

Abstract
Mammalian target of rapamycin complex 1 (mTORC1) inhibitors are extensively used as immunosuppressants to prevent transplant rejection and in treatment of certain cancers. In patients, chronic treatment with rapamycin or its analogues (rapalogues) has been reported to lead to sensory hypersensitivity and pain conditions via an unknown mechanism. Here, we show that pharmacological or genetic inhibition of mTORC1 activates the extracellular signal-regulated kinase (ERK) pathway in sensory neurons via suppression of S6K1 to insulin receptor substrate 1 negative feedback loop. As a result, increased ERK activity induces sensory neuron sensitization, mechanical hypersensitivity, and spontaneous pain. The clinically available adenosine monophosphate-activated protein kinase activator, metformin, which is an antidiabetic drug, prevents rapamycin-induced ERK activation and the development of mechanical hypersensitivity and spontaneous pain. Taken together, our findings demonstrate that activation of the ERK pathway in sensory neurons as a consequence of mTORC1 inhibition leads to the development of pain. Importantly, this effect is abolished by co-treatment with metformin, thus providing a potential treatment option for rapalogue-evoked pain. Our findings highlight the physiological relevance of feedback signaling through mTORC1 inhibition and have important implications for development of pain therapeutics that target the mTOR pathway.
Keywords: Rapamycin, Sensory neurons, mTOR, ERK, Nav1.7, AMPK
1. Introduction
Mammalian target of rapamycin (mTOR) is an evolutionarily conserved serine/threonine kinase that regulates growth and proliferation, translation, autophagy, and cytoskeleton organization. mTOR has recently emerged as a potential target for the treatment of pain because mTOR activation has been linked to pathology underlying neuropathic pain at both peripheral and spinal levels [2,19,28,34]. mTOR complex 1 (mTORC1) exerts its effect via phosphorylation of its 2 key substrates, p70 S6 kinase (S6K; of which there are 2 members, S6K1 and S6K2) and eIF4E-binding proteins. When phosphorylated by mTORC1, S6K1 stimulates mRNA translation by phosphorylating proteins that control translation initiation and elongation [53]. As part of a negative feedback loop, S6K1 also phosphorylates and inhibits insulin receptor substrate 1 (IRS-1), leading to inhibition of AKT (protein kinase B)-mTOR and extracellular signal-regulated kinase (ERK) signaling. With long-term mTORC1 inhibition, decreased S6K1 activity leads to disinhibition of IRS-1, resulting in enhanced AKT [7,22,33] and ERK [16] activity in cells. Because increased ERK activity is a well-known sensitizer of peripheral nociceptors and is also involved in sensitization of spinal cord neurons in the pain pathway [20,27], we reasoned that this feedback activation of ERK might be physiologically relevant for therapeutics targeting mTORC1 in the pain pathway. Interestingly, chronic treatment of patients with mTORC1 inhibitors is associated with increases in the incidence of pain [6,26], including the possible development of complex regional pain syndrome (CRPS) [24,31,47]; however, mechanistic data are lacking concerning whether and how rapalogues are linked to the development of pain in patients treated with these drugs.mTORC1 can be inhibited by rapalogues, some of which have been shown to negatively regulate pain when given locally [15,19], but it is also negatively regulated by the energy-sensing kinase, adenosine monophosphate-activated protein kinase (AMPK). Importantly, AMPK activation also leads to phosphorylation of IRS-1 at a site associated with decreased receptor tyrosine kinase signaling efficacy [42]. We have recently shown that AMPK activators completely reverse neuropathic allodynia following peripheral nerve injury [28], and are effective for the treatment of postsurgical pain in mice [41]. We therefore reasoned that AMPK activators might block feedback activation of ERK in sensory neurons, revealing distinct advantages of this pharmacological approach for the treatment of pain, including pain potentially caused by clinical use of rapalogues.
Using pharmacological and genetic approaches, we demonstrate that mTORC1 inhibition evokes spontaneous pain, mechanical hypersensitivity, and increased sensory neuron excitability. These effects are mediated by ERK activation in sensory neurons and blocked by AMPK activators. Hence, we provide insight into basic signaling of the mTOR pathway in sensory neurons, a further rationale for the development of AMPK activators as a novel class of pain therapeutics and a potential mechanistic rationale for rapalogue-evoked pain.
2. Materials and methods
2.1. Surgery and behavioral testing
Male ICR and C57BL/6 mice (20–25 g; Harlan, Indianapolis, IN, USA) and male Sprague-Dawley rats (250–300 g; Harlan) were used. S6K1 and S6K2 double-knockout mice (S6K DKO) on a mixed 129/SveJ × C57Bl/6 background were generated as described by Pende et al. [36] and kindly provided by Dr. Thomas (University of Cincinnati). All animal procedures were approved by the Institutional Animal Care and Use Committee of The University of Arizona or McGill University and were in accordance with International Association for the Study of Pain, National Institutes of Health, and Canadian Council on Animal Care guidelines. Prior to surgery, all animals were assessed for mechanical withdrawal thresholds [8]. Spared nerve injury (SNI) was performed on the mice as described previously [5]. Spinal nerve ligation (SNL) was done on rats by tight ligation of the L5 and L6 spinal nerves as described by Kim and Chung [21]. Sham control animals underwent the same surgery and handling as the experimental animals but without the SNL or SNI. All animals were allowed to recover for 14 days and all testing commenced day 14 postsurgery. Following nerve injury, only animals that developed paw withdrawal thresholds <1 g for SNI and <4.7 g for SNL by day 14 postsurgery were used. Animals were placed in acrylic boxes with wire mesh floors and allowed to habituate for 1 hour. Predrug mechanical thresholds were recorded and the animals received intraperitoneal injections of vehicle, rapamycin, or metformin (doses indicated in text). S6K DKO and their respective wild-type controls were subjected to a battery of nociceptive tests as described elsewhere [30], that included determination of mechanical (von Frey filaments, automated von Frey, and tail clip) and thermal thresholds (radiant heat paw withdrawal and tail withdrawal). Specifically, calibrated von Frey filaments (Stoelting, Wood Dale, IL, USA) were used for mechanical stimulation of the plantar surface of the left hind paw and withdrawal thresholds were calculated using the up-down method [8]. In SNI mice, mechanical stimulation was applied to the dermatome innervated by the spared fibers. To determine rapamycin-induced hypersensitivity, the animals were allowed to acclimate in the acrylic box for 1 hour and baseline thresholds were recorded. The animals were then treated with rapamycin or co-injected with rapamycin and an AMPK activator (metformin 200 mg/kg or A769662 30 mg/kg). For Mouse Grimace Scale experiments, mice received injections and were placed in a plexiglass box for observation. Between 2 hours, 15 minutes and 2 hours, 45 minutes after injection, digital recordings were made and facial expression was scored from 80 images taken over this time period. Images were randomly chosen for scoring as described in detail previously [23,25]. In all behavioral experiments the experimenter was blinded to the conditions of the experiment. For Western blotting studies, tissues were harvested 17 days post SNL or 3 hours after treatment of mice with rapamycin alone or in combination with AMPK activators.
2.2. Viral-mediated Raptor deletion
Raptor floxed mice on a C57BL/6 background were provided by D. Sabatini (Whitehead Institute for Biomedical Research, Cambridge, MA, USA). Long-term herpes simplex viral vector expressing Cre under Cytomegalovirus (CMV) promoter (LT HSV-Cre) was generated by R. Neve (Viral Gene Transfer Core, MIT, Cambridge, MA, USA). Under isoflurane anesthesia, 5 μL of solution containing viral particles (3 × 108 transducing units/mL) were injected subcutaneously into the middle of the plantar surface of the left hind paw of the Raptor floxed mice (Raptorflox/flox) and the experiments were conducted at least 2 weeks after the injection. Raptor floxed mice injected with HSV absent Cre and wild-type C57BL/6 mice injected with LT HSV-Cre were used as controls.
2.3. Primary neuronal cultures
Mouse dorsal root or trigeminal ganglia (DRG or TG) from ICR mice were excised aseptically and placed in Hank’s Balanced Salt Solution (Invitrogen, Grand Island, NY, USA) on ice. The ganglia were dissociated enzymatically with collagenase A (1 mg/mL, 25 minutes, Roche, Indianapolis, IN, USA) and collagenase D (1 mg/mL, Roche) with papain (30 U/mL, Roche) for 20 minutes at 37°C. To eliminate debris, 70 μm (BD) cell strainers were used. The dissociated cells were resuspended in Dulbecco’s Modified Eagle Medium/F12 (Invitrogen) containing 1× pen-strep (Invitrogen), 1× GlutaMax, 3 μg/mL 5-FDU (Sigma, St. Louis, MO, USA), 7 μg/mL uridine (Sigma), and 10% fetal bovine serum (Thermo Hyclone, Rockford, IL, USA). The cells were plated in 6-well plates (BD Biosciences, San Jose, CA, USA) and incubated at 37°C in a humidified 95% air/5% CO2 incubator. On day 5 the cells were washed in Dulbecco’s Modified Eagle Medium/F12 media for 30 minutes, followed by treatment.
2.4. Western blotting
Proteins were extracted from the cells and tissue in lysis buffer (50 mM Tris HCl, 1% Triton X-100, 150 mM NaCl, and 1 mM ethylenediaminetetraacetic acid at pH 7.4) containing protease and phosphatase inhibitor mixtures (Sigma) with an ultrasonicator on ice, and cleared of cellular debris and nuclei by centrifugation at 14,000 relative centrifugal force for 15 minutes at 4°C. Fifteen micrograms of protein per well were loaded and separated by standard 7.5% or 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis. Proteins were transferred to Immobilon-P membranes (Millipore, Billerica, MA, USA) and then blocked with 5% dry milk for 3 hours at room temperature. The blots were incubated with primary antibody overnight at 4°C and detected the following day with donkey anti-rabbit antibody conjugated to horseradish peroxidase (Jackson Immunoresearch, West Grove, PA, USA). Signal was detected by enhanced chemiluminescence on chemiluminescent films. Each phosphoprotein was normalized to the expression of the corresponding total protein on the same membrane. Densitometric analyses were performed with Image J software (National Institutes of Health, Bethesda, MD, USA).
2.5. Co-immunoprecipitations
After protein extraction, 50 μg protein was incubated overnight with 1:100 Nav1.7 antibody (NeuroMab, UC Davis, CA, USA). The samples were incubated with Protein G conjugated Sepharose beads (Sigma) for 3 hours. The beads were then pelleted and washed twice with lysis buffer. After a final centrifugation, the pellet was suspended in 1× Laemmli Sample Buffer containing 5% v/v β-mercaptoethanol. The co-immunoprecipitated total ERK (Cell Signaling, Beverly, MA, USA) was detected by Western blotting.
2.6. Patch clamp electrophysiology
Whole-cell patch clamp experiments were performed on isolated mouse TG, cultured as described above with experiments performed 24 hours after isolation, using a MultiClamp 700B (Axon Instruments, Foster City, CA, USA) patch-clamp amplifier and pClamp10 acquisition software (Axon Instruments). Recordings were sampled at 5 kHz and filtered at 1 kHz (Digidata 1322A, Axon Instruments). Pipettes (outer diameter 1.5 mm, inner diameter 0.86 mm; Sutter Instrument Company, Novato, CA, USA) were pulled using a P-97 puller (Sutter) and heat polished to 2.5–4 MΩ resistance using a microforge (MF-83; Narishige International USA Inc, East Meadow, NY, USA). Pipette offsets were zeroed automatically before seal formation and liquid junction potentials were not corrected. Pipette capacitance neutralization and bridge balance were adjusted automatically in current-clamp mode. All recordings were performed at room temperature on cells with capacitance ranging from 20–30 pF, corresponding to small-diameter neurons (~20–30 μm). Data were analyzed using Clampfit 10 (Molecular Devices, Sunnyvale, CA, USA) and Origin 8 (OriginLab Corporation, Northampton, MA, USA). Pipette solution contained (in mM) 140 KCl, 11 ethylene glycol tetraacetic acid, 2 MgCl2, 10 NaCl, 10 HEPES, 1 CaCl2 pH 7.3 (adjusted with N-methyl glucamine), and was 320 mOsm. External solution contained (in mM) 135 NaCl, 2 CaCl2, 1 MgCl2, 5 KCl, 10 glucose and 10 HEPES, pH 7.4 (adjusted with N-methyl glucamine), and was 320 mOsm. The cells were treated with relevant drug for 1 hour before patch clamp studies were carried out. Ramp stimulus protocols for Nav1.7 were performed as described previously [40], as these protocols have been shown to preferentially elicit function from Nav1.7 channels [11]. The viability of the neurons was verified by eliciting currents in response to a voltage step protocol before and after each current ramp protocol was carried out.
2.7. Drugs and primary antibodies
U0126, U0124, and PF-4708671 were from Tocris (Ellisville, MO, USA) and metformin was from LKT Laboratories (St. Paul, MN, USA). The following rabbit polyclonal antibodies were obtained from Cell Signaling: p-IRS-1 (Ser636/639, cat# 2388; Ser789, cat# 2389; and Tyr895, cat# 3070), total IRS, p-ERK (Thr202/Tyr204, cat# 4377), total ERK, p-AKT (Ser473, cat# 4058), total AKT, p-rS6 (Ser240/244, cat#2215); and total rS6. Nav1.7 mouse monoclonal antibody was from University of California Davis/National Institutes of Health NeuroMab Facility. Rapamycin, CCI-779, and A769662 were from LC Laboratories (Woburn, MA, USA).
2.8. Statistical analysis and data presentation
Data are shown as means ± SEM of n = 8 independent cell culture wells, n = 6 tissue samples (for in vivo Western blotting, and co-immunoprecipitations), or n = 6 animals (for behavioral studies) per group. Graph plotting and statistical analysis used GraphPad Prism Version 5.03 (Graph Pad Software, Inc. San Diego, CA, USA). Statistical evaluation was performed by one- or two-way analysis of variance, followed where appropriate by Dunnett post hoc analysis. The a priori level of significance was considered to be P < 0.05.
3. Results
3.1. Pharmacological or genetic inhibition of mTORC1 results in mechanical allodynia
Previous studies suggested that acute local, intrathecal, or systemic mTORC1 inhibition with rapamycin alleviates neuropathic pain [2,15,19]. Consistent with this, treatment of rats having received SNL with rapamycin (1 mg/kg/day for 9 days) resulted in a partial, but significant, reduction in SNL-induced allodynia (Fig. 1a). However, sham-operated animals developed profound tactile hypersensitivity as a result of rapamycin treatment (Fig. 1a). Inhibition of mTORC1 and, in turn, its downstream target, S6K1, can lead to disinhibition of IRS-1 signaling, resulting in activation of ERK [16] and AKT [53] pathways. However, this feedback signaling has not been demonstrated in vivo and its physiological relevance is unknown. To study whether this feedback signaling occurred in vivo as a result of rapamycin treatment, sciatic nerves were collected from SNL or sham rats treated with rapamycin (1 mg/kg/day) for 9 days. Continuous rapamycin treatment evoked an increase in p-ERK at Thr202/Tyr204 and p-AKT at S473 [53] in sham-operated animals (Fig. 1b).
Fig. 1.

Rapamycin treatment induces allodynia and increases AKT and extracellular signal-regulated kinase (ERK) phosphorylation.(a) Daily intraperitoneal injection of rapamycin in rats with spinal nerve ligation (SNL) (treatment started 14 days post-SNL) leads to a partial reversal of mechanical allodynia but causes allodynia in sham-operated animals. (b) Treatment with rapamycin causes significant increases in ERK (Thr202/Tyr204) and AKT (S473) phosphorylation in the sciatic nerve of sham-operated animals and rapamycin fails to inhibit these kinases in SNL rats. (c) Daily intraperitoneal injection of rapamycin in mice with spared nerve injury (SNI) partially reverses mechanical allodynia on day 7 (#, P < 0.05), however, sham-operated animals develop allodynia within 3 hours after rapamycin injection. (d) Daily injection with metformin (Met, 200 mg/kg) for 7 days in mice with SNI 7 weeks postsurgery reverses neuropathic allodynia for at least 61 days after the cessation of the treatment. (e) Naïve mice develop allodynia when treated with rapamycin in a dose dependent manner and CCI-779 treatment induces allodynia in naïve mice. (f) Area-over-the-curve (AOC) analysis demonstrates dose-related induction of allodynia in response to rapalogue treatment. n = 6 per condition. *P < 0.05; **P < 0.01; ***P < 0.001.
In addition, we investigated the efficacy of rapamycin in mice with SNI. Similar to a previous report with the same dose [34], systemic administration of rapamycin partially reversed SNI-induced mechanical allodynia. However, daily treatment with rapamycin (20 mg/kg/day) produced allodynia in sham animals (Fig. 1c). In contrast, we have previously shown that AMPK activators, metformin and A769662, effectively reverse mechanical allodynia induced by SNI [28]. Here we show that even after long-term establishment of neuropathic pain (7 weeks after surgery), metformin completely reversed neuropathic allodynia in C57BL/6 mice, and this effect persisted following the cessation of treatment for at least 2 months (Fig. 1d), highlighting the efficacy of AMPK activators for the potential treatment of neuropathic pain. Metformin had no effect on mechanical thresholds in sham animals (Fig. 1d).
We then asked whether mTORC1 inhibition would evoke mechanical allodynia in naïve mice. Systemic administration of escalating doses of rapamycin (2, 6, and 20 mg/kg) caused an acute and dose-related allodynia that peaked between 2 and 3 hours and subsided by 4 hours (Fig. 1e and f). Systemic treatment with the rapalogue CCI-779 (20 mg/kg) produced a similar effect (Fig. 1e and f). Daily rapamycin dosing of naïve rats (1 mg/kg) also caused allodynia within 4 days that persisted through at least day 7 (data not shown). Therefore, our data demonstrate that although systemic treatment with rapalogues reduces neuropathic allodynia, these drugs also cause tactile hypersensitivity.
Although rapamycin and CCI-779 are thought to be selective mTORC1 inhibitors, nonspecific effects cannot be excluded, especially at higher doses. Moreover, systemic treatment may not selectively target sensory neurons. Thus, we used a Cre/loxP system to delete a critical component of mTORC1 complex, Raptor, selectively in sensory neurons (Fig. 2a). Raptor floxed mice were injected in the hind paw with neurotrophic herpes simplex viral vector expressing Cre recombinase (HSV-Cre). Two weeks after the infection, the levels of Raptor and the downstream mTORC1 target, ribosomal S6 protein phosphorylation, were significantly decreased in DRG of the floxed mice injected with HSV-Cre (Fig. 2a), as compared to Raptor floxed mice injected with HSV (without Cre) or non-floxed mice injected with HSV-Cre. Remarkably, deletion of Raptor induced mechanical allodynia but had no effect on thermal thresholds (Fig. 2b), similar to the effects observed with rapamycin treatment in mice and rats (Fig. 1a, c, e, and f).
Fig. 2.

Raptor deletion leads to tactile allodynia. Raptor floxed mice were injected into the hind paw with herpes simplex viral (HSV) vector expressing Cre. (a) The levels of Raptor and rS6 phosphorylation decreased in the dorsal root ganglia (DRG) of Raptor floxed mice (Raptorflox/flox) 2 weeks after the injection compared to the floxed mice injected with HSV without Cre or C57BL/6 mice injected with HSV-Cre. (b) Raptor deletion leads to mechanical allodynia, whereas thermal thresholds are not altered (n = 8 per group). **P < 0.01.
S6K1/2 are direct targets of mTORC1 and phosphorylate IRS-1 as part of a negative feedback loop [53]. Thus, we hypothesized that loss of S6K1/2 activity should recapitulate mTORC1 inhibition-mediated allodynia. To inhibit S6 kinases we used both genetic and pharmacological approaches. Double-knockout mice for S6K1/2 (S6K DKO) exhibited tactile hypersensitivity, whereas thermal thresholds were not altered (Fig. 3a). Similarly, inhibition of S6K1 with the specific inhibitor PF-4708671 led to a decrease in mechanical thresholds (Fig. 3b). Taken together, our data indicate that inhibition of mTORC1 with either pharmacological or genetic approaches leads to tactile allodynia via decreased S6K signaling. Importantly, and consistent with effects observed with rapamycin treatment in vivo, deletion of Raptor and S6K1/2 led to enhanced ERK and AKT phosphorylation in DRG neurons (Fig. 3c and d), whereas the direct target of S6K1/2, ribosomal S6 protein phosphorylation, was absent (Fig. 3d).
Fig. 3.

Reduction of S6K activity leads to enhanced tactile sensation. (a) Mechanical sensitivity in automated von Frey and tail clip tests is increased in S6K1/2 double-knockout (DKO) mice as compared to wild-type (WT) littermates (n = 8 per group). No change in thermal sensitivity was measured in radiant heat paw withdrawal and hot plate tests. (b) Daily intraperitoneal administration of S6K1 inhibitor PF 4708671 (50 mg/kg) leads to reduction of mechanical threshold (left panel; n = 4 per group), whereas no change in thermal sensation was observed (right panel; n = 6). Phosphorylation of AKT and extracellular signal-regulated kinase (ERK) is increased in sciatic nerves following Raptor deletion (c, n = 6) and in dorsal root ganglia of S6K1/2 DKO mice, while insulin receptor substrate (IRS) S636 phosphorylation is decreased in these mice (d, n = 6). *P < 0.05; **P < 0.01; ***P < 0.001.
Phosphorylation of S636/639 residues on IRS-1 was also reduced in S6K DKO mice (Fig. 3d). Since this is an inhibitory site on IRS, reduced phosphorylation would be associated with enhanced signaling.
3.2. mTORC1 inhibition promotes ERK activity resulting in sensory neuron hyperexcitability
We hypothesized that mTORC1-mediated disinhibition of ERK signaling may lead to sensory neuron hyperexcitability. Rapamycin treatment of cultured mouse sensory neurons resulted in a concentration-related increase in p-ERK and p-AKT (Fig. 4a), recapitulating changes observed in vivo (Fig. 1b). Moreover, to ensure an inhibitory effect of rapamycin on mTORC1, we determined that rapamycin suppressed phosphorylation of ribosomal S6 protein (Fig. 4a).
Fig. 4.

Rapamycin promotes extracellular signal-regulated kinase (ERK)-dependent hyperexcitability of sensory neurons. (a) Treatment of mouse primary sensory neurons in culture with rapamycin for 1 hour results in concentration-related increases in p-ERK and p-AKT (S473). Rapamycin inhibits the phosphorylation rS6, a downstream target of mammalian target of rapamycin complex 1 (mTORC1)-S6K (n = 6). (b) Co-immunoprecipitation analysis reveals that the treatment of primary sensory neurons in culture with rapamycin (100 nM) for 1 hour enhances the association of ERK1 with Nav1.7 (n = 6). ERK2 does not immunoprecipitate with Nav1.7. Rapamycin-enhanced association of ERK1 with Nav1.7 is blocked by U0126 (10 μM) but not affected by U0124 (10 μM) treatment. (c) Patch clamp analysis of mouse sensory neurons in culture treated with rapamycin (n = 13) for 1 hour demonstrates that: rapamycin dose-dependently (d) increases the number of action potentials evoked by ramp currents and (e) reduces the latency to first action potential in response to escalating ramp currents. (f and g) Rapamycin-induced hyperexcitability is blocked by U0126 (n = 10) but unaffected by U0124 (n = 11). Color code of the stars: red (30 nm Rap vs vehicle), orange (100 nM Rap vs vehicle), brown (100 nM Rap + 10 μM U0124 vs vehicle), and green (100 nM Rap + 10 μM U0126 vs 100 nM Rap). *P < 0.05; **P < 0.01; ***P < 0.001.
The voltage-gated sodium channel Nav1.7 is a threshold channel that augments weak stimuli and exhibits gating properties modulated by ERK phosphorylation [40]. ERK1 phosphorylation of specific residues on the intracellular L1 loop of Nav1.7 mediates an increase in action potential firing and a decrease in latency to first action potential in sensory neurons stimulated with depolarizing ramp current injections [40]. Importantly, Nav1.7 is linked to human pain conditions wherein loss-of-function results in insensitivity to pain, while gain-of-function causes erythromelalgia and paroxysmal extreme pain disorder [12]. Using co-immunoprecipitation, we demonstrate that treatment of primary sensory neurons in culture with rapamycin (100 nM) for 1 hour enhances the association of ERK1 but not ERK2 (which did not immunoprecipitate) with Nav1.7 (Fig. 4b). Moreover, inhibition of ERK activation by co-treatment of the cultures with rapamycin and the mitogen-activated protein kinase kinase (MEK) inhibitor U0126 (10 μM) prevents the association of ERK1 with Nav1.7, whereas the inactive analogue U0124 has no effect (Fig. 4b).
We then used patch clamp electrophysiology to study the effect of rapamycin on the excitability of small-diameter (20–30 μm) sensory neurons. Rapamycin treatment caused a dose-related increase in the number of action potentials evoked in response to ramp currents (Fig. 4c and d) and a decrease in latency to first action potential firing (Fig. 4c and e), an effect consistent with phosphorylation of Nav1.7. Co-treatment of cultured neurons with rapamycin (100 nM) and the MEK inhibitor U0126 (10 μM) completely blocked these effects, whereas U0124 (10 μM) was inactive (Fig. 4c, f, and g). Thus, rapamycin enhances excitability of sensory neurons in a manner consistent with ERK-mediated modulation of Nav1.7-dependent action potential generation, highlighting the potential importance of mTORC1-IRS-1 crosstalk in rapamycin-evoked hypersensitivity.
3.3. AMPK activators block the behavioral, biochemical, and electrophysiological effects of mTORC1 inhibition via IRS-1
Phosphorylation of serine 636/639 and 789 residues is associated with decreased signaling from IRS-1. IRS-1 S636/639 residues are phosphorylated by S6K [35,43], while the S789 site is phosphorylated by AMPK [42]. Treatment of primary afferent neurons in culture with rapamycin for 1 hour results in decreased phosphorylation of S636/639 residues on IRS-1 (Fig. 5a), consistent with decreased S6K activity. Decreased phosphorylation of S636/639 residues allows for enhanced signaling through IRS-1 [53]. Treatment of either DRG or TG neurons in culture with rapamycin enhanced IRS-1 Y895 phosphorylation (Fig. 5a and b), which is required for the recruitment of the adaptor protein Grb2 that is critical for activation of the ERK pathway [44]. Consistent with this, decreased S636/639 phosphorylation and enhanced Y895 phosphorylation of IRS-1 are linked to augmented ERK activity following rapamycin treatment (Fig. 5a and b). In contrast, treatment of sensory neuron cultures with the AMPK activator A769662 (200 μM) enhanced S789 phosphorylation and attenuated rapamycin-induced changes in Y895 phosphorylation of IRS-1 (Fig. 5a and b). This increase in IRS-1 S789 phosphorylation and blockade of IRS-1 Y895 phosphorylation was associated with an AMPK-mediated block of ERK activation by rapamycin treatment (Fig. 5a and b). Hence, rapamycin treatment leads to phosphorylation of IRS-1 at specific sites that promote ERK activity; however, co-treatment with A769662, a positive allosteric modulator of AMPK [10], blocks the effects of rapamycin.
Fig. 5.

Rapamycin activates extracellular signal-regulated kinase (ERK) by suppressing insulin receptor substrate (IRS)-1 phosphorylation, while adenosine monophosphate-activated protein kinase (AMPK) activator A769662 inhibits the ERK activation by enhancing the phosphorylation of IRS-1. Treatment of dorsal root ganglion (a) or trigeminal ganglia (b) neurons in culture with rapamycin (100 nM) for 1 hour suppresses the phosphorylation of IRS-1 on S636. Treatment of the cultures with A769662 (200 μM) enhances S789 phosphorylation on IRS-1. The net outcome of these phosphorylation events is enhanced Y895 phosphorylation of IRS-1 in response to rapamycin (100 nM) treatment alone. Phosphorylation of Y895 is associated with the activation of ERK (n = 6). (b) AMPK activator A769662 blocks rapamycin-enhanced excitability of sensory neurons. Patch-clamp electrophysiology reveals that the treatment of sensory neurons in culture (n = 10) with rapamycin (100 nM) for 1 hour results in an increase in the number of action potentials evoked by ramp currents and reduces latency to first action potential in response to ramp currents. Co-treatment of sensory neurons in culture (n = 10 per group) with A769662 (200 μM) and rapamycin (100 nM) profoundly attenuates the number of action potential evoked by ramp currents (c) and increases the time to first action potential in response to ramp currents (d). *P < 0.05; **P < 0.01; ***P < 0.001.
Next we used patch clamp electrophysiology to study the effect of AMPK activation on rapamycin-evoked hyperexcitability in small-diameter sensory neurons. The selective AMPK activator A769662 [10] completely reversed the rapamycin-induced increase in neuronal excitability (Fig. 5c–e), similar to the effect of the MEK inhibitor U0126. To determine whether AMPK activators prevent mTORC1-inhibition-induced allodynia and ERK activation in vivo, we co-injected naïve mice with rapamycin (20 mg/kg) and either metformin (200 mg/kg) or A769662 (30 mg/kg). Rapamycin-induced tactile hypersensitivity was prevented by both metformin and A768662 (Fig. 6a). Concomitantly, in the sciatic nerve, we observed a significant increase in p-ERK 3 hours after rapamycin treatment, and this was prevented by both metformin and A769662 (Fig. 6b). To determine if mTORC1 inhibition leads to the development of spontaneous pain, we used the Mouse Grimace Scale [23,25]. Injection of rapamycin (20 mg/kg) led to an increase in Mouse Grimace Scale scores consistent with the development of spontaneous pain after rapamycin injection (Fig. 6c). This effect was completely blocked by treatment with metformin (200 mg/kg), consistent with findings using mechanical sensitivity as an end point (Fig. 6c). Next, we asked whether metformin treatment would similarly reverse mechanical allodynia induced by genetic disruption of mTORC1 activity. To test this, we treated mice with sensory neuron deletion of raptor with 200 mg/kg metformin. Consistent with effects observed with acute rapamycin treatment, metformin also reversed allodynia in mice lacking Raptor (Fig. 6d), and no effect was seen on thermal nociception (Fig. 6e). Therefore, mTORC1 inhibition leads to mechanical allodynia and spontaneous pain in an AMPK activator reversible fashion.
Fig. 6.

Adenosine monophosphate-activated protein kinase (AMPK) activators block rapamycin-induced behavioral and biochemical changes. (a) Treatment of naïve mice with rapamycin (20 mg/kg) induces mechanical allodynia that peaks at 3 hours and subsides by 4 hours. Co-treatment with rapamycin and either metformin (200 mg/kg) or A769662 (30 mg/kg) blocks the development of rapamycin-induced allodynia. (b) Sciatic nerves from these animals show a significant increase in p-ERK (extracellular signal-regulated kinase) and its blockade with either metformin or A769662 treatment at 3 hours posttreatment. (c) Rapamycin (20 mg/kg) treatment causes a significant increase in Mouse Grimace Scale (MGS) score at 2–3 hours after systemic administration of drug. This effect is completely blocked by co-administration of metformin (200 mg/kg, Sal = saline). (d) Control (open circles) and Raptor knockout mice (Raptorflox/flox + HSV-Cre; filled circles) were injected daily with metformin (200 mg/kg). Tactile allodynia, exhibited by Raptor-deleted mice, was resolved after 4 days of treatment with metformin. (e) No change in thermal threshold was measured after 4 days of metformin treatment. n = 6 per group. *P < 0.05; **P < 0.01; ***P < 0.001.
4. Discussion
The findings described herein add substantially to our understanding of mTOR signaling in sensory neurons and how this pathway might be therapeutically manipulated for the treatment of pain. First, consistent with studies in cell lines [16,53] and in cancers obtained from patients treated with rapalogues [7,33], we observed that inhibition of mTORC1 leads to feedback activation of AKT and ERK in DRG and TG neurons. This is recapitulated by inducible knockout of raptor in DRG neurons and by either knockout or inhibition of S6K1/2. Physiologically, this leads to increased sensory neuron excitability, mechanical allodynia, and spontaneous pain. Importantly, we show that activation of AMPK is capable of blocking these effects, including rapalogue-evoked spontaneous pain, likely via modulation of IRS-1 phosphorylation. Furthermore, we show that the AMPK activator metformin leads to a long-lasting reduction in neuropathic allodynia, whereas rapalogue effects on neuropathic pain are less efficacious and transient. Hence, we conclude that mTORC1 inhibition with rapalogues as a therapeutic target for pain is likely to be limited by feedback activation or ERK in sensory neurons. AMPK activators do not promote this mechanism, while blocking mTORC1 activity in sensory neurons [28,41], further strengthening the case for development of AMPK activators for the treatment of pain.
A number of previous studies have assessed the role of mTORC1 in a variety of preclinical pain models utilizing either intrathecal or local injection of rapamycin. Rapamycin, when given intrathecally (spinally), inhibits formalin-induced pain [37] and dorsal horn excitability [1], and reduces neuropathic pain via action on sensory afferents [15] or spinal cord dorsal horn neurons [2]. Spinal mTORC1 has also been linked to inflammatory [32,49] and bone cancer-induced pain [39]. Similarly, several studies have suggested a role of mTORC1 in neuropathic or algogen-induced pain via a peripheral mechanism of action through local administration of rapamycin [3,19,27,34]. However, only one study has assessed how systemically administered rapalogues influence neuropathic pain, finding a small but significant reduction in neuropathic allodynia after repeated or single systemic administration [34]. Interestingly, these authors did not find any change in mechanical threshold in sham mice treated with CCI-779 [34], in direct contrast to the findings reported here. We found that rapamycin or CCI-779 treatment induced allodynia and spontaneous pain (in the case of rapamycin) in either sham or naïve mice treated with these compounds. This could be recapitulated through pharmacological inhibition of S6K1/2 or global knockout of S6K1/2. Moreover, selective deletion of raptor in sensory afferents using HSV-Cre led to the development of mechanical allodynia, replicating our findings with rapamycin or CCI-779 and strongly implicating inhibition of mTORC1 in sensory afferents as the cause of allodynia and spontaneous pain resulting from mTORC1 inhibition. Why, though, does systemic treatment with rapalogues cause this pain state, whereas local treatment, especially intrathecal treatment, does not? This may be due to pharmacokinetic factors, as local treatment might lead to a short duration of mTORC1 inhibition with relatively rapid washout of local drug, whereas systemic treatment would lead to sustained inhibition of mTORC1, allowing for the engagement of feedback signaling and activation of ERK and AKT. In support of this notion, the doses of rapalogues used in this study, and a previous study showing efficacy in mouse neuropathic pain models [34], are substantially higher than doses used in long-term studies examining the role of mTORC1 in longevity [17,29,46], for instance. Another possibility, which we cannot exclude, is that these differences in local vs systemic treatment arise simply from differences in local concentration of drug, depending on delivery route dosing.
Interestingly, numerous clinical studies have reported pain as a side effect of treatment with rapamycin and rapalogues [6,9,24,26,31,47]; however, the molecular mechanism underlying pain induction by mTORC1 inhibition remains elusive. Here, we found that disruption of mTORC1 signaling in sensory neurons causes suppression of IRS-1-mediated negative feedback, leading to AKT and ERK activation in TG and DRG as well as sciatic nerves. Enhanced ERK activity increases sensory neuron excitability and is linked to mechanical hypersensitivity and spontaneous pain. Hence, we provide a potential molecular mechanism for rapalogue-induced pain, highlighting the potential physiological ramifications of feedback signaling where mTORC1 is inhibited pharmacologically or genetically. Clearly, not all patients develop pain when treated with rapalogues [6,13,26], however, some severe cases of pain have been reported with these drugs, including the possibility that they may provoke CRPS in certain susceptible individuals [24,31,45]. CRPS symptoms have been shown to resolve after rapalogue withdrawal [31,45], suggesting a causal relationship. It is possible that rapalogues have a different drug disposition or pharmacokinetic profile in certain individuals, leading to sufficient accumulation of drug to stimulate feedback activation of ERK. Such speculation will have to await further clinical examination.
Acute treatment with neurotrophic factors and proinflammatory cytokines increases excitability of sensory neurons and induces hyperalgesia, in part, via activation of ERK/MAP kinase [18,27,50]. ERK activity has been linked to increased neuronal excitability via reduction of voltage-gated potassium conductance (Kv4.2) [51], an increase in voltage-gated calcium conductance (Cav2.2) [14,48], and modulation of sodium channel (Nav1.7) gating properties [40]. We show that rapalogues activate ERK signaling and lead to increased sensory neuron excitability, spontaneous pain, and allodynia, consistent with the previously established role of ERK as modulator of neuron excitability. Though modulation of calcium and potassium currents by enhanced ERK activity could contribute to the observed effects, several lines of evidence indicate that changes in Nav1.7 channel gating properties have a major role. First, in immunoprecipitation experiments, we found that rapamycin treatment increased the association of Nav1.7 with ERK1. Second, mTORC1 inhibition decreases the latency to first action potential and number of action potentials evoked by ramp depolarization in small-diameter sensory neurons. These first 2 findings are consistent with the fact that phosphorylation of Nav1.7 channel by ERK modulates its gating properties without increasing current density [40], in contrast to the reported effect of ERK on potassium and calcium channels current density [14,48,51]. Third, selective ablation of mTORC1 in sensory neurons recapitulated the pharmacological effect of rapalogues and/or S6K1 inhibitors, suggesting that the locus of action is in sensory afferents, where Nav1.7 is predominantly expressed. Finally, injection of rapamycin induced spontaneous pain in mice, consistent with gain of function phenotypes of Nav1.7 mutations found in humans that induce spontaneous pain [12]. Hence, although we cannot exclude possible effects on other channels or at other anatomical sites, our findings point to mTORC1 inhibition-mediated modulation of Nav1.7 in sensory afferents as an important mediator of mTORC1 inhibition-mediated pain.
Cells utilize the endogenous energy-sensing kinase, AMPK, to regulate mTORC1 activity under conditions of low nutrient availability. Activated AMPK also phosphorylates IRS-1 at Ser789, a site associated with decreased IRS-1 downstream signaling [42,52]. Importantly, we demonstrate that rapalogue-induced and IRS-1-associated activation of AKT and ERK is prevented when Ser789 is phosphorylated in the presence of AMPK activators, and that rapalogue-triggered phosphorylation of ERK and AKT is blocked in vitro and in vivo (summarized in Fig. 7). Activation of AMPK also rescued the increase in sensory neuron excitability, spontaneous pain, and allodynia, suggesting that AMPK activators might be utilized to alleviate pain-promoting signaling engaged by rapalogues in clinical situations where rapalogue treatment is warranted. The AMPK activator metformin, which is widely used as a first-line therapy for type 2 diabetes, is a remarkably well-tolerated drug with low incidence of side effects [4,38]. We previously showed a beneficial effect of AMPK activators, including metformin, in preclinical models of neuropathic and postsurgical pain [28,41]. The present findings, with AMPK activators, further support the case for the continued development of this line of drugs as pain therapeutics with potential disease-modifying attributes.
Fig. 7.
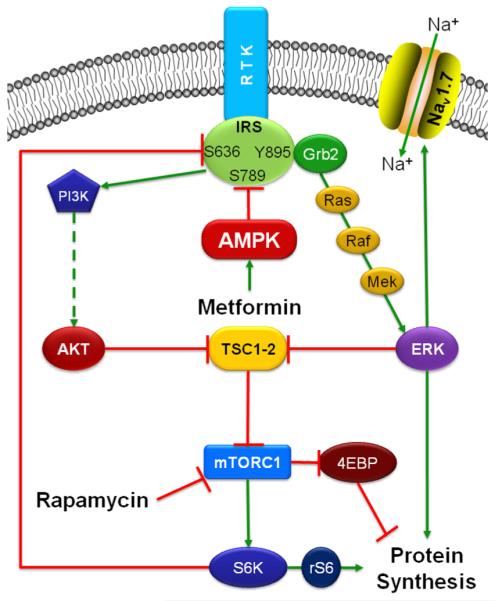
Schematic diagram summarizing the signaling pathways relevant to this study. Activation of receptor tyrosine kinases (RTK) results in the recruitment of the adaptor protein insulin receptor substrate (IRS), which results in the activation of PI3K-AKT- mammalian target of rapamycin (mTOR) and Ras-Raf-Mek-ERK (extracellular signal-regulated kinase) pathways. Serine phosphorylation of IRS on the 636/639 and 789 residues is associated with reduced signaling from IRS, whereas Y895 phosphorylation is associated with enhanced signaling. Blockade of mTOR complex 1 (mTORC1) with rapamycin results in the inhibition of S6K, which leads to reduced IRS S636/639 phosphorylation, enhanced phosphorylation of IRS Y895 residue, and the recruitment of the adaptor protein Grb2, resulting in the activation of the ERK pathway. Activated ERK1 associates and phosphorylates Nav1.7, leading to hyperexcitability of sensory neurons. Activation of adenosine monophosphate-activated protein kinase (AMPK) in response to metformin treatment, leads to enhanced IRS S789 phosphorylation (which is an inhibitory phospho site) and reduced phosphorylation of IRS Y895 (which is a phospho site associated with enhanced IRS activity) residue, which suppresses the recruitment of Grb2, thus reversing rapamycin-induced activation of the ERK pathway.
In summary, the present findings use pharmacological and genetic mechanisms to elucidate a novel physiological role of mTORC1 inhibition-mediated feedback activation of ERK, causing sensory neuron hyperexcitability, spontaneous pain, and mechanical allodynia. Moreover, our results identify AMPK activation as a preferable therapeutic avenue for the treatment of neuropathic pain, as this mechanism is devoid of liabilities clearly engaged by direct inhibition of mTORC1.
Acknowledgements
This work was supported by funds from The Rita Allen Foundation (T.J.P.), The Louise and Alan Edwards Foundation (J.S.M.), National Institutes of Health grants NS065926 (T.J.P.), NS072204 (G.D.), CA149258 (S.G.), and Canadian Institutes for Health Research grant MOP-114994 (NS). T.J.P. is a Rita Allen Foundation Scholar in Pain.
Footnotes
Conflict of interest statement The authors do not have any conflicts of interest to report.
References
- [1].Asante CO, Wallace VC, Dickenson AH. Formalin-induced behavioural hypersensitivity and neuronal hyperexcitability are mediated by rapid protein synthesis at the spinal level. Mol Pain. 2009;5:27. doi: 10.1186/1744-8069-5-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Asante CO, Wallace VC, Dickenson AH. Mammalian target of rapamycin signaling in the spinal cord is required for neuronal plasticity and behavioral hypersensitivity associated with neuropathy in the rat. J Pain. 2010;11:1356–67. doi: 10.1016/j.jpain.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Asiedu MN, Tillu DV, Melemedjian OK, Shy A, Sanoja R, Bodell B, Ghosh S, Porreca F, Price TJ. Spinal protein kinase M zeta underlies the maintenance mechanism of persistent nociceptive sensitization. J Neurosci. 2011;31:6646–53. doi: 10.1523/JNEUROSCI.6286-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bailey CJ, Turner RC. Metformin. N Engl J Med. 1996;334:574–9. doi: 10.1056/NEJM199602293340906. [DOI] [PubMed] [Google Scholar]
- [5].Bourquin AF, Suveges M, Pertin M, Gilliard N, Sardy S, Davison AC, Spahn DR, Decosterd I. Assessment and analysis of mechanical allodynia-like behavior induced by spared nerve injury (SNI) in the mouse. PAIN®. 2006;122:e11–4. doi: 10.1016/j.pain.2005.10.036. [DOI] [PubMed] [Google Scholar]
- [6].Budde K, Becker T, Arns W, Sommerer C, Reinke P, Eisenberger U, Kramer S, Fischer W, Gschaidmeier H, Pietruck F. Everolimus-based, calcineurin-inhibitor-free regimen in recipients of de-novo kidney transplants: an open-label, randomised, controlled trial. Lancet. 2011;377:837–47. doi: 10.1016/S0140-6736(10)62318-5. [DOI] [PubMed] [Google Scholar]
- [7].Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, Papa A, Nardella C, Cantley LC, Baselga J, Pandolfi PP. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–74. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- [9].Christopher JEW. Sirolimus (rapamycin) in clinical transplantation. Transplant Rev. 2001;15:165–77. [Google Scholar]
- [10].Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, Zhao G, Marsh K, Kym P, Jung P, Camp HS, Frevert E. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403–16. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- [11].Cummins TR, Howe JR, Waxman SG. Slow closed-state inactivation: a novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J Neurosci. 1998;18:9607–19. doi: 10.1523/JNEUROSCI.18-23-09607.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. Sodium channels in normal and pathological pain. Annu Rev Neurosci. 2010;33:325–47. doi: 10.1146/annurev-neuro-060909-153234. [DOI] [PubMed] [Google Scholar]
- [13].Ekberg H, Kyllonen L, Madsen S, Grave G, Solbu D, Holdaas H. Increased prevalence of gastrointestinal symptoms associated with impaired quality of life in renal transplant recipients. Transplantation. 2007;83:282–9. doi: 10.1097/01.tp.0000251923.14697.f5. [DOI] [PubMed] [Google Scholar]
- [14].Fitzgerald EM. Regulation of voltage-dependent calcium channels in rat sensory neurones involves a Ras-mitogen-activated protein kinase pathway. J Physiol. 2000;527(Pt. 3):433–44. doi: 10.1111/j.1469-7793.2000.00433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Geranton SM, Jimenez-Diaz L, Torsney C, Tochiki KK, Stuart SA, Leith JL, Lumb BM, Hunt SP. A rapamycin-sensitive signaling pathway is essential for the full expression of persistent pain states. J Neurosci. 2009;29:15017–27. doi: 10.1523/JNEUROSCI.3451-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ghosh S, Tergaonkar V, Rothlin CV, Correa RG, Bottero V, Bist P, Verma IM, Hunter T. Essential role of tuberous sclerosis genes TSC1 and TSC2 in NF-kappaB activation and cell survival. Cancer Cell. 2006;10:215–26. doi: 10.1016/j.ccr.2006.08.007. [DOI] [PubMed] [Google Scholar]
- [17].Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–5. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ji RR, Gereau RW, 4th, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev. 2009;60:135–48. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jimenez-Diaz L, Geranton SM, Passmore GM, Leith JL, Fisher AS, Berliocchi L, Sivasubramaniam AK, Sheasby A, Lumb BM, Hunt SP. Local translation in primary afferent fibers regulates nociception. PLoS One. 2008;3:e1961. doi: 10.1371/journal.pone.0001961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Karim F, Wang CC, Gereau RW., 4th Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J Neurosci. 2001;21:3771–9. doi: 10.1523/JNEUROSCI.21-11-03771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. PAIN®. 1992;50:355–63. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- [22].Kinkade CW, Castillo-Martin M, Puzio-Kuter A, Yan J, Foster TH, Gao H, Sun Y, Ouyang X, Gerald WL, Cordon-Cardo C, Abate-Shen C. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J Clin Invest. 2008;118:3051–64. doi: 10.1172/JCI34764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Langford DJ, Bailey AL, Chanda ML, Clarke SE, Drummond TE, Echols S, Glick S, Ingrao J, Klassen-Ross T, Lacroix-Fralish ML, Matsumiya L, Sorge RE, Sotocinal SG, Tabaka JM, Wong D, van den Maagdenberg AM, Ferrari MD, Craig KD, Mogil JS. Coding of facial expressions of pain in the laboratory mouse. Nat Methods. 2010;7:447–9. doi: 10.1038/nmeth.1455. [DOI] [PubMed] [Google Scholar]
- [24].Massard C, Fizazi K, Gross-Goupil M, Escudier B. Reflex sympathetic dystrophy in patients with metastatic renal cell carcinoma treated with everolimus. Invest New Drugs. 2010;28:879–81. doi: 10.1007/s10637-009-9295-8. [DOI] [PubMed] [Google Scholar]
- [25].Matsumiya LC, Sorge RE, Sotocinal SG, Tabaka JM, Wieskopf JS, Zaloum A, King OD, Mogil JS. Using the Mouse Grimace Scale to reevaluate the efficacy of postoperative analgesics in laboratory mice. J Am Assoc Lab Anim Sci. 2012;51:42–9. [PMC free article] [PubMed] [Google Scholar]
- [26].McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, Nakata K, Barker AF, Chapman JT, Brantly ML, Stocks JM, Brown KK, Lynch JP, Goldberg HJ, Young LR, Kinder BW, Downey GP, Sullivan EJ, Colby TV, McKay RT, Cohen MM, Korbee L, Taveira-Dasilva AM, Lee HS, Krischer JP, Trapnell BC. National Institutes of Health Rare Lung Diseases Consortium; MILES Trial Group. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011;364:1595–606. doi: 10.1056/NEJMoa1100391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Melemedjian OK, Asiedu MN, Tillu DV, Peebles KA, Yan J, Ertz N, Dussor GO, Price TJ. IL-6- and NGF-induced rapid control of protein synthesis and nociceptive plasticity via convergent signaling to the eIF4F complex. J Neurosci. 2010;30:15113–23. doi: 10.1523/JNEUROSCI.3947-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Melemedjian OK, Asiedu MN, Tillu DV, Sanoja R, Yan J, Lark A, Khoutorsky A, Johnson J, Peebles KA, Lepow T, Sonenberg N, Dussor G, Price TJ. Targeting adenosine monophosphate-activated protein kinase (AMPK) in preclinical models reveals a potential mechanism for the treatment of neuropathic pain. Mol Pain. 2011;7:70. doi: 10.1186/1744-8069-7-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF, Orihuela CJ, Pletcher S, Sharp ZD, Sinclair D, Starnes JW, Wilkinson JE, Nadon NL, Strong R. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2011;66:191–201. doi: 10.1093/gerona/glq178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mogil JS, Wilson SG, Bon K, Lee SE, Chung K, Raber P, Pieper JO, Hain HS, Belknap JK, Hubert L, Elmer GI, Chung JM, Devor M. Heritability of nociception I: responses of 11 inbred mouse strains on 12 measures of nociception. PAIN®. 1999;80:67–82. doi: 10.1016/s0304-3959(98)00197-3. [DOI] [PubMed] [Google Scholar]
- [31].Molina MG, Diekmann F, Burgos D, Cabello M, Lopez V, Oppenheimer F, Navarro A, Campistol J. Sympathetic dystrophy associated with sirolimus therapy. Transplantation. 2008;85:290–2. doi: 10.1097/TP.0b013e3181601230. [DOI] [PubMed] [Google Scholar]
- [32].Norsted Gregory E, Codeluppi S, Gregory JA, Steinauer J, Svensson CI. Mammalian target of rapamycin in spinal cord neurons mediates hypersensitivity induced by peripheral inflammation. Neuroscience. 2010;169:1392–402. doi: 10.1016/j.neuroscience.2010.05.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].O’Reilly KE, Rojo F, She Q-B, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N. MTOR inhibition induces upstream receptor tyrosine kinase signaling and activates AKT. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Obara I, Tochiki KK, Geranton SM, Carr FB, Lumb BM, Liu Q, Hunt SP. Systemic inhibition of the mammalian target of rapamycin (mTOR) pathway reduces neuropathic pain in mice. PAIN®. 2011;152:2582–95. doi: 10.1016/j.pain.2011.07.025. [DOI] [PubMed] [Google Scholar]
- [35].Ozes ON, Akca H, Mayo LD, Gustin JA, Maehama T, Dixon JE, Donner DB. A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proc Natl Acad Sci. 2001;98:4640–5. doi: 10.1073/pnas.051042298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, Mueller M, Fumagalli S, Kozma SC, Thomas G. S6K1(−/−)/S6K2(−/−) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol. 2004;24:3112–24. doi: 10.1128/MCB.24.8.3112-3124.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Price TJ, Rashid MH, Millecamps M, Sanoja R, Entrena JM, Cervero F. Decreased nociceptive sensitization in mice lacking the fragile X mental retardation protein: role of mGluR1/5 and mTOR. J Neurosci. 2007;27:13958–67. doi: 10.1523/JNEUROSCI.4383-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Reece EA, Leguizamon G, Wiznitzer A. Gestational diabetes: the need for a common ground. Lancet. 2009;373:1789–97. doi: 10.1016/S0140-6736(09)60515-8. [DOI] [PubMed] [Google Scholar]
- [39].Shih MH, Kao SC, Wang W, Yaster M, Tao YX. Spinal cord NMDA receptor-mediated activation of mammalian target of rapamycin is required for the development and maintenance of bone cancer-induced pain hypersensitivities in rats. J Pain. 2012;13:338–49. doi: 10.1016/j.jpain.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Stamboulian S, Choi JS, Ahn HS, Chang YW, Tyrrell L, Black JA, Waxman SG, Dib-Hajj SD. ERK1/2 mitogen-activated protein kinase phosphorylates sodium channel Na(v)1.7 and alters its gating properties. J Neurosci. 2010;30:1637–47. doi: 10.1523/JNEUROSCI.4872-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tillu DV, Melemedjian OK, Asiedu MN, Qu N, De Felice M, Dussor G, Price TJ. Resveratrol engages AMPK to attenuate ERK and mTOR signaling in sensory neurons and inhibits incision-induced acute and chronic pain. Mol Pain. 2012;8:5. doi: 10.1186/1744-8069-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Tzatsos A, Tsichlis PN. Energy depletion inhibits phosphatidylinositol 3-kinase/Akt signaling and induces apoptosis via AMP-activated protein kinase-dependent phosphorylation of IRS-1 at Ser-794. J Biol Chem. 2007;282:18069–82. doi: 10.1074/jbc.M610101200. [DOI] [PubMed] [Google Scholar]
- [43].Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–5. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- [44].Valverde AM, Mur C, Pons S, Alvarez AM, White MF, Kahn CR, Benito M. Association of insulin receptor substrate 1 (IRS-1) Y895 with Grb-2 mediates the insulin signaling involved in IRS-1-deficient brown adipocyte mitogenesis. Mol Cell Biol. 2001;21:2269–80. doi: 10.1128/MCB.21.7.2269-2280.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Vanacker A, Vandewiele I, Aerts P, De Groof J, Maes B. Reflex sympathetic dystrophy in a renal transplant patient treated with sirolimus. Nephrol Dial Transplant. 2007;22:2709–12. doi: 10.1093/ndt/gfm358. [DOI] [PubMed] [Google Scholar]
- [46].Wilkinson JE, Burmeister L, Brooks SV, Chan CC, Friedline S, Harrison DE, Hejtmancik JF, Nadon N, Strong R, Wood LK, Woodward MA, Miller RA. Rapamycin slows aging in mice. Aging Cell. 2012;11:675–82. doi: 10.1111/j.1474-9726.2012.00832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Witzig TE, Geyer SM, Ghobrial I, Inwards DJ, Fonseca R, Kurtin P, Ansell SM, Luyun R, Flynn PJ, Morton RF, Dakhil SR, Gross H, Kaufmann SH. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol. 2005;23:5347–56. doi: 10.1200/JCO.2005.13.466. [DOI] [PubMed] [Google Scholar]
- [48].Woodall AJ, Richards MA, Turner DJ, Fitzgerald EM. Growth factors differentially regulate neuronal Cav channels via ERK-dependent signalling. Cell Calcium. 2008;43:562–75. doi: 10.1016/j.ceca.2007.10.001. [DOI] [PubMed] [Google Scholar]
- [49].Xu Q, Fitzsimmons B, Steinauer J, O’Neill A, Newton AC, Hua XY, Yaksh TL. Spinal phosphinositide 3-kinase-Akt-mammalian target of rapamycin signaling cascades in inflammation-induced hyperalgesia. J Neurosci. 2011;31:2113–24. doi: 10.1523/JNEUROSCI.2139-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yan J, Melemedjian OK, Price TJ, Dussor G. Sensitization of dural afferents underlies migraine-related behavior following meningeal application of interleukin-6 (IL-6) Mol Pain. 2012;8:6. doi: 10.1186/1744-8069-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Yuan LL, Adams JP, Swank M, Sweatt JD, Johnston D. Protein kinase modulation of dendritic K+ channels in hippocampus involves a mitogen-activated protein kinase pathway. J Neurosci. 2002;22:4860–8. doi: 10.1523/JNEUROSCI.22-12-04860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zakikhani M, Blouin MJ, Piura E, Pollak MN. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast Cancer Res Treat. 2010;123:271–9. doi: 10.1007/s10549-010-0763-9. [DOI] [PubMed] [Google Scholar]
- [53].Zoncu R, Efeyan A, Sabatini DM. MTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]