaud M, Durand JM, Buchs N, et al. Human interleukin-17: a T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis & Rheumatism. 1999;42(5):963–970. doi: 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 76.Lin AM, Rubin CJ, Khandpur R, et al. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. Journal of Immunology. 2011;187(1):490–500. doi: 10.4049/jimmunol.1100123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tzartos JS, Friese MA, Craner MJ, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. American Journal of Pathology. 2008;172(1):146–155. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Res PCM, Piskin G, de Boer OJ, et al. Overrepresentation of IL-17A and IL-22 producing CD8 T cells in lesional skin suggests their involvement in the pathogenesis of psoriasis. PLoS ONE. 2010;5(11) doi: 10.1371/journal.pone.0014108.e14108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cai Y, Shen X, Ding C, et al. Pivotal Role of Dermal IL-17-Producing γδ T Cells in Skin Inflammation. Immunity. 2011;35(4):596–610. doi: 10.1016/j.immuni.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pantelyushin S, Haak S, Ingold B, et al. Rorγt+ innate lymphocytes and γδ T cells initiate psoriasiform plaque formation in mice. Journal of Clinical Investigation. 2012;122:2252–2256. doi: 10.1172/JCI61862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Petermann F, Rothhammer V, Claussen MC, et al. γδ T cells enhance autoimmunity by restraining regulatory T cell responses via an interleukin-23-dependent mechanism. Immunity. 2010;33(3):351–363. doi: 10.1016/j.immuni.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. Journal of Immunology. 2003;171(11):6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- 83.Yang XO, Seon HC, Park H, et al. Regulation of inflammatory responses by IL-17F. Journal of Experimental Medicine. 2008;205(5):1063–1075. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bush KA, Farmer KM, Walker JS, Kirkham BW. Reduction of joint inflammation and bone erosion in rat adjuvant arthritis by treatment with interleukin-17 receptor IgG1 Fc fusion protein. Arthritis and Rheumatism. 2002;46(3):802–805. doi: 10.1002/art.10173. [DOI] [PubMed] [Google Scholar]
- 85.Komiyama Y, Nakae S, Matsuki T, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. Journal of Immunology. 2006;177(1):566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 86.Hofstetter HH, Ibrahim SM, Koczan D, et al. Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cellular Immunology. 2005;237(2):123–130. doi: 10.1016/j.cellimm.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 87.McGeachy MJ, Bak-Jensen KS, Chen Y, et al. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain TH-17 cell-mediated pathology. Nature Immunology. 2007;8(12):1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 88.Croxford AL, Mair F, Becher B. IL-23: one cytokine in control of autoimmunity. European Journal of Immunology. 2012;42:2263–2273. doi: 10.1002/eji.201242598. [DOI] [PubMed] [Google Scholar]
- 89.Kotake S, Udagawa N, Takahashi N, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. Journal of Clinical Investigation. 1999;103(9):1345–1352. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Singh R, Aggarwal A, Misra R. Th1/Th17 cytokine profiles in patients with reactive arthritis/ undifferentiated spondyloarthropathy. Journal of Rheumatology. 2007;34(11):2285–2290. [PubMed] [Google Scholar]
- 91.Shen H, Goodall JC, Hill Gaston JS. Frequency and phenotype of peripheral blood Th17 cells in ankylosing spondylitis and rheumatoid arthritis. Arthritis and Rheumatism. 2009;60(6):1647–1656. doi: 10.1002/art.24568. [DOI] [PubMed] [Google Scholar]
- 92.Yang J, Chu Y, Yang X, et al. Th17 and natural treg cell population dynamics in systemic lupus erythematosus. Arthritis and Rheumatism. 2009;60(5):1472–1483. doi: 10.1002/art.24499. [DOI] [PubMed] [Google Scholar]
- 93.Kurasawa K, Hirose K, Sano H, et al. Increased interleukin-17 production in patients with systemic sclerosis. Arthritis & Rheumatism. 2000;43(11):2455–2463. doi: 10.1002/1529-0131(200011)43:11<2455::AID-ANR12>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 94.Lubberts E, Joosten LAB, van de Loo FAJ, Schwarzenberger P, Kolls J, van den Berg WB. Overexpression of IL-17 in the knee joint of collagen type II immunized mice promotes collagen arthritis and aggravates joint destruction. Inflammation Research. 2002;51(2):102–104. doi: 10.1007/BF02684010. [DOI] [PubMed] [Google Scholar]
- 95.Lubberts E, Koenders M, van den Berg WB. The role of T cell interleukin-17 in conducting destructive arthritis: lessons from animal models. Arthritis Research and Therapy. 2005;7(1):29–37. doi: 10.1186/ar1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sato K, Suematsu A, Okamoto K, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. Journal of Experimental Medicine. 2006;203(12):2673–2682. doi: 10.1084/jem.20061775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lubberts E, Koenders MI, Oppers-Walgreen B, et al. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis and Rheumatism. 2004;50(2):650–659. doi: 10.1002/art.20001. [DOI] [PubMed] [Google Scholar]
- 98.Wong CK, Lit LCW, Tam LS, Li EKM, Wong PTY, Lam CWK. Hyperproduction of IL-23 and IL-17 in patients with systemic lupus erythematosus: implications for Th17-mediated inflammation in auto-immunity. Clinical Immunology. 2008;127(3):385–393. doi: 10.1016/j.clim.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 99.Noordenbos T, Yeremenko N, Gofita I, et al. Interleukin-17-positive mast cells contribute to synovial inflammation in spondylarthritis. Arthritis and Rheumatism. 2012;64(1):99–109. doi: 10.1002/art.33396. [DOI] [PubMed] [Google Scholar]
- 100.Kenna TJ, Brown MA. The role of IL-17-secreting mast cells in inflammatory joint disease. Nature Reviews Rheumatology. 2013;9(6):375–379. doi: 10.1038/nrrheum.2012.205. [DOI] [PubMed] [Google Scholar]
- 101.Appel H, Maier R, Wu P, et al. Analysis of IL-17+ cells in facet joints of patients with spondyloarthritis suggests that the innate immune pathway might be of greater relevance than the Th17-mediated adaptive immune response. Arthritis Research and Therapy. 2011;13(3, article R95) doi: 10.1186/ar3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hueber AJ, Asquith DL, Miller AM, et al. Cutting edge: mast cells express IL-17A in rheumatoid arthritis synovium. Journal of Immunology. 2010;184(7):3336–3340. doi: 10.4049/jimmunol.0903566. [DOI] [PubMed] [Google Scholar]
- 103.Hirota K, Yoshitomi H, Hashimoto M, et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. Journal of Experimental Medicine. 2007;204(12):2803–2812. doi: 10.1084/jem.20071397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.van Hamburg JP, Asmawidjaja PS, Davelaar N, et al. Th17 cells, but not Th1 cells, from patients with early rheumatoid arthritis are potent inducers of matrix metalloproteinases and proinflammatory cytokines upon synovial fibroblast interaction, including autocrine interleukin-17A production. Arthritis and Rheumatism. 2011;63(1):73–83. doi: 10.1002/art.30093. [DOI] [PubMed] [Google Scholar]
- 105.Yago T, Nanke Y, Ichikawa N, et al. IL-17 induces osteoclastogenesis from human monocytes alone in the absence of osteoblasts, which is potently inhibited by anti-TNF-α antibody: a novel mechanism of osteoclastogenesis by IL-17. Journal of Cellular Biochemistry. 2009;108(4):947–955. doi: 10.1002/jcb.22326. [DOI] [PubMed] [Google Scholar]
- 106.Braun T, Zwerina J. Positive regulators of osteoclastogenesis and bone resorption in rheumatoid arthritis. Arthritis Research and Therapy. 2011;13(4, article 235) doi: 10.1186/ar3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gaffen SL. Biology of recently discovered cytokines: interleukin-17—a unique inflammatory cytokine with roles in bone biology and arthritis. Arthritis Research and Therapy. 2004;6(6):240–247. doi: 10.1186/ar1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shen F, Ruddy MJ, Plamondon P, Gaffen SL. Cytokines link osteoblasts and inflammation: microarray analysis of interleukin-17- and TNF-α-induced genes in bone cells. Journal of Leukocyte Biology. 2005;77(3):388–399. doi: 10.1189/jlb.0904490. [DOI] [PubMed] [Google Scholar]
- 109.Tokuda H, Kanno Y, Ishisaki A, Takenaka M, Harada A, Kozawa O. Interleukin (IL)-17 enhances tumor necrosis factor-α-stimulated IL-6 synthesis via p38 mitogen-activated protein kinase in osteoblasts. Journal of Cellular Biochemistry. 2004;91(5):1053–1061. doi: 10.1002/jcb.20004. [DOI] [PubMed] [Google Scholar]
- 110.Ruddy MJ, Shen F, Smith JB, Sharma A, Gaffen SL. Interleukin-17 regulates expression of the CXC chemokine LIX/CXCL5 in osteoblasts: implications for inflammation and neutrophil recruitment. Journal of Leukocyte Biology. 2004;76(1):135–144. doi: 10.1189/jlb.0204065. [DOI] [PubMed] [Google Scholar]
- 111.Koenders MI, Kolls JK, Oppers-Walgreen B, et al. Interleukin-17 receptor deficiency results in impaired synovial expression of interleukin-1 and matrix metalloproteinases 3, 9, and 13 and prevents cartilage destruction during chronic reactivated streptococcal cell wall-induced arthritis. Arthritis and Rheumatism. 2005;52(10):3239–3247. doi: 10.1002/art.21342. [DOI] [PubMed] [Google Scholar]
- 112.Chabaud M, Garnero P, Dayer JM, Guerne PA, Fossiez F, Miossec P. Contribution of interleukin 17 to synovium matrix destruction in rheumatoid arthritis. Cytokine. 2000;12(7):1092–1099. doi: 10.1006/cyto.2000.0681. [DOI] [PubMed] [Google Scholar]
- 113.Onishi RM, Gaffen SL. Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology. 2010;129(3):311–321. doi: 10.1111/j.1365-2567.2009.03240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hot A, Miossec P. Effects of interleukin (IL)-17A and IL-17F in human rheumatoid arthritis synoviocytes. Annals of the Rheumatic Diseases. 2011;70(5):727–732. doi: 10.1136/ard.2010.143768. [DOI] [PubMed] [Google Scholar]
- 115.Goldberg M, Nadiv O, Luknar-Gabor N, Agar G, Beer Y, Katz Y. Synergism between tumor necrosis factor alpha and interleukin-17 to induce IL-23 p19 expression in fibroblast-like synoviocytes. Molecular Immunology. 2009;46(8-9):1854–1859. doi: 10.1016/j.molimm.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 116.Chabaud M, Fossiez F, Taupin JL, Miossec P. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. Journal of Immunology. 1998;161(1):409–414. [PubMed] [Google Scholar]
- 117.Hot A, Zrioual S, Lenief V, Miossec P. IL-17 and tumour necrosis factor α combination induces a HIF-1α-dependent invasive phenotype in synoviocytes. Annals of the Rheumatic Diseases. 2012;71:1393–1401. doi: 10.1136/annrheumdis-2011-200867. [DOI] [PubMed] [Google Scholar]
- 118.Fossiez F, Djossou O, Chomarat P, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. Journal of Experimental Medicine. 1996;183(6):2593–2603. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zrioual S, Ecochard R, Tournadre A, Lenief V, Cazalis MA, Miossec P. Genome-wide comparison between IL-17A- and IL-17F-induced effects in human rheumatoid arthritis synoviocytes. Journal of Immunology. 2009;182(5):3112–3120. doi: 10.4049/jimmunol.0801967. [DOI] [PubMed] [Google Scholar]
- 120.Park YE, Woo YJ, Park SH, et al. IL-17 increases cadherin-11 expression in a model of autoimmune experimental arthritis and in rheumatoid arthritis. Immunology Letters. 2011;140(1-2):97–103. doi: 10.1016/j.imlet.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 121.Chabaud M, Lubberts E, Joosten L, van den Berg W, Miossec P. IL-17 derived from juxta-articular bone and synovium contributes to joint degradation in rheumatoid arthritis. Arthritis Research. 2001;3(3):168–177. doi: 10.1186/ar294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kato H, Endres J, Fox DA. The roles of IFN-γ versus IL-17 in pathogenic effects of human Th17 cells on synovial fibroblasts. Modern Rheumatology. 2013 doi: 10.1007/s10165-012-0811-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Toh ML, Gonzales G, Koenders MI, et al. Role of interleukin 17 in arthritis chronicity through survival of synoviocytes via regulation of synoviolin expression. PLoS ONE. 2010;5(10) doi: 10.1371/journal.pone.0013416.e13416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lefèvre S, Knedla A, Tennie C, et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nature Medicine. 2009;15(12):1414–1420. doi: 10.1038/nm.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ryu S, Lee JH, Kim SI. IL-17 increased the production of vascular endothelial growth factor in rheumatoid arthritis synoviocytes. Clinical Rheumatology. 2006;25(1):16–20. doi: 10.1007/s10067-005-1081-1. [DOI] [PubMed] [Google Scholar]
- 126.Honorati MC, Neri S, Cattini L, Facchini A. Interleukin-17, a regulator of angiogenic factor release by synovial fibroblasts. Osteoarthritis and Cartilage. 2006;14(4):345–352. doi: 10.1016/j.joca.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 127.Shalom-Barak T, Quach J, Lotz M. Interleukin-17-induced gene expression in articular chondrocytes is associated with activation of mitogen-activated protein kinases and NF-κB. Journal of Biological Chemistry. 1998;273(42):27467–27473. doi: 10.1074/jbc.273.42.27467. [DOI] [PubMed] [Google Scholar]
- 128.Martel-Pelletier J, Mineau F, Jovanovic D, Di Battista JA, Pelletier JP. Mitogen-activated protein kinase and nuclear factor kappaB together regulate interleukin-17-induced nitric oxide production in human osteoarthritic chondrocytes: possible role of transactivating factor mitogen-activated protein kinase-activated proten kinase (MAPKAPK) Arthritis & Rheumatism. 1999;42:2399–2409. doi: 10.1002/1529-0131(199911)42:11<2399::AID-ANR19>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 129.Honorati MC, Bovara M, Cattini L, Piacentini A, Facchini A. Contribution of interleukin 17 to human cartilage degradation and synovial inflammation in osteoarthritis. Osteoarthritis and Cartilage. 2002;10(10):799–807. doi: 10.1053/joca.2002.0829. [DOI] [PubMed] [Google Scholar]
- 130.Dudler J, Renggli-Zulliger N, Busso N, Lotz M, So A. Effect of interleukin 17 on proteoglycan degradation in murine knee joints. Annals of the Rheumatic Diseases. 2000;59(7):529–532. doi: 10.1136/ard.59.7.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Koenders MI, Marijnissen RJ, Devesa I, et al. Tumor necrosis factor-interleukin-17 interplay induces S100A8, interleukin-1β, and matrix metalloproteinases, and drives irreversible cartilage destruction in murine arthritis: rationale for combination treatment during arthritis. Arthritis and Rheumatism. 2011;63(8):2329–2339. doi: 10.1002/art.30418. [DOI] [PubMed] [Google Scholar]
- 132.Romero-Sánchez C, Jaimes DA, Londoño J, et al. Association between Th-17 cytokine profile and clinical features in patients with spondyloarthritis. Clinical and Experimental Rheumatology. 2011;29(5):828–834. [PubMed] [Google Scholar]
- 133.Mei Y, Pan F, Gao J, et al. Increased serum IL-17 and IL-23 in the patient with ankylosing spondylitis. Clinical Rheumatology. 2011;30(2):269–273. doi: 10.1007/s10067-010-1647-4. [DOI] [PubMed] [Google Scholar]
- 134.Wang X, Lin Z, Wei Q, Jiang Y, Gu J. Expression of IL-23 and IL-17 and effect of IL-23 on IL-17 production in ankylosing spondylitis. Rheumatology International. 2009;29(11):1343–1347. doi: 10.1007/s00296-009-0883-x. [DOI] [PubMed] [Google Scholar]
- 135.Sherlock JP, Joyce-Shaikh B, Turner SP, et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+CD4-CD8- entheseal resident T cells. Nature Medicine. 2012;18:1069–1076. doi: 10.1038/nm.2817. [DOI] [PubMed] [Google Scholar]
- 136.Schaeverbeke T, Truchetet MÉ, Richez C. When and where does rheumatoid arthritis begin? Joint Bone Spine. 2012;79(6):550–554. doi: 10.1016/j.jbspin.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 137.Manoury-Schwartz B, Chiocchia G, Bessis N, et al. High susceptibility to collagen-induced arthritis in mice lacking IFN-γ receptors. Journal of Immunology. 1997;158(11):5501–5506. [PubMed] [Google Scholar]
- 138.Vermeire K, Heremans H, Vandeputte M, Huang S, Billiau A, Matthys P. Accelerated collagen-induced arthritis in IFN-γ receptor-deficient mice. Journal of Immunology. 1997;158(11):5507–5513. [PubMed] [Google Scholar]
- 139.Hirota K, Hashimoto M, Yoshitomi H, et al. T cell self-reactivity forms a cytokine milieu for spontaneous development of IL-17+ Th cells that cause autoimmune arthritis. Journal of Experimental Medicine. 2007;204(1):41–47. doi: 10.1084/jem.20062259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Koenders MI, Devesa I, Marijnissen RJ, et al. Interleukin-1 drives pathogenic Th17 cells during spontaneous arthritis in interleukin-1 receptor antagonist-deficient mice. Arthritis and Rheumatism. 2008;58(11):3461–3470. doi: 10.1002/art.23957. [DOI] [PubMed] [Google Scholar]
- 141.Horai R, Nakajima A, Habiro K, et al. TNF-α is crucial for the development of autoimmune arthritis in IL-1 receptor antagonist-deficient mice. Journal of Clinical Investigation. 2004;114(11):1603–1611. doi: 10.1172/JCI20742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nakae S, Saijo S, Horai R, Sudo K, Mori S, Iwakura Y. IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(10):5986–5990. doi: 10.1073/pnas.1035999100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ogura H, Murakami M, Okuyama Y, et al. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity. 2008;29(4):628–636. doi: 10.1016/j.immuni.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 144.Wu HJ, Ivanov II, Darce J, et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32(6):815–827. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Miossec P. IL-17 and Th17 cells in human inflammatory diseases. Microbes and Infection. 2009;11(5):625–630. doi: 10.1016/j.micinf.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 146.Peters A, Pitcher LA, Sullivan JM, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity. 2011;35(6):986–996. doi: 10.1016/j.immuni.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pöllinger B, Junt T, Metzler B, et al. Th17 cells, not IL-17+ γδ T cells, drive arthritic bone destruction in mice and humans. Journal of Immunology. 2011;186(4):2602–2612. doi: 10.4049/jimmunol.1003370. [DOI] [PubMed] [Google Scholar]
- 148.Metawi SA, Abbas D, Kamal MM, Ibrahim MK. Serum and synovial fluid levels of interleukin-17 in correlation with disease activity in patients with RA. Clinical Rheumatology. 2011;30(9):1201–1207. doi: 10.1007/s10067-011-1737-y. [DOI] [PubMed] [Google Scholar]
- 149.Zhang L, Li YG, Li YH, et al. Increased frequencies of th22 cells as well as th17 cells in the peripheral blood of patients with ankylosing spondylitis and rheumatoid arthritis. PLoS ONE. 2012;7(4) doi: 10.1371/journal.pone.0031000.e31000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chen J, Li J, Gao H, et al. Comprehensive evaluation of different T-helper cell subsets differentiation and function in rheumatoid arthritis. Journal of Biomedicine and Biotechnology. 2012;2012:6 pages. doi: 10.1155/2012/535361.535361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Moran EM, Mullan R, McCormick J, et al. Human rheumatoid arthritis tissue production of IL-17A drives matrix and cartilage degradation: synergy with tumour necrosis factor-α, Oncostatin M and response to biologic therapies. Arthritis Research and Therapy. 2009;11(4, article R113) doi: 10.1186/ar2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ito Y, Usui T, Kobayashi S, et al. Gamma/delta T cells are the predominant source of interleukin-17 in affected joints in collagen-induced arthritis, but not in rheumatoid arthritis. Arthritis and Rheumatism. 2009;60(8):2294–2303. doi: 10.1002/art.24687. [DOI] [PubMed] [Google Scholar]
- 153.Gatto M, Zen M, Ghirardello A, et al. Emerging and critical issues in the pathogenesis of lupus. Autoimmunity Reviews. 2013;12(4):523–536. doi: 10.1016/j.autrev.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 154.Borchers AT, Naguwa SM, Shoenfeld Y, Gershwin ME. The geoepidemiology of systemic lupus erythematosus. Autoimmunity Reviews. 2010;9(5):A277–A287. doi: 10.1016/j.autrev.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 155.Nalbandian A, Crispín JC, Tsokos GC. Interleukin-17 and systemic lupus erythematosus: current concepts. Clinical and Experimental Immunology. 2009;157(2):209–215. doi: 10.1111/j.1365-2249.2009.03944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Edgerton C, Crispín JC, Moratz CM, et al. IL-17 producing CD4+ T cells mediate accelerated ischemia/reperfusion-induced injury in autoimmunity-prone mice. Clinical Immunology. 2009;130(3):313–321. doi: 10.1016/j.clim.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kang HK, Liu M, Datta SK. Low-dose peptide tolerance therapy of lupus generates plasmacytoid dendritic cells that cause expansion of autoantigen-specific regulatory T cells and contraction of inflammatory Th17 cells. Journal of Immunology. 2007;178(12):7849–7858. doi: 10.4049/jimmunol.178.12.7849. [DOI] [PubMed] [Google Scholar]
- 158.Hsu HC, Yang P, Wang J, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nature Immunology. 2008;9(2):166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 159.Craft JE. Follicular helper T cells in immunity and systemic autoimmunity. Nature Reviews Rheumatology. 2012;8:337–347. doi: 10.1038/nrrheum.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chen DY, Chen YM, Wen MC, Hsieh TY, Hung WT, Lan JL. The potential role of Th17 cells and Th17-related cytokines in the pathogenesis of lupus nephritis. Lupus. 2012;21(13):1385–1396. doi: 10.1177/0961203312457718. [DOI] [PubMed] [Google Scholar]
- 161.Nakou M, Papadimitraki ED, Fanouriakis A, et al. Interleukin-21 is increased in active systemic lupus erythematosus patients and contributes to the generation of plasma B cells. Clinical and Experimental Rheumatology. 2013;31(2):172–179. [PubMed] [Google Scholar]
- 162.Chen DY, Chen YM, Lan JL, Lin CC, Chen HH, Hsieh CW. Potential role of Th17 cells in the pathogenesis of adult-onset Still’s disease. Rheumatology. 2010;49(12):2305–2312. doi: 10.1093/rheumatology/keq284. [DOI] [PubMed] [Google Scholar]
- 163.Shah K, Lee WW, Lee SH, et al. Correction: dysregulated balance of Th17 and Th1 cells in systemic lupus erythematosus. Arthritis Research and Therapy. 2010;12(2, article R53) doi: 10.1186/ar2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Crispín JC, Oukka M, Bayliss G, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. Journal of Immunology. 2008;181(12):8761–8766. doi: 10.4049/jimmunol.181.12.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wen Z, Xu L, Xu W, Yin Z, Gao X, Xiong S. Interleukin-17 expression positively correlates with disease severity of lupus nephritis by increasing anti-double-stranded DNA antibody production in a lupus model induced by activated lymphocyte derived DNA. Plos One. 2013;8(3) doi: 10.1371/journal.pone.0058161.e58161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Yu B, Guan M, Peng Y, et al. Copy number variations of interleukin-17F, interleukin-21, and interleukin-22 are associated with systemic lupus erythematosus. Arthritis and Rheumatism. 2011;63(11):3487–3492. doi: 10.1002/art.30595. [DOI] [PubMed] [Google Scholar]
- 167.Chizzolini C, Brembilla NC, Montanari E, Truchetet ME. Fibrosis and immune dysregulation in systemic sclerosis. Autoimmunity Reviews. 2011;10(5):276–281. doi: 10.1016/j.autrev.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 168.Shapira Y, Agmon-Levin N, Shoenfeld Y. Geoepidemiology of autoimmune rheumatic diseases. Nature Reviews Rheumatology. 2010;6(8):468–476. doi: 10.1038/nrrheum.2010.86. [DOI] [PubMed] [Google Scholar]
- 169.McMahan ZH, Hummers LK. Systemic sclerosis—challenges for clinical practice. Nature Reviews Rheumatology. 2013;9:90–100. doi: 10.1038/nrrheum.2012.191. [DOI] [PubMed] [Google Scholar]
- 170.Katsumoto TR, Whitfield ML, Connolly MK. The pathogenesis of systemic sclerosis. Annual Review of Pathology. 2011;6:509–537. doi: 10.1146/annurev-pathol-011110-130312. [DOI] [PubMed] [Google Scholar]
- 171.Dieudé P, Boileau C, Allanore Y. Immunogenetics of systemic sclerosis. Autoimmunity Reviews. 2011;10(5):282–290. doi: 10.1016/j.autrev.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 172.Dieudé P, Guedj M, Truchetet ME, et al. Association of the CD226 Ser307 variant with systemic sclerosis. Arthritis and Rheumatism. 2011;63(4):1097–1105. doi: 10.1002/art.30204. [DOI] [PubMed] [Google Scholar]
- 173.Prescott RJ, Freemont AJ, Jones CJP, Hoyland J, Fielding P. Sequential dermal microvascular and perivascular changes in the development of scleroderma. Journal of Pathology. 1992;166(3):255–263. doi: 10.1002/path.1711660307. [DOI] [PubMed] [Google Scholar]
- 174.Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nature Reviews Immunology. 2004;4:583–594. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ferrarini M, Steen V, Medsger TA, Jr., Whiteside TL. Functional and phenotypic analysis of T lymphocytes cloned from the skin of patients with systemic sclerosis. Clinical and Experimental Immunology. 1990;79(3):346–352. doi: 10.1111/j.1365-2249.1990.tb08094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Truchetet ME, Brembilla NC, Montanari E, Allanore Y, Chizzolini C. Increased frequency of circulating Th22 in addition to Th17 and Th2 lymphocytes in systemic sclerosis: association with interstitial lung disease. Arthritis Research & Therapy. 2011;13(5, article R166) doi: 10.1186/ar3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Truchetet ME, Brembilla NC, Montanari E, et al. Interleukin-17A+ cell counts are increased in systemic sclerosis skin and their number is inversely correlated with the extent of skin involvement. Arthritis & Rheumatism. 2013;65(5):1347–1356. doi: 10.1002/art.37860. [DOI] [PubMed] [Google Scholar]
- 178.Wilson MS, Madala SK, Ramalingam TR, et al. Bleomycin and IL-1β-mediated pulmonary fibrosis is IL-17A dependent. Journal of Experimental Medicine. 2010;207(3):535–552. doi: 10.1084/jem.20092121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Okamoto Y, Hasegawa M, Matsushita T, et al. Potential roles of interleukin-17A in the development of skin fibrosis in mice. Arthritis & Rheumatism. 2012;64(11):3726–3735. doi: 10.1002/art.34643. [DOI] [PubMed] [Google Scholar]
- 180.Yoshizaki A, Yanaba K, Iwata Y, et al. Cell adhesion molecules regulate fibrotic process via Th1/Th2/Th17 cell balance in a bleomycin-induced scleroderma model. Journal of Immunology. 2010;185(4):2502–2515. doi: 10.4049/jimmunol.0901778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Meloni F, Solari N, Cavagna L, Morosini M, Montecucco CM, Fietta AM. Frequency of Th1, Th2 and Th17 producing T lymphocytes in bronchoalveolar lavage of patients with systemic sclerosis. Clinical and Experimental Rheumatology. 2009;27(5):765–772. [PubMed] [Google Scholar]
- 182.Truchetet EM, Allanore Y, Montanari E, Chizzolini C, Brembilla NC. Prostaglandin I(2) analogues enhance already exuberant Th17 cell responses in systemic sclerosis. Annals of the Rheumatic Diseases. 2012;71(12):2044–2050. doi: 10.1136/annrheumdis-2012-201400. [DOI] [PubMed] [Google Scholar]
- 183.Laurence A, O’Shea JJ. T(H)-17 differentiation: of mice and men. Nature Immunology. 2007;8:903–905. doi: 10.1038/ni0907-903. [DOI] [PubMed] [Google Scholar]
- 184.Gladman DD, Mease PJ, Krueger G, et al. Outcome measures in psoriatic arthritis. Journal of Rheumatology. 2005;32(11):2262–2269. [PubMed] [Google Scholar]
- 185.Liu Y, Helms C, Liao W, et al. A genome-wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLoS Genetics. 2008;4(3) doi: 10.1371/journal.pgen.1000041.e1000041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Nair RP, Ruether A, Stuart PE, et al. Polymorphisms of the IL12B and IL23R genes are associated with psoriasis. Journal of Investigative Dermatology. 2008;128(7):1653–1661. doi: 10.1038/sj.jid.5701255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Szodoray P, Alex P, Chappell-Woodward CM, et al. Circulating cytokines in Norwegian patients with psoriatic arthritis determined by a multiplex cytokine array system. Rheumatology. 2007;46(3):417–425. doi: 10.1093/rheumatology/kel306. [DOI] [PubMed] [Google Scholar]
- 188.Raychaudhuri SP, Raychaudhuri SK, Genovese MC. IL-17 receptor and its functional significance in psoriatic arthritis. Molecular and Cellular Biochemistry. 2012;359(1-2):419–429. doi: 10.1007/s11010-011-1036-6. [DOI] [PubMed] [Google Scholar]
- 189.Hüffmeier U, Uebe S, Ekici AB, et al. CoMon variants at TRAF3IP2 are aSociated with susceptibility to psoriatic arthritis and psoriasis. Nature Genetics. 2010;42(11):996–999. doi: 10.1038/ng.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Doyle MS, Collins ES, Fitzgerald OM, Pennington SR. New insight into the functions of the interleukin-17 receptor adaptor protein Act1 in psoriatic arthritis. Arthritis Research & Therapy. 2012;14(5, article 226) doi: 10.1186/ar4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Leipe J, Grunke M, Dechant C, et al. Role of Th17 cells in human autoimmune arthritis. Arthritis and Rheumatism. 2010;62(10):2876–2885. doi: 10.1002/art.27622. [DOI] [PubMed] [Google Scholar]
- 192.van Tubergen A, Weber U. Diagnosis and classification in spondyloarthritis: identifying a chameleon. Nature Reviews Rheumatology. 2012;8(5):253–261. doi: 10.1038/nrrheum.2012.33. [DOI] [PubMed] [Google Scholar]
- 193.Warde N. Spondyloarthropathies: HLA-B27 and ERAP1 contribute to ankylosing spondylitis via aberrant peptide processing and presentation. Nature Reviews Rheumatology. 2011;7(9, article 498) doi: 10.1038/nrrheum.2011.112. [DOI] [PubMed] [Google Scholar]
- 194.Tam LS, Gu J, Yu D. Pathogenesis of ankylosing spondylitis. Nature Reviews Rheumatology. 2010;6(7):399–405. doi: 10.1038/nrrheum.2010.79. [DOI] [PubMed] [Google Scholar]
- 195.Rahman P, Inman RD, Gladman DD, Reeve JP, Peddle L, Maksymowych WP. Association of interleukin-23 receptor variants with ankylosing spondylitis. Arthritis and Rheumatism. 2008;58(4):1020–1025. doi: 10.1002/art.23389. [DOI] [PubMed] [Google Scholar]
- 196.Yoshitomi H, Sakaguchi N, Kobayashi K, et al. A role for fungal β-glucans and their receptor Dectin-1 in the induction of autoimmune arthritis in genetically susceptible mice. Journal of Experimental Medicine. 2005;201(6):949–960. doi: 10.1084/jem.20041758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ruutu M, Thomas G, Steck R, et al. β-glucan triggers spondylarthritis and Crohn’s disease-like ileitis in SKG mice. Arthritis & Rheumatism. 2012;64(7):2211–2222. doi: 10.1002/art.34423. [DOI] [PubMed] [Google Scholar]
- 198.Bowness P, Ridley A, Shaw J, et al. Th17 cells expressing KIR3DL2+ and responsive to HLA-B27 homodimers are increased in ankylosing spondylitis. Journal of Immunology. 2011;186(4):2672–2680. doi: 10.4049/jimmunol.1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Genovese MC, van den Bosch F, Roberson SA, et al. LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: a phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis and Rheumatism. 2010;62(4):929–939. doi: 10.1002/art.27334. [DOI] [PubMed] [Google Scholar]
- 200.Elloso MM, Gomez-Angelats M, Fourie AM. Targeting the Th17 pathway in psoriasis. Journal of Leukocyte Biology. 2012;92:1187–1197. doi: 10.1189/jlb.0212101. [DOI] [PubMed] [Google Scholar]
- 201.Patel DD, Lee DM, Kolbinger F, Antoni C. Effect of IL-17A blockade with secukinumab in autoimmune diseases. Annals of the Rheumatic Diseases. 2013;72(supplement 2):iii116–iii123. doi: 10.1136/annrheumdis-2012-202371. [DOI] [PubMed] [Google Scholar]
- 202.Papp KA, Leonardi C, Menter A, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. New England Journal of Medicine. 2012;366(13):1181–1189. doi: 10.1056/NEJMoa1109017. [DOI] [PubMed] [Google Scholar]
- 203.Garber K. Anti-IL-17 mAbs herald new options in psoriasis. Nature Biotechnology. 2012;30:475–477. doi: 10.1038/nbt0612-475. [DOI] [PubMed] [Google Scholar]
- 204.Yeilding N, Szapary P, Brodmerkel C, et al. Development of the IL-12/23 antagonist ustekinumab in psoriasis: past, present, and future perspectives. Annals of the New York Academy of Sciences. 2011;1222(1):30–39. doi: 10.1111/j.1749-6632.2011.05963.x. [DOI] [PubMed] [Google Scholar]
- 205.Gottlieb A, Menter A, Mendelsohn A. Ustekinumab, a human interleukin 12/23 monoclonal antibody, for psoriatic arthritis: randomised, double-blind, placebo-controlled, crossover trial. The Lancet. 2009;373:633–640. doi: 10.1016/S0140-6736(09)60140-9. [DOI] [PubMed] [Google Scholar]
- 206.Kavanaugh A, Menter A, Mendelsohn A, Shen YK, Lee S, Gottlieb AB. Effect of ustekinumab on physical function and health-related quality of life in patients with psoriatic arthritis: a randomized, placebo-controlled, phase II trial. Current Medical Research and Opinion. 2010;26(10):2385–2392. doi: 10.1185/03007995.2010.515804. [DOI] [PubMed] [Google Scholar]
- 207.Williams SCP. New biologic drugs get under the skin of psoriasis. Nature Medicine. 2012;18(article 638) doi: 10.1038/nm0512-638. [DOI] [PubMed] [Google Scholar]
- 208.Bangert E, Laimer D, Riedl E, et al. Anti-IL-23p19 (MK-3222): effects on the hallmarks of inflammation in psoriasis. Journal of Investigative Dermatology. 2012;132:S50–S65. [Google Scholar]
- 209.Ash Z, Emery P. The role of tocilizumab in the management of rheumatoid arthritis. Expert Opinion on Biological Therapy. 2012;12:1277–1289. doi: 10.1517/14712598.2012.707178. [DOI] [PubMed] [Google Scholar]
- 210.Mihara M, Kasutani K, Okazaki M, et al. Tocilizumab inhibits signal transduction mediated by both mIL-6R and sIL-6R, but not by the receptors of other members of IL-6 cytokine family. International Immunopharmacology. 2005;5(12):1731–1740. doi: 10.1016/j.intimp.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 211.Emery P, Keystone E, Tony HP, et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Annals of the Rheumatic Diseases. 2008;67(11):1516–1523. doi: 10.1136/ard.2008.092932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Genovese MC, McKay JD, Nasonov EL, et al. Interleukin-6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to disease-modifying antirheumatic drugs: the tocilizumab in combination with traditional disease-modifying antirheumatic drug therapy study. Arthritis and Rheumatism. 2008;58(10):2968–2980. doi: 10.1002/art.23940. [DOI] [PubMed] [Google Scholar]
- 213.Maini RN, Taylor PC, Szechinski J, et al. Double-blind randomized controlled clinical trial of the interleukin-6 receptor antagonist, tocilizumab, in European patients with rheumatoid arthritis who had an incomplete response to methotrexate. Arthritis and Rheumatism. 2006;54(9):2817–2829. doi: 10.1002/art.22033. [DOI] [PubMed] [Google Scholar]
- 214.Woodrick RS, Ruderman EM. IL-6 inhibition for the treatment of rheumatoid arthritis and other conditions. Bulletin of the NYU Hospital for Joint Diseases. 2012;70(3):195–199. [PubMed] [Google Scholar]
- 215.Lekpa FK, Poulain C, Wendling D, et al. Is IL-6 an appropriate target to treat spondyloarthritis patients refractory to anti-TNF therapy? A multicentre retrospective observational study. Arthritis Research and Therapy. 2012;14(2, article R53) doi: 10.1186/ar3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Mease P, Strand V, Shalamberidze L, et al. A phase II, double-blind, randomised, placebo-controlled study of BMS945429 (ALD518) in patients with rheumatoid arthritis with an inadequate response to methotrexate. Annals of the Rheumatic Diseases. 2012;71(7):1183–1189. doi: 10.1136/annrheumdis-2011-200704. [DOI] [PubMed] [Google Scholar]
- 217.Dinarello CA, Simon A, van der Meer JWM. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nature Reviews Drug Discovery. 2012;11:633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Huh JR, Leung MWL, Huang P, et al. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORγ3t activity. Nature. 2011;472(7344):486–490. doi: 10.1038/nature09978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Solt LA, Kumar N, Nuhant P, et al. Suppression of TH 17 differentiation and autoimmunity by a synthetic ROR ligand. Nature. 2011;472(7344):491–494. doi: 10.1038/nature10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Xu T, Wang X, Zhong B, Nurieva RI, Ding S, Dong C. Ursolic acid suppresses interleukin-17 (IL-17) production by selectively antagonizing the function of RORγt protein. Journal of Biological Chemistry. 2011;286(26):22707–22710. doi: 10.1074/jbc.C111.250407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Zhang X, Jin J, Peng X, Ramgolam VS, Markovic-Plese S. Simvastatin inhibits IL-17 secretion by targeting multiple IL-17-regulatory cytokines and by inhibiting the expression of IL-17 transcription factor RORC in CD4+ lymphocytes. Journal of Immunology. 2008;180(10):6988–6996. doi: 10.4049/jimmunol.180.10.6988. [DOI] [PubMed] [Google Scholar]
- 222.Ju JH, Heo YJ, Cho ML, et al. Modulation of STAT-3 in rheumatoid synovial T cells suppresses Th17 differentiation and increases the proportion of Treg cells. Arthritis & Rheumatism. 2012;64:3543–3552. doi: 10.1002/art.34601. [DOI] [PubMed] [Google Scholar]
- 223.Sen M, Thomas SM, Kim S, et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: implications for cancer therapy. Cancer Discovery. 2012;2(8):694–705. doi: 10.1158/2159-8290.CD-12-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Burmester GR, Blanco R, Charles-Schoeman C, et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet. 2013;381(9865):451–460. doi: 10.1016/S0140-6736(12)61424-X. [DOI] [PubMed] [Google Scholar]
- 225.Maeshima K, Yamaoka K, Kubo S, et al. The JAK inhibitor tofacitinib regulates synovitis through inhibition of interferon-γ and interleukin-17 production by human CD4+ T cells. Arthritis & Rheumatism. 2012;64(6):1790–1798. doi: 10.1002/art.34329. [DOI] [PubMed] [Google Scholar]
- 226.Tanaka Y, Yamaoka K. JAK inhibitor tofacitinib for treating rheumatoid arthritis: from basic to clinical. Modern Rheumatology. 2013;23(3):415–424. doi: 10.1007/s10165-012-0799-2. [DOI] [PubMed] [Google Scholar]
- 227.Tanaka Y, Maeshima Y, Yamaoka K. In vitro and in vivo analysis of a JAK inhibitor in rheumatoid arthritis. Annals of the Rheumatic Diseases. 2012;71(supplement 2):i70–i74. doi: 10.1136/annrheumdis-2011-200595. [DOI] [PubMed] [Google Scholar]
- 228.Rommel C, Camps M, Ji H. PI3K delta and PI3K gamma: partners in crime in inflammation in rheumatoid arthritis and beyond? Nature Reviews Immunology. 2007;7:191–201. doi: 10.1038/nri2036. [DOI] [PubMed] [Google Scholar]
- 229.Kim KW, Cho ML, Park MK, et al. Increased interleukin-17 production via a phosphoinositide 3-kinase/Akt and nuclear factor kappaB-dependent pathway in patients with rheumatoid arthritis. Arthritis Research & Therapy. 2005;7(1):R139–R148. doi: 10.1186/ar1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Toyama S, Tamura N, Haruta K, et al. Inhibitory effects of ZSTK474, a novel phosphoinositide 3-kinase inhibitor, on osteoclasts and collagen-induced arthritis in mice. Arthritis Research and Therapy. 2010;12(3, article R92) doi: 10.1186/ar3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Camps M, Rückle T, Ji H, et al. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nature Medicine. 2005;11:936–943. doi: 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- 232.Hayer S, Pundt N, Peters MA, et al. PI3Kgamma regulates cartilage damage in chronic inflammatory arthritis. FASEB Journal. 2009;23(12):4288–4298. doi: 10.1096/fj.09-135160. [DOI] [PubMed] [Google Scholar]
- 233.Marwick JA, Caramori G, Stevenson CS, et al. Inhibition of PI3Kδ restores glucocorticoid function in smoking-induced airway inflammation in mice. American Journal of Respiratory and Critical Care Medicine. 2009;179(7):542–548. doi: 10.1164/rccm.200810-1570OC. [DOI] [PubMed] [Google Scholar]
- 234.Banham-Hall E, Clatworthy MR, Okkenhaug K. The therapeutic potential for PI3K inhibitors in autoimmune rheumatic diseases. Open Rheumatology Journal. 2012;6:245–258. doi: 10.2174/1874312901206010245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Wu C, Yosef N, Thalhamer T, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496:513–517. doi: 10.1038/nature11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Kleinewietfeld M, Manzel A, Titze J, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496:518–522. doi: 10.1038/nature11868. [DOI] [PMC free article] [PubMed] [Google Scholar]