

Abstract
5′-Methylthioadenosine/S-adenosylhomocysteine nucleosidase (MTAN) is a dual substrate bacterial enzyme involved in S-adenosylmethionine (SAM)-related quorum sensing pathways that regulates virulence in many bacterial species. MTANs from many bacteria are directly involved in the quorum sensing mechanism by regulating the synthesis of autoinducer molecules that are used by bacterial communities to communicate. In humans, 5′-methylthioadenosine phosphorylase (MTAP) is involved in polyamine biosynthesis as well as in purine and SAM salvage pathways and thus has been identified as an anticancer target. Previously we have described the synthesis and biological activity of several aza-C-nucleoside mimics with a sulfur atom at the 5′ position that are potent E. coli MTAN and human MTAP inhibitors. Because of the possibility that the sulfur may affect bioavailability we were interested in synthesizing “sulfur-free” analogues. Herein we describe the preparation of a series of “sulfur-free” transition state analogues inhibitors, of E. coli MTAN and human MTAP that have low nano- to pico-molar dissociation constants and are potentially novel bacterial anti-infective and anti-cancer drug candidates.
Introduction
The determination of enzymatic transition states is a powerful technique used to provide information on both the geometric and electronic features of the transition state and thus provides a blueprint for putative enzyme inhibitors incorporating transition state features.1 This process has been applied to a number of N-ribosyl transferases; including purine nucleoside phosphorylase (PNP*),2 5′-methylthioadenosine phosphorylase (MTAP)3 and 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase (MTAN),4,5,6 and has led to the development of extremely potent inhibitors that incorporate transition state features.1,7,8,9
MTAN is a dual substrate enzyme that is found in bacteria but not mammals and is involved in polyamine and S-adenosylmethionine (SAM) quorum sensing pathways. Many bacteria utilize quorum sensing signaling molecules known as autoinducers (AIs) to communicate with each other. The production and detection of AIs co-ordinates some gene expression and regulates processes beneficial to microbial communities. Quorum sensing is a potentially useful target for bacterial anti-infective agents as it has been shown that several mutant bacterial strains defective in quorum sensing are less virulent,10,11 and as inhibition of quorum sensing is non-lethal to bacteria the potential for the development of resistance is reduced when compared to more traditional anti-infective agents.
MTAN is a major route for the metabolism of methylthioadenosine (MTA) and S-adenosylhomocysteine (SAH) in bacteria. MTA is a by-product of the formation of the AI-1 family of autoinducers (acylhomoserine lactones, for example hydroxybutanoyl-L-homoserine lactone) that are believed to be responsible for intraspecies communication.12 MTAN subsequently catalyses the hydrolysis of MTA to adenine and 5-methylthio-D-ribose, which in turn is recycled into SAM. The MTAN-catalyzed hydrolysis of SAH leads to S-ribosylhomocysteine (SRH) which is a biochemical precursor to AI-2, a family of AIs important in interspecies bacterial communications.13 Thus inhibition of MTAN is expected to cause the accumulation of MTA, resulting in product inhibition of AI-1 production, and directly block the formation of SRH, the precursor of AI-2.
Human MTAP effects phosphorolysis of MTA and is involved in polyamine biosynthesis and purine salvage pathways. MTA is a by-product formed during the biosynthesis of the polyamines putrescine, spermidine and spermine, which have an important role in growth-related processes.14 MTAP is the only known enzyme involved in the recycling of MTA in humans and it has been proposed that inhibition of MTAP could increase MTA concentration to cause feedback inhibition of polyamine biosynthesis resulting in potential antiproliferative activity, which has made MTAP a target for the design of novel anticancer drugs.15,16 We have previously reported a variety of transition state inhibitors of MTAP, one of which is currently in preclinical trials for the treatment of head and neck tumours.8,17
The transition states for the hydrolysis of MTA by Escherichia coli MTAN and phosphorolysis of MTA by human MTAP have been solved using a combination of kinetic isotope effects and computational modeling. In the case of E. coli it has been shown to have a late dissociative SN1 transition state with extensive cleavage of the bond between adenine and the ribosyl moiety,4 and for human MTAP it has been shown to be late dissociative SN1 with a zero N-glycosidic bond order.3 In MTAP the nucleophile responsible for the cleavage of the N-glycosidic bond is phosphate while for E. coli MTAN the nucleophile is water (Figure 1).
Figure 1.

The reaction catalyzed by MTAP and MTANs with MTA as the substrate. In the case of MTAP the attacking nucleophile is phosphate, whereas in MTANs it is water.
Previously we have reported the synthesis of potent transition state analogue inhibitors, based on a chiral 4-substitued-3-hydroxypyrrolidine motif, of which several exhibit binding affinities in the femtomolar range against E. coli MTAN and picomolar against MTAP.18,19 Recently it has been shown that three of these compounds, MT-, EtT- and BuT-DADMe-Immucillin (3R,4S)-1, (3R,4S)-2 and (3R,4S)-3, respectively (Figure 2), disrupt AI production in Vibrio cholerae and E. coli and cause reduction in biofilm formation.20
Figure 2.

Known enantiopure transition state analogue inhibitors of MTANs (3R,4S)-1,(3R,4S)-2 and (3R,4S)-3 and target racemic “sulfur-free” inhibitors (±)-4.
In vivo screening of some of these chiral femtomolar inhibitors indicated that the sulfur atom may adversely affect bioavailability.21 Herein we report the synthesis of a new family of transition state analogue inhibitors of MTAN and MTAP in which the sulfur atom has been removed. These racemic “sulfur-free” DADMe-Immucillins (±)-4 (Figure 2) have also been found to be potent inhibitors of the E. coli MTAN and human MTAP with dissociation constants in the low nano- to pico-molar range and show potential as new anti-infective and anti-cancer drug candidates. These simplified targets are easier and cheaper to prepare than the previously reported chiral compounds and the removal of the sulfur atom may offer improved pharmacological properties.
Results and Discussion
Synthesis
Compounds (±)-4 should be readily available by a Mannich reaction between a suitably substituted hydroxypyrrolidine, 9-DAA and formaldehyde as described for the corresponding chiral immucillins.22 Hydroxypyrrolidines of the type required for the racemic “sulfur-free” DADMe-Immucillins (±)-4 have been previously reported to be formed via the nucleophile opening of epoxypyrrolidines.23,24 We decided to use a modification of the procedure described by Hansen and Bols,23 as this would allow us to rapidly generate a series of hydroxypyrrolidines, via the addition of Grignard reagents to epoxides meso-10 and meso-11. Diallyl amine was protected as either its tert-butylcarbamate 6 or benzyloxycarbamate 7 and subjected to a ring closing metathesis reaction using Grubb’s 1st generation catalyst.25,26 Pyrrolines 8 and 9 were converted into the corresponding epoxides meso-10 and meso-11 via a two step bromohydrin/epoxidation sequence, in good yields (Scheme 1).
Scheme 1a.

aReagents: (a) Boc2O, MeOH, 0 °C; (b) Benzyl chloroformate, Et3N, CH2Cl2, 0 °C → room temperature; (c) (PCy3)2Cl2Ru=CHPh, CH2Cl2, room temperature; (d) (i) NBS, DMSO, H2O, room temperature, (ii) NaOH, MeOH, 0 °C → room temperature.
With the key epoxypyrrolidines in hand, ring-opening of the epoxide ring with various alkyl, cycloalkyl and aryl Grignard reagents was investigated. Pyrrolidine sub-units (±)-20 - (±)-22, (±)-24, (±)-26 were prepared by the copper(I) catalyzed addition of the corresponding Grignard reagent into epoxide meso-11, followed by hydrogenolysis of the N-protecting group. The deprotected amines were treated with formaldehyde and 9-DAA to afford the corresponding Mannich products (±)-28 - (±)-30, (±)-32 and (±)-34. Compounds (±)-15, (±)-17 and (±)-19 could not be isolated cleanly from the ring-opening reactions and were therefore used as crude compounds and purified after subsequent steps to generate immucillins (±)-31, (±)-33 and (±)-35 (Scheme 2).
Scheme 2a.

aReagents: (a) RMgX, CuBr·DMS, THF, −30 °C; (b) Pd (10 wt% on carbon), H2, MeOH, room temperature; (c) Formaldehyde, 9-DAA, 1,4-dioxane, H2O, room temperature.
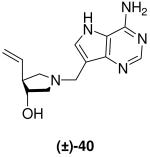
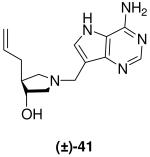
The vinyl and allyl-substituted DADMe-Immucillins (±)-40 and (±)-41 were prepared in an analogous manner, by addition of vinylmagnesium bromide or allylmagnesium chloride, in the presence of copper(I) bromide dimethylsulfide complex to N-Boc protected epoxide meso-10 to give pyrrolidines (±)-36 and (±)-37. Subsequent deprotection with 36% aq. HCl in methanol, afforded the corresponding pyrrolidine sub-units (±)-38 and (±)-39, which were coupled to 9-DAA to generate (±)-40 and (±)-41 respectively (Scheme 3).
Scheme 3a.

aReagents: (a) RMgX, CuBr·DMS, THF, −30 °C; (b) 36% aq. HCl, MeOH, room temperature; (c) Formaldehyde, 9-DAA, 1,4-dioxane, H2O, room temperature.
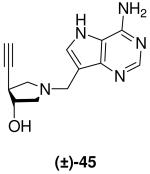
Ethynyl DADMe-Immucillin (±)-45 was synthesized in 4 steps from epoxide meso-10. Addition of trimethylsilylacetylene to meso-10 in the presence of n-butyllithium and boron trifluoride etherate,27 gave (±)-42. Treatment with methanolic HCl gave (±)-43, which underwent a Mannich with 9-DAA to give crude (±)-44 which was directly deprotected under Zemplén conditions to give the desired DADMe-Immucillin (±)-45 after purification (Scheme 4).
Scheme 4a.

aReagents: (a) Trimethylsilylacetylene, n-BuLi, BF3·OEt2, THF, −78 °C → room temperature; (b) 36% aq. HCl, MeOH, room temperature; (c) Formaldehyde, 9-DAA, 1,4-dioxane, H2O, room temperature; (d) NaOMe, MeOH, room temperature.
Having prepared a series of alkyl, cycloalkyl and aromatic substituted immucillins we investigated what other biologically relevant substituents would be tolerated by the target enzymes, for example 1,4-disubstituted 1-H-1,2,3-triazoles.28
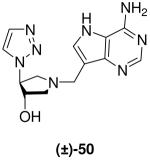
Tsuzuki and co-workers reported the ring-opening of epoxide meso-10 with sodium azide.29 We repeated this and subjected the resulting azide (±)-46 and trimethylsilylacetylene to the Sharpless copper(I) catalyzed azide-alkyne cycloaddition (CuAAC) conditions (CuSO4, sodium ascorbate, t-BuOH, H2O, room temperature),30,31 but this was unsuccessful. However, the desired transformation could be achieved by simply refluxing the reagents in toluene. The crude mixture was treated with tetrabutylammonium fluoride to give TMS-triazole (±)-47 and the desired desilylated triazole (±)-48 in good yield. Removal of the nitrogen protecting group was achieved under our standard conditions (HCl/MeOH) and then pyrrolidine (±)-49 was subjected to the Mannich reaction to afford DADMe-Immucillin (±)-50 (Scheme 5).
Scheme 5a.

aReagents: (a) NaN3, 1,4-dioxane, H2O, 100 °C; (b) (i) Trimethylsilylacetylene, toluene, 110 °C, (ii) TBAF (1M in THF), THF, room temperature; (c) 36% aq. HCl, MeOH; (d) Formaldehyde, 9-DAA, 1,4-dioxane, H2O, room temperature.
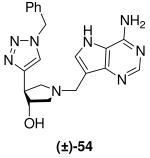
With ethynyl pyrrolidine (±)-42 in hand we attempted the formation of the NH-1,2,3-triazol-4-ylpyrrolidine subunit; an isomer of triazole sub-unit (±)-48. Selective removal of the TMS group in (±)-42 with tetrabutylammonium fluoride gave (±)-51. Attempts to form the NH-1,2,3-triazol-4-yl compound from the resulting terminal alkyne present in (±)-51 using recently reported methodology with either sodium azide32 or ‘protected’ azides33 were unsuccessful. However, the N-substituted triazol-4-ylpyrrolidine (±)-52 was prepared by reaction of (±)-51 with benzyl azide under Sharpless CuAAC conditions.31 Removal of the Boc protecting group gave (±)-53 which was coupled to 9-DAA using the standard Mannich conditions to afford (±)-54 (Scheme 6).
Scheme 6a.

aReagents: (a) TBAF (1M in THF), THF, room temperature; (b) Benzyl azide, sodium ascorbate, CuSO4, t-BuOH, H2O, room temperature; (c) 36% aq. HCl, MeOH, room temperature; (d) Formaldehyde, 9-DAA, 1,4-dioxane, H2O, room temperature.
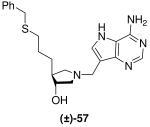
As we had easy access to allyl pyrrolidine (±)-37 we investigated the thiol-ene “click reaction”.34 Although counter to our major objective of eliminating sulfur from our putative inhibitors this reaction would allow us to probe the effect of increasing the length of the linker between the sulfur moiety and the pyrrolidine ring of the DADMe-Immucillins and also demonstrate another synthetic strategy to capable of generating large libraries of compounds via “click chemistry”. Thus allyl pyrrolidine (±)-37 was treated with benzyl mercaptan and 1,1′-azobis(cyanocyclohexane) in 1,4-dioxane at 90 °C,35 to give (±)-55. The crude product was treated with 36% aq. HCl in methanol to afford (±)-56, which underwent a subsequent Mannich reaction to give the desired DADMe-Immucillin (±)-57 (Scheme 7).
Scheme 7a.

aReagents: (a) 1,1′-Azobis(cyanocyclohexane), benzyl mercaptan, 1,4-dioxane, 90 °C; (b) 36% aq. HCl, MeOH, room temperature; (c) Formaldehyde, 9-DAA, 1,4-dioxane, H2O, room temperature.
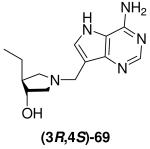
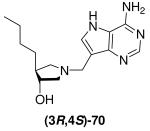
The use of copper salts to assist in the ring-opening of epoxides is known in some cases to give a mixture of the cis and the expected trans isomers,23,36 thus it was necessary to confirm the relative stereochemistry of our pyrrolidines. X-ray crystallography was not applicable as none of the relevant intermediates or final DADMe-Immucillin compounds were crystalline, therefore, known chiral amine (3R,4R)-58,37 was converted into Et-DADMe-Immucillin (3R,4S)-69 via a modification of a previously reported route.19 This provided material with known relative stereochemistry between the 3- and 4-position for direct comparison with the corresponding racemic compound (±)-28. Selective protection of the secondary hydroxyl group of amine (3R,4R)-58 was achieved in three steps. Oxidation of the primary hydroxyl with Dess-Martin periodinane reagent gave aldehyde (3R,4S)-62 which was methylenated under standard Wittig conditions. Hydrogenation of the olefinic group, followed by concomitant removal of both protecting groups gave enantiopure amine (3R,4S)-67. A Mannich reaction with 9-DAA gave the enantiopure DADMe-Immucillin (3R,4S)-69. As Bu-DADMe-Immucillin (±)-29 was the most potent racemic inhibitor of both E. coli MTAN and human MTAP the same sequence was used to prepare enantiopure Bu-DADMe-Immucillin (3R,4S)-70, by substituting n-propyltriphenyl phosphonium bromide as the Wittig reagent (Scheme 8).
Scheme 8a.

aReagents: (a) (Bu)2SnO, toluene, 110 °C then BzCl, 5 °C → room temperature; (b) TBDMSCl, imidazole, DMF, room temperature; (c) NaOMe, MeOH, room temperature; (d) Dess-Martin periodinane, CH2Cl2, room temperature; (e) Ph3PCH3Br or Ph3PCH2CH2CH3Br, n-BuLi, THF, −78 °C → room temperature; (f) Pd (10 wt% on carbon) or Pd(OH)2 (20 wt% on carbon), H2 (1 atm), EtOH, room temperature; (g) TFA or 36% aq. HCl in MeOH, room temperature; (h) Formaldehyde, 9-DAA, 1,4-dioxane, H2O, room temperature (for (3R,4S)-69) or 85 °C (for (3R,4S)-70).
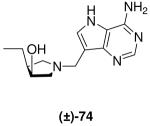
The 1H and 13C NMR data for enantiopure amine (3R,4S)-67 and DADMe-Immucillin (3R,4S)-69 were identical to those of their racemic equivalents, compounds (±)-20 and (±)-28 respectively. To further confirm the relative stereochemical assignment of the C-3 and C-4 substituents in (±)-20 and (±)-28, the hydroxyl group in pyrrolidine (±)-12 was subjected to a Mitsunobu inversion protocol,38 by treatment with benzoic acid in the presence of DIAD and triphenylphosphine to give inverted ester (±)-71 in good yield. Saponification of the benzoate followed by hydrogenation of the Cbz group gave pyrrolidine (±)-73 which was converted into racemic cis-Et-DADMe-Immucillin (±)-74 (Scheme 9). Comparison of the NMR data of pyrrolidine cis-(±)-73 with trans-(±)-20 and (3R,4S)-67 as well as DADMe-Immucillin cis-(±)-74 with trans-(±)-28 and (3R,4S)-69 showed clear differences. In particular the 13C NMR resonance for C-3 in trans-Et-DADMe-Immucillin (±)-28 (77.7 ppm) was further downfield than that for the cis isomer (±)-74 (72.5 ppm). All the DADMe-Immucillins generated via the copper(I)-catalyzed Grignard ring-opening of epoxides meso-10 or meso-11 have the C-3 resonance between 76.7 - 78.1 ppm, indicating a trans arrangement of substituents at C-3 and C-4.
Scheme 9a.

aReagents: (a) Benzoic acid, DIAD, PPh3, THF, 0 °C → room temperature; (b) K2CO3, EtOH, H2O, 100 °C; (c) Pd (10 wt% on carbon), H2, MeOH, room temperature; (d) Formaldehyde, 9-DAA, 1,4-dioxane, H2O, room temperature.
Inhibition Studies
The inhibition of Escherichia coli MTAN and human MTAP was evaluated with racemic immucillins (±)-28 - (±)-35, (±)-40, (±)-41, (±)-45, (±)-50, (±)-54, (±)-57 and (±)-74 and enantiopure immucillins (3R,4S)-69 and (3R,4S)-70 (Table 1).
Table 1.
Inhibition constants for the interaction of immucillins with E. coli MTAN and human MTAPa and the ratio of the two values in order to provide a qualitative indication of the therapeutic window for the treatment of an E. coli infection.
| Compound |
Ki for E. coli MTAN (nM) |
Ki for Human MTAP (nM) |
Ki Human MTAP / Ki E. coli MTAN |
|---|---|---|---|
|
0.84 ± 0.06 | *3.2 ± 0.2 | 4 |
|
*0.009 ± 0.001 | 0.8 ± 0.1 | 89 |
|
*0.047 ± 0.009 | *1.7 ± 0.3 | 36 |
|
0.7 ±0.1 | 18 ± 2 | 26 |
|
0.063 ± 0.005 | *2.1 ± 0.3 | 33 |
|
*0.013 ± 0.001 | *2.6 ± 0.4 | 200 |
|
0.059 ± 0.008 | 14 ± 2 | 237 |
|
*0.03 ± 0.002 | 8 ± 1 | 267 |
|
0.65 ± 0.03 | 3.2 ± 0.4 | 4.9 |
|
0.35 ± 0.03 | 3.0 ± 0.3 | 8.6 |
|
0.39 ± 0.02 | 31 ± 3 | 80 |
|
2.0 ± 0.2 | 59 ± 8 | 30 |
|
*0.064 + 0.005 | -b | selective |
|
*0.054 ± 0.005 | 71 ± 5 | 1314 |
|
0.31 ± 0.02 | *0.7 ± 0.2 | 2 |
|
0.0034 ± 0.0009 | *0.55 ± 0.07 | 162 |
|
1.8 ± 0.3 | 34 ± 3 | 19 |
Ki is the dissociation constant for the first step in E + I ↔ EI ↔ EI*, the two-step binding characteristic of slow-onset tight-binding inhibition.
indicates slow onset binding and is theoverall dissociation constant for E + I ↔ EI*. In cases where no * is shown then slow-onset inhibition was not observed.
indicates that no inhibition was observed at 2.5 μM.
E. coli MTAN showed broad tolerance for a variety of substituents in the C-4 position of the pyrrolidine ring and bound most of the compounds tightly with dissociation constants in the picomolar range. The diversity of functional groups accommodated at this position reaffirms the structural flexibility of the E. coli MTAN 5′ binding pocket.9 The Bu-DADMe compound (±)-29 gave the tightest binding (Ki* value of 9 pM) and was equivalent to its sulfur isostere (Compound 2, Ki* = 8 pM).18 Thus, the presence of sulfur is not necessary for activity against E. coli MTAN. Shorter substituents (e.g., alkyl (±)-28, olefinic (±)-40 and (±)-41, and alkynyl (±)-45), as well as the highly branched alkyl group (±)-31 bound less well. The N-linked triazole group (±)-50 (Ki = 2 nM) did not bind as well as the C-linked benzyl group (±)-54 (Ki = 64 pM).
For human MTAP, Bu-DADMe-Immucillin (±)-29 was the best inhibitor in this family of compounds (Ki = 0.8 nM) with a comparable affinity to its sulfur-containing counterpart (Ki* = 1.4 nM), also suggesting that sulfur was not a necessary component of human MTAP inhibitors. Although MTAN prefers bulky substituents, human MTAP preferred the short alkyl and olefinic groups (±)-28, (±)-40 and (±)-41. Comparisons of inhibitors for MTAN and MTAP are best made from the Km/Ki ratio to provide comparative substrate and inhibitor interaction energies. For racemic Et-DADMe-Immucillin (±)-28, the Ki values were 0.84 nM and 1.7 nM for E. coli MTAN and human MTAP, respectively. The respective Km/Ki ratios are 512 and 3105 for E. coli MTAN and human MTAP. Thus, the inhibitor is a better competitor for the catalytic site of human MTAP than for E. coli MTAN by a factor of 6. The same is true for inhibitors with short olefinic groups (±)-40 and (±)-41. For (±)-40, the Km/Ki ratios are 662 and 1649, while they are 1228 and 1759 for (±)-41, favored more by human MTAP than by E. coli MTAN. For the rest of the inhibitors, the Km/Ki ratio suggests that E. coli MTAN has a higher affinity for these compounds relative to substrate than does human MTAP.
Single enantiomers are usually more potent than racemic mixtures,39 as is the case for the inhibition constants for compound (3R,4S)-69 and (±)-28 and (3R,4S)-70 and (±)-29 with E. coli MTAN and human MTAP. Likewise, the trans-Et-DADMe-Immucillin (±)-28 is a better inhibitor of both E. coli MTAN and human MTAP than cis-Et-DADMe-Immucillin (±)-74 which confirms the trans stereochemistry is preferred as a transition state analogue.
DADMe-Immucillin (±)-57 is a relatively potent inhibitor of bacterial MTAN, however increasing the linker between the benzyl thiol moiety and the pyrrolidine ring from one carbon (Ki* = 0.5 pM)18 to three carbons as in (±)-57 (Ki* = 54 pM) is detrimental to activity against E. coli MTAN. Although a similar reduction in activity against human MTAP was also observed, the magnitude of the Km/Ki ratio of (±)-57 (7963 for MTAN vs. 74 for MTAP) reveals that this is one of the more selective inhibitors for the bacterial enzyme.
In terms of a therapeutic window a highly selective bacterial MTAN inhibitor is the benzyl-triazole compound (±)-54 which inhibited E. coli MTAN with a Ki* of 64 pM without inhibiting human MTAP even at 2.5 μM concentration. Interestingly, the N-linked triazole compound (±)-50 was a weaker inhibitor than (±)-54 for the bacterial enzyme but a better inhibitor for human MTAP. Again, this suggests that the E. coli enzyme 5′-binding pocket is able to accommodate bulkier aromatic groups much better than its human ortholog or than other MTAN isozymes.9 Exploiting these enzymatic subtleties may prove useful in designing clinically selective antibacterials.
Conclusions
A series of “sulfur-free” transition state analogues of E. coli MTAN and human MTAP have been designed and synthesized based on their dissociative transition state structures. These racemic compounds can be rapidly prepared in a simple manner. We have demonstrated that the removal of the sulfur moiety from the transition state structures is not detrimental to their activity against these enzymes. Further investigation in to the bioavailability and other pharmacological properties of these potent “sulfur-free” inhibitors will be conducted and reported in due course.
Experimental Section
General
NMR spectra were recorded on a Bruker Avance III spectrometer at 500 MHz (1H) or 125 MHz (13C). Spectra were measured in either CDCl3 or CD3OD using Me4Si as internal reference. High resolution accurate mass determinations were performed on a Waters Q-Tof Premier Mass Spectrometer under electrospray ionization conditions. Analytical HPLC was performed on an Agilent 1100 instrument, eluting with 0.1 %w/w TFA in H2O : MeOH gradient through a Phenomenex® Synergi 4 polar-RP 80A, 250 × 4.6 mm column. Preparative HPLC was performed using a Gilson 321 Pump eluting 0.1 %w/w TFA in H2O : MeOH gradient through a Phenomenex® Synergi 4 polar-RP 80A, 250 × 30 mm column and Agilent 1100 photo diode array detector. Thin layer chromatography was performed on glass-backed silica gel plates (Merck). Column chromatography was performed on silica gel (Davisil LC60A 40-63 micron). Anhydrous solvents were obtained from Aldrich. Methyltriphenylphosphonium bromide was azeotropically dried with toluene and stored in vacuo over P2O5 prior to use. All other reagents were used as received. All final compounds had a purity of >95% as assessed by analytical HPLC.
Chemistry
tert-Butyl diallylcarbamate (6)25
Di-tert-butyl dicarbonate (42.2g, 193 mmol) was added, portion-wise, to a solution of diallylamine (20 mL, 162 mmol) in methanol (500 mL) at 0 °C. After complete addition the reaction mixture was allowed to warm to room temperature, stirred for 1 h then concentrated under reduced pressure. Dry flash chromatography of the residue (gradient 0 to 50% EtOAc in Petrol) afforded 6 as a colorless oil (31.9 g, 99%). 1H NMR (500 MHz, CDCl3): δ = 5.80 - 5.72 (m, 2H), 5.13 - 5.09 (m, 4H), 3.80 (br. s, 4H) and 1.45 ppm (s, 9H).
tert-Butyl 3-pyrroline-1-carboxylate (8)23
Grubb’s 1st generation catalyst (920 mg, 1.1 mmol) was added to a solution of tert-butyl diallylcarbamate (6) (31.9 g, 162 mmol) in CH2Cl2 (370 mL). The reaction mixture was stirred for 17 h and then concentrated under reduced pressure. Dry flash chromatography of the residue (gradient: 0 to 70% EtOAc in Petrol) afforded 8 as a pale yellow oil (19.9 g, 73%). 1H NMR (500 MHz, CDCl3): δ = 5.80 - 5.74 (m, 2 H), 4.14 - 4.08 (m, 4H) and 1.48 ppm (s, 9H). 13C NMR (125 MHz, CDCl3): δ = 154.3, 125.9, 125.8, 79.3, 53.1, 52.8 (rotamers) and 28.5 ppm. ESI-HRMS for C9H15NONa [MNa]+ calcd, 192.1000; found, 192.0997.
tert-Butyl 3,4-epoxypyrrolidine-1-carboxylate (meso-10)23
N-Bromosuccinimide (8.05 g, 45 mmol) was added portion-wise, over 10 min, to a solution of olefin (8) (6.4 g, 38 mmol) in DMSO (40 mL) and water (2 mL) at 0 °C. The reaction mixture was warmed to room temperature, stirred for 2.5 h and then further N-bromosuccinimide (2 g, 11 mmol) was added. After stirring for a further 2 h the reaction was quenched by the addition of water (50 mL) and then extracted into EtOAc (4 × 50 mL). The combined organic phase was washed with brine (2 × 50 mL), dried (MgSO4), filtered and concentrated under reduced pressure. The residue was dissolved in MeOH (100 mL), cooled to 0 °C and then an aqueous solution of NaOH (23 mL, 46 mmol, 2 M) was added in one portion. The reaction was warmed to room temperature, stirred for 17.5 h and then the MeOH was removed under reduced pressure. The residue was partitioned between water (100 mL) and EtOAc (100 mL) extracted with EtOAc (3 × 100 mL). The combined organic phase was washed with brine (2 × 100 mL), dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography of the residue (1 : 9 then 2 : 8, EtOAc : Petrol) afforded meso-10 as a pale yellow oil (4.54 g, 64% over the two steps). 1H NMR (500 MHz, CDCl3): δ = 3.81 (d, J = 12.9 Hz, 1H), 3.74 (d, J = 12.8 Hz, 1H), 3.66 (d, J = 5.3 Hz, 2H), 3.31 (dd, J = 12.6, 6.2 Hz, 2H) and 1.44 ppm (s, 9H). 13C NMR (125 MHz, CDCl3): δ = 154.8, 79.8, 55.6, 55.1 (rotamers), 47.3, 46.9 (rotamers) and 28.4 ppm. ESI-HRMS for C9H15NO3Na [MNa]+ calcd, 208.0950; found, 208.0955.
Benzyl diallylcarbamate (7)26
A solution of diallylamine (15 mL, 120 mmol) in CH2Cl2 (150 mL) was cooled to 0 °C and Et3N (23 mL, 160 mmol) then benzyl chloroformate (20 mL, 140 mmol) were added drop-wise. The reaction mixture was slowly allowed to warm to room temperature and stirred for 16 h and then quenched with water (200 mL). The phases were separated and the aqueous phase was extracted with CH2Cl2 (3 × 200 mL). The combined organic phase was washed with brine (200 mL), dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography (gradient: 2 to 10% EtOAc in Petrol) of the residue afforded 7 as a pale yellow oil (25.9 g, 92%). 1H NMR (500 MHz, CDCl3): δ = 7.35 - 7.29 (m, 5H), 5.77 (br. s, 2H), 5.17 - 5.13 (br. m, 6H) and 3.88 ppm (br. s, 4H).
Benzyl 3-pyrroline-1-carboxylate (9)26
Grubb’s 1st generation catalyst (173 mg, 0.21 mmol) was added to a solution of benzyl diallylcarbamate (7) (6.78 g, 29 mmol) in CH2Cl2 (300 mL). The reaction mixture was stirred for 16 h and then further catalyst was added (340 mg, 0.4 mmol). The reaction mixture was stirred for a further 16 h and then concentrated under reduced pressure. Flash chromatography (1 : 9 then 2 : 8, EtOAc : Petrol) afforded 9 as a yellow oil (5.74 g, 94%). 1H NMR (500 MHz, CDCl3): δ = 7.39 - 7.29 (m, 5 H), 5.82 - 5.80 (m, 1H), 5.77 - 5.75 (m, 1H), 5.17 (s, 2H) and 4.22 - 4.18 ppm (m, 4H).
Benzyl 3,4-epoxypyrrolidine-1-carboxylate (meso-11)26
N-Bromosuccinimide (5.47 g, 31 mmol) was added to a solution of olefin (9) (5.02 g, 25 mmol) in DMSO (65 mL) and water (3.4 mL) at 0 °C. The reaction mixture was warmed to room temperature, stirred for 1.5 h then further N-bromosuccinimide (1.1 g, 6.2 mmol) was added. After stirring for a further 3.5 h the reaction was quenched by the addition of water (150 mL) and then extracted into EtOAc (3 × 150 mL). The combined organic phase was washed with brine (3 × 100 mL), dried (MgSO4), filtered and concentrated under reduced pressure. The residue was dissolved in MeOH (80 mL), cooled to 0 °C and then an aqueous solution of NaOH (37 mL, 37 mmol, 1 M) was added in one portion. The reaction was warmed to room temperature, stirred for 5 h then the MeOH was removed under reduced pressure. The residue was diluted with water (100 mL) and extracted with EtOAc (3 × 200 mL). The combined organic phases were washed with brine (200 mL), dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography of the residue (3 : 7 then 4 : 6, EtOAc : Petrol) afforded meso-11 as a pale yellow oil (3.69 g, 68% over the 2 steps). 1H NMR (500 MHz, CDCl3): δ = 7.35 - 7.30 (m, 5H), 5.11 (dd, J = 17.9, 12.4 Hz, 2H), 3.87 (dd, J = 25.0, 12.9 Hz, 2H), 3.68 (d, J = 5.1 Hz, 2H) and 3.39 ppm (ddd, J = 12.8, 7.6, 0.8 Hz, 2H). 13C NMR (125 MHz, CDCl3): δ = 155.3, 136.6, 128.5, 128.0, 127.9, 67.0, 55.5, 54.9 (rotamers), 47.5 and 47.2 (rotamers) ppm. ESI-HRMS for C12H13NO3Na [MNa]+ calcd, 242.0793; found, 242.0791.
(±)-Benzyl trans-4-ethyl-3-hydroxypyrrolidine-1-carboxylate [(±)-12]
A solution of epoxide (meso-11) (467 mg, 2.13 mmol) and CuBr·DMS (53 mg, 0.26 mmol) in THF (20 mL) was cooled to −30 °C. Ethylmagnesium bromide (10 mL, 10 mmol, 1 M solution in THF) was added drop-wise over 20 min, keeping the internal temperature below −30 °C. After complete addition the reaction was allowed to warm to −15 °C over 50 min and then quenched with 10% aqueous NH4Cl solution (20 mL) and EtOAc (40 mL). The mixture was stirred at room temperature for 45 min and then the layers were separated. The aqueous phase was extracted with EtOAc (3 × 50 mL). The combined organic phases were dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography of the residue (3 : 7 then 4 : 6, EtOAc : Petrol) afforded (±)-12 as a pale yellow oil (370 mg, 70%). 1H NMR (500 MHz, CDCl3): δ = 7.35 - 7.28 (m, 5H), 5.11 (s, 2H), 3.99 (br. s, 1H,), 3.69 - 3.60 (m, 2H), 3.29 (td, J = 11.3, 4.1 Hz, 1H), 3.16 - 3.08 (m, 2H), 1.99 - 1.92 (m, 1H), 1.53 - 1.47 (m, 1H), 1.24 - 1.17 (m, 1H) and 0.93 ppm (dd, J = 12.4, 7.3 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ = 155.3, 136.9, 128.5, 128.0, 127.9, 67.0, 66.9 (rotamers), 53.1, 52.7 (rotamers), 49.4, 49.2 (rotamers), 48.0, 47.4 (rotamers), 24.4 and 12.3 ppm. ESI-HRMS for C14H19NO3Na [MNa]+ calcd, 272.1263; found, 272.1269.
(±)-trans-4-Ethyl-3-hydroxypyrrolidine [(±)-20]
Palladium (20 mg, 0.02 mmol, 10 wt% on carbon) was added to a solution of (±)-12 (270 mg, 1.1 mmol) in MeOH (10 mL) under argon. The reaction mixture was placed under a hydrogen atmosphere and stirred for 1 h and then filtered through Celite® and concentrated under reduced pressure. Flash chromatography of the residue (5 : 4 : 1, CH2Cl2 : MeOH : 28% aq. NH4OH) afforded (±)-20 as a yellow oil (70 mg, 56%). 1H NMR (500 MHz, CD3OD): δ = 3.94 - 3.91 (m, 1H), 3.23 - 3.19 (m, 1H), 2.98 - 2.95 (m, 1H), 2.78 - 2.76 (m, 1H), 2.51 - 2.47 (m, 1H), 1.87 - 1.81 (m, 1H), 1.54 - 1.46 (m, 1H), 1.32 - 1.23 (m, 1H) and 0.96 ppm (t, J = 7.4, 3H). 13C NMR (125 MHz, CD3OD): δ = 78.1, 54.7, 51.6, 51.0, 26.3 and 12.9 ppm. ESI-HRMS for C6H14NO [MH]+ calcd, 116.1075; found, 116.1070.
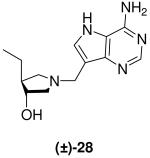
(±)-trans-1-[(9-Deaza-adenin-9-yl)methyl]-4-ethyl-3-hydroxypyrrolidine [(±)-28]
Formaldehyde (120 L, 1.5 mmol, 37 wt% solution in water) followed by 9-deazaadenine (102 mg, 0.9 mmol) were added to a solution of (±)-20 (116 mg, 0.87 mmol) in 1,4-dioxane (1.6 mL) and water (3.2 mL). The reaction mixture was stirred at room temperature for 15 h, absorbed onto silica and eluted down a silica column using a gradient 5 - 30% (7 N NH3 in MeOH) in CH2Cl2. The crude product was collected, concentrated and subjected to flash chromatography (5 : 4.9 : 0.1 then 5 : 4.8 : 0.2, CH2Cl2 : MeOH : 28% aq. NH4OH) to afford (±)-28 as a pale yellow solid (175 mg, 77%). 1H NMR (500 MHz, CD3OD): δ = 8.16 (s, 1H), 7.49 (s, 1H), 3.87 - 3.78 (m, 3H), 3.07 (dd, J = 9.6, 8.1 Hz, 1H), 2.76 (dd, J = 10.4, 6.3 Hz, 1H), 2.70 (dd, J = 10.4, 4.0 Hz, 1H), 2.19 (dd, J = 9.7, 7.9 Hz, 1H), 1.91 - 1.84 (m, 1H), 1.59 - 1.50 (m, 1H), 1.38 - 1.28 (m, 1H) and 0.92 ppm (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CD3OD): δ = 152.1, 151.0, 147.0, 130.1, 115.1, 112.5, 77.4, 62.4, 59.4, 50.4, 49.1, 27.1 and 12.9 ppm; ESI-HRMS for C13H20N5O [MH]+ calcd, 262.1668; found, 262.1664.
(±)-Benzyl trans-4-butyl-3-hydroxypyrrolidine-1-carboxylate [(±)-13]
A solution of epoxide (meso-11) (80 mg, 0.4 mmol) and CuBr·DMS (7 mg, 0.03 mmol) in THF (3 mL) was cooled to −30 °C. n-Butylmagnesium chloride (1 mL, 2 mmol, 2 M solution in THF) was added drop-wise over 10 min, keeping the internal temperature below −25 °C. After complete addition the reaction was allowed to warm to −15 °C over 45 min and then quenched with 10% aqueous NH4Cl solution (10 mL) and EtOAc (20 mL). The mixture was stirred at room temperature for 75 min then the layers were separated. The aqueous phase was extracted with EtOAc (2 × 20 mL) and the combined organic extracts were dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography of the residue (4 : 6 then 1 : 1, EtOAc : Petrol) afforded (±)-13 as a pale yellow oil (72 mg, 71%). 1H NMR (500 MHz, CDCl3): δ = 7.38 - 7.32 (m, 5H), 5.13 (s, 2H) 4.06 - 4.03 (m, 1H), 3.70 - 3.65 (m, 2H), 3.34 - 3.27 (m, 1H), 3.19 - 3.12 (m, 1H), 2.08 - 1.98 (m, 1H), 1.64 (br. s, 1H), 1.51 - 1.44 (m, 1H), 1.36 - 1.15 (m, 5H) and 0.91 - 0.88 ppm (m, 3H). 13C NMR (125 MHz, CDCl3): δ = 155.1, 136.8, 128.5, 127.9, 127.8, 75.4, 74.6 (rotamers), 66.8, 53.0, 52.6 (rotamers), 49.6, 49.4 (rotamers), 46.2, 45.7 (rotamers), 31.2, 29.9, 29.7 (rotamers), 22.7 and 13.9 ppm. ESI-HRMS for C16H23NO3Na [MNa]+ calcd, 300.1576; found, 300.1584.
(±)-trans-4-Butyl-3-hydroxypyrrolidine [(±)-21]
Palladium (10 mg, 0.01 mmol, 10 wt% on carbon) was added to a solution of (±)-13 (70 mg, 0.3 mmol) in MeOH (4 mL) under argon. The reaction mixture was placed under a hydrogen atmosphere and stirred for 1.5 h and then filtered through Celite® and concentrated under reduced pressure. Flash chromatography of the residue (5 : 4.8 : 0.2 then 5 : 4.5 : 0.5, CH2Cl2 : MeOH : 28% aq. NH4OH) afforded (±)-21 as a yellow oil (20 mg, 54%). 1H NMR (500 MHz, CD3OD): δ = 4.19 - 4.17 (m, 1H), 3.55 (dd, J = 11.7, 7.3 Hz, 1H), 3.41 (dd, J = 12.3, 5.1 Hz, 1H), 3.16 (dd, J = 12.3, 3.1 Hz, 1H), 3.02 (dd, J = 11.7, 5.8 Hz, 1H), 2.23 - 2.17 (m, 1H), 1.55 - 1.49 (m, 1H), 1.40 - 1.28 (m, 5H) and 0.95 - 0.92 ppm (m, 3H). 13C NMR (125 MHz, CD3OD): δ = 75.3, 52.4, 50.0, 47.3, 31.9, 31.0, 23.6 and 14.3 ppm. ESI-HRMS for C8H18NO [MH]+ calcd, 144.1388; found, 144.1390.
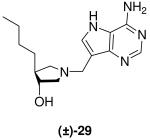
(±)-trans-4-Butyl-1-[(9-deaza-adenin-9-yl)methyl]-3-hydroxypyrrolidine [(±)-29]
Formaldehyde (95 L, 1.2 mmol, 37 wt% solution in water) followed by 9-deazaadenine (100 mg, 0.7 mmol) were added to a solution of (±)-21 (88 mg, 0.66 mmol) in 1,4-dioxane (1.2 mL) and water (2.5 mL). The reaction mixture was stirred at room temperature for 66 h, absorbed onto silica and eluted down a silica column using a gradient 10 - 20% (7 N NH3 in MeOH) in CH2Cl2. The crude product was collected, concentrated and subjected to flash chromatography (5 : 4.8 : 0.2, CH2Cl2 : MeOH : 28% aq. NH4OH) to afford (±)-29 as a pale yellow solid (89 mg, 47%). 1H NMR (500 MHz, CD3OD): δ = 8.16 (s, 1H), 7.5 (s, 1H), 3.86 - 3.79 (m, 3H), 3.07 (dd, J = 9.5, 8.0 Hz, 1H), 2.76 (dd, J = 10.4, 6.3 Hz, 1H), 2.71 (dd, J = 10.4, 4.0 Hz, 1H), 2.19 (dd, J = 9.7, 8.0 Hz, 1H), 1.98 - 1.91 (m, 1H), 1.55 - 1.48 (m, 1H), 1.35 - 1.25 (m, 5H) and 0.89 ppm (t, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CD3OD): δ = 152.1, 151.0, 147.0, 130.1, 115.2, 112.4, 77.7, 62.3, 59.7, 49.0, 48.5, 34.0, 31.5, 23.8 and 14.3 ppm; ESI-HRMS for C15H24N5O [MH]+ calcd, 290.1981; found, 290.1989.
(±)-Benzyl trans-3-hydroxy-4-isobutylpyrrolidine-1-carboxylate [(±)-14]
A solution of epoxide (meso-11) (203 mg, 0.93 mmol) and CuBr·DMS (30 mg, 0.15 mmol) in THF (8 mL) was cooled to −30 °C. iso-Butylmagnesium bromide (2.3 mL, 4.6 mmol, 2 M solution in THF) was added drop-wise over 10 min, keeping the internal temperature below −27 °C. After complete addition the reaction was allowed to warm to −15 °C over 80 min and then quenched with 10% aqueous NH4Cl solution (20 mL) and EtOAc (50 mL). The mixture was stirred at room temperature for 45 min and then the layers were separated. The aqueous phase was extracted with EtOAc (2 × 50 mL) and the combined organic layers were dried (MgSO4), filtered and concentrated under reduced pressure to afford crude (±)-14 (193 mg, 75%). This material was used in the next step without further purification. 1H NMR (300 MHz, CDCl3): δ = 7.38 - 7.29 (m, 5H), 5.13 (s, 2H), 4.04 - 3.98 (m, 1H), 3.74 - 3.64 (m, 2H), 3.34 - 3.25 (m, 1H), 3.17 - 3.09 (m, 1H), 2.17 - 2.06 (m, 1H), 1.70 - 1.58 (m, 2H), 1.36 - 1.25 (m, 1H), 1.20 - 1.08 (m, 1H) and 0.91 ppm (t, J = 6.6 Hz, 6H). 13C NMR (125 MHz, CDCl3): δ = 155.1, 136.9, 128.5, 127.9, 127.9, 75.8, 75.0 (rotamers), 52.9, 52.5 (rotamers), 49.7, 49.5 (rotamers), 44.2, 43.6 (rotamers), 40.7, 26.3, 26.2 (rotamers), 23.2 and 22.1 ppm. ESI-HRMS for C16H23NO3Na [MNa]+ calcd, 300.1576; found, 300.1575.
(±)-trans-3-Hydroxy-4-isobutylpyrrolidine [(±)-22]
Palladium (20 mg, 0.02 mmol, 10 wt% on carbon) was added to a solution of crude (±)-14 (190 mg, 0.7 mmol) in MeOH (10 mL) under argon. The reaction mixture was placed under a hydrogen atmosphere and stirred for 2 h and then filtered through Celite® and concentrated under reduced pressure. Flash chromatography of the residue (gradient 5 : 4.8 : 0.2 to 5 : 4 : 1, CH2Cl2 : MeOH : 28% aq. NH4OH) afforded (±)-22 as a yellow oil (50 mg, 49%). 1H NMR (500 MHz, CD3OD): δ = 4.16 - 4.13 (m, 1H), 3.57 (dd, J = 11.7, 7.3 Hz, 1H), 3.41 (dd, J = 12.3, 5.1 Hz, 1H), 3.16 (dd, J = 12.3, 3.1 Hz, 1H), 3.00 (dd, J = 11.7, 5.8 Hz, 1H), 2.32 - 2.26 (m, 1H), 1.69 - 1.61 (m, 1H), 1.39 - 1.34 (m, 1H), 1.23 - 1.18 (m, 1H) and 0.95 ppm (dd, J = 10.5, 6.6 Hz, 6H). 13C NMR (125 MHz, CD3OD): δ = 75.6, 52.5, 50.2, 45.3, 41.4, 27.4, 23.2 and 22.6 ppm. ESI-HRMS for C8H18NO [MH]+ calcd, 144.1388; found, 144.1382.
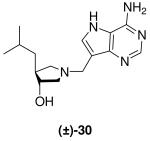
(±)-trans-1-[(9-Deaza-adenin-9-yl)methyl]-3-hydroxy-4-isobutylpyrrolidine [(±)-30]
Formaldehyde (75 L, 0.9 mmol, 37 wt% solution in water) followed by 9-deazaadenine (52 mg, 0.4 mmol) were added to a solution of (±)-22 (49 mg, 0.34 mmol) in 1,4-dioxane (0.6 mL) and water (1.2 mL). The reaction mixture was stirred at room temperature for 17 h, absorbed onto silica and eluted down a silica column with 10% (7 N NH3 in MeOH) in CH2Cl2. The crude product was collected, concentrated and subjected to flash chromatography (5 : 4.9 : 0.1, CH2Cl2 : MeOH : 28% aq. NH4OH) to afford (±)-30 as an off-white solid (42 mg, 42%). 1H NMR (500 MHz, CD3OD): δ = 8.16 (s, 1H), 7.49 (s, 1H), 3.86 - 3.78 (m, 3H), 3.09 - 3.05 (m, 1H), 2.75 (dd, J = 10.4, 6.3 Hz, 1H), 2.70 (dd, J = 10.3, 3.9 Hz, 1H), 2.18 - 2.14 (m, 1H), 2.09 - 2.02 (m, 1H), 1.61 - 1.52 (m, 1H), 1.42 - 1.36 (m, 1H), 1.23 - 1.17 (m, 1H) and 0.88 ppm (t, J = 6.7 Hz, 6H). 13C NMR (125 MHz, CD3OD): δ = 152.1, 150.1, 147.0, 130.1, 115.1, 112.6, 78.1, 62.3, 59.8, 49.1, 46.2, 43.9, 27.7, 23.6 and 22.7 ppm. ESI-HRMS for C15H24N5O [MH]+ calcd, 290.1981; found, 290.1979.
(±)-Benzyl trans-3-hydroxy-4-(penta-3-yl)pyrrolidine-1-carboxylate [(±)-15]
A solution of epoxide (meso-11) (230 mg, 1.05 mmol) and CuBr·DMS (28 mg, 0.22 mmol) in THF (10 mL) was cooled to −30 °C (internal temperature). 3-Pentylmagnesium bromide (2.3 mL, 4.6 mmol, 2 M solution in ether) was added drop-wise over 10 min. After complete addition the reaction was allowed to warm to −20 °C over 30 min and then quenched with 10% aqueous NH4Cl solution (40 mL) and EtOAc (40 mL). The mixture was stirred at room temperature for 1 h then the layers were separated. The aqueous phase was extracted with EtOAc (3 × 40 mL) and the combined organic layers were dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography of the residue (2 : 8, EtOAc : Petrol) afforded (±)-15 and an unknown co-running minor impurity as a pale yellow oil (195 mg). 1H NMR (500 MHz, CDCl3): δ = 7.37 - 7.29 (m, 5H), 5.11 (s, 2H), 4.20 - 4.14 (m, 1H), 3.73 - 3.69, (m, 1H), 3.67 - 3.59 (m, 1H), 3.29 - 3.23 (m, 1H), 3.19 - 3.14 (m, 1H), 2.14 - 2.07 (m, 1H), 1.42 - 1.38 (m, 3H), 1.30 - 1.20 (m, 2H) and 0.90 - 0.85 ppm (m, 6H). 13C NMR (125 MHz, CDCl3): δ = 154.9, 136.9, 128.5, 127.9, 127.8, 73.3, 72.5 (rotamers), 67.4, 66.8 (rotamers), 53.5, 53.1 (rotamers), 48.4, 47.7, 47.4 (rotamers), 41.2, 23.1, 22.4 (rotamers) and 10.9 ppm. ESI-HRMS for C17H25NO3Na [MNa]+ calcd, 314.1732; found, 314.1731.*
(±)-trans-3-Hydroxy-4-(penta-3-yl)pyrrolidine [(±)-23]
Palladium (35 mg, 0.03 mmol, 10 wt% on carbon) was added to a solution of crude (±)-15 (195 mg, 0.9 mmol) in MeOH (10 mL) under argon. The reaction mixture was placed under a hydrogen atmosphere and stirred for 1 h and then filtered through Celite® and concentrated under reduced pressure. Flash chromatography of the residue (5 : 4.9 : 0.1 then 5 : 4.8 : 0.2, CH2Cl2 : MeOH : 28% aq. NH4OH) afforded (±)-23 as a pale yellow gum (27 mg, 16% over 2 steps). 1H NMR (500 MHz, CD3OD): δ = 4.07 (m, 1H), 3.15 (dd, J = 11.5, 8.2 Hz, 1H), 2.89 (dd, J = 12.1, 5.8 Hz, 1H), 2.80 (dd, J = 12.1, 3.3 Hz, 1H), 2.50 (dd, J = 11.5, 8.5 Hz, 1H), 1.94 (q, J = 8.2, 4.4 Hz, 1H), 1.54 - 1.39 (m, 3H), 1.32 - 1.21 (m, 2H) and 0.91 ppm (dt, J = 12.9, 7.4 Hz, 6H). 13C NMR (125 MHz, CD3OD): δ = 76.7, 56.0, 52.1, 50.5, 43.4, 24.4, 23.6, 11.4 and 11.0 ppm. ESI-HRMS for C9H20NO [MH]+ calcd, 158.1545; found, 158.1540.
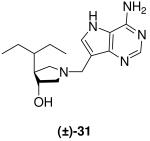
(±)-trans-1-[(9-Deaza-adenin-9-yl)methyl]-3-hydroxy-4-(penta-3-yl)pyrrolidine [(±)-31]
Formaldehyde (25 L, 0.3 mmol, 37 wt% solution in water) followed by 9-deazaadenine (30 mg, 0.22 mmol) were added to a solution of (±)-23 (27 mg, 0.17 mmol) in 1,4-dioxane (0.6 mL) and water (0.6 mL). The reaction mixture was stirred at room temperature for 17 h, absorbed onto silica and eluted down a silica column with a gradient of 5% to 20% (7 N NH3 in MeOH) in CH2Cl2. The crude product was collected, concentrated and subjected to flash chromatography (5 : 4.9 : 0.1, CH2Cl2 : MeOH : 28% aq. NH4OH) to afford (±)-31 as a pale yellow solid (24 mg, 33%). 1H NMR (500 MHz, CD3OD): δ = 8.16 (s, 1H), 7.50 (s, 1H), 3.99 - 3.96 (m, 1H), 3.84 (d, J = 13.4 Hz, 1H), 3.77 (d, J = 13.4 Hz, 1H), 3.04 (t, J = 8.8 Hz, 1H), 2.79 (dd, J = 10.4, 2.3 Hz, 1H), 2.62 (dd, J = 10.4, 6.4 Hz, 1H), 2.17 (t, J = 9.5 Hz, 1H), 2.04 - 1.98 (m, 1H), 1.51 - 1.37 (m, 3H), 1.27 - 1.19 (m, 2H), 0.89 (t, J = 7.4, 3H) and 0.84 ppm (t, J = 7.1, 3H). 13C NMR (125 MHz, CD3OD): δ = 152.1, 151.0, 147.0, 130.1, 115.2, 112.5, 75.8, 63.3, 58.2, 51.3, 49.1, 44.3, 24.0, 23.6, 11.3 and 10.7 ppm. ESI-HRMS for C16H26N5O [MH]+ calcd, 304.2137; found, 304.2137.
(±)-Benzyl trans-4-cyclopropyl-3-hydroxypyrrolidine-1-carboxylate [(±)-16]
A solution of epoxide (meso-11) (283 mg, 1.3 mmol) and CuBr·DMS (43 mg, 0.21 mmol) in THF (10 mL) was cooled to −30 °C (internal temperature. Cyclopropylmagnesium bromide (10 mL, 5.0 mmol, 0.5 M solution in THF) was added drop-wise over 25 min. After complete addition the reaction was allowed to slowly warm to −15 °C over 45 min and then quenched with 10% aqueous NH4Cl solution (20 mL) and EtOAc (50 mL). The mixture was stirred at room temperature for 40 min and then the layers were separated. The aqueous phase was extracted with EtOAc (3 × 50 mL) and the combined organic phases were dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography of the residue (3 : 7 then 4 : 6, EtOAc: Petrol) afforded (±)-16 as a pale yellow oil (311 mg, 92%). 1H NMR (500 MHz, CDCl3): δ = 7.38 - 7.29 (m, 5H), 5.13 (s, 2H), 4.25 - 4.22 (m, 1H), 3.75 - 3.70 (m, 1H), 3.69 - 3.63 (m, 1H), 3.40 - 3.28 (m, 1H), 1.94 - 1.85 (m, 1H), 1.48 - 1.43 (m, 1H), 0.60 - 0.44 (m, 3H), 0.30 - 0.25 (m, 1H) and 0.16 - 0.10 ppm (m, 1H). 13C NMR (125 MHz, CDCl3): δ = 155.1, 136.9, 128.5, 128.0, 127.9, 75.8, 75.0 (rotamers), 66.9, 53.3, 52.8 (rotamers), 51.3, 50.5 (rotamers), 49.2, 49.1 (rotamers), 12.4, 12.2 (rotamers), 3.4, 3.4 (rotamers), 3.1 and 3.0 (rotamers) ppm. ESI-HRMS for C15H19NO3Na [MNa]+ calcd, 284.1263; found, 284.1260.
(±)-trans-4-cyclopropyl-3-hydroxypyrrolidine [(±)-24]
Palladium (25 mg, 0.02 mmol, 10 wt% on carbon) was added to a solution of (±)-16 (310 mg, 1.2 mmol) in MeOH (20 mL) under argon. The reaction mixture was placed under a hydrogen atmosphere and stirred for 1 h and then filtered through Celite® and concentrated under reduced pressure. Flash chromatography of the residue (5 : 4.6 : 0.4, CH2Cl2 : MeOH : 28% aq. NH4OH) afforded (±)-24 as an off-white solid (126 mg, 84%). 1H NMR (500 MHz, CD3OD): δ = 4.30 (dt, J = 5.0, 2.7 Hz, 1H), 3.51 (dd, J = 11.7, 7.3 Hz, 1H), 3.43 (dd, J = 12.4, 4.7 Hz, 1H), 3.17 (dd, J = 12.3, 2.2 Hz, 1H), 3.11 (dd, J = 11.7, 4.6 Hz, 1H), 1.61 - 1.55 (m, 1H), 0.71 - 0.63 (m, 1H), 0.57 - 0.48 (m, 1H), 0.33 - 0.29 (m, 1H) and 0.21 - 0.17 ppm (m, 1H). 13C NMR (125 MHz, CD3OD): δ = 76.4, 52.9, 52.6, 50.0, 13.2, 4.2 and 3.6 ppm. ESI-HRMS for C7H14NO [MH]+ calcd, 128.1075; found, 128.1082.
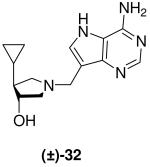
(±)-trans-4-Cyclopropyl-1-[(9-deaza-adenin-9-yl)methyl]-3-hydroxypyrrolidine [(±)-32]
Formaldehyde (50 L, 0.6 mmol, 37 wt% solution in water) followed by 9-deazaadenine (60 mg, 0.45 mmol) were added to a solution of (±)-24 (51 mg, 0.40 mmol) in 1,4-dioxane (1 mL) and water (1 mL). The reaction mixture was stirred at room temperature for 41 h, absorbed onto silica and eluted down a silica column with a gradient of 5% to 30% (7 N NH3 in MeOH) in CH2Cl2. The crude product was collected, concentrated and subjected to flash chromatography (5 : 4.5 : 0.5, CH2Cl2 : MeOH : 28% aq. NH4OH) to afford (±)-32 as an off-white solid (59 mg, 54%). 1H NMR (500 MHz, CD3OD): δ = 8.16 (s, 1H), 7.45 (s, 1H), 4.05 (dt, J = 6.4, 4.1 Hz, 1H), 3.85 (d, J = 13.4 Hz, 1H), 3.80 (d, J = 13.4 Hz, 1H), 3.02 (dd, J = 9.6, 8.1 Hz, 1H), 2.82 (dd, J = 10.5, 6.4 Hz, 1H), 2.68 (dd, J = 10.4, 3.9 Hz, 1H), 2.36 (dd, J = 9.8, 7.5 Hz, 1H), 1.38 (ddd, J = 16.9, 7.8, 4.3 Hz, 1H), 0.75 - 0.68 (m, 1H), 0.46 - 0.37 (m, 2H), 0.28 - 0.24 (m, 1H) and 0.08 - 0.03 ppm (m, 1H). 13C NMR (125 MHz, CD3OD): δ = 152.1, 151.0, 147.0, 130.1, 115.2, 112.5, 77.4, 62.6, 59.1, 53.6, 49.1, 14.5, 3.8 and 3.5 ppm. ESI-HRMS for C14H20N5O [MH]+ calcd, 274.1668; found, 274.1666.
(±)-Benzyl trans-4-cyclopentyl-3-hydroxypyrrolidine-1-carboxylate [(±)-17]
Cyclopentyl bromide (0.5 mL, 4.7 mmol) was added drop-wise to a suspension of magnesium (221 mg, 9 mmol) in THF (10 mL), activated with 1,2-dibromoethane. After complete addition the reaction mixture was stirred for 1 h at room temperature and then added drop-wise, over 10 min, to a solution of epoxide (meso-11) (230 mg, 1 mmol) and CuBr·DMS (55 mg, 0.3 mmol) in THF (10 mL) at −30 °C (internal temperature). The reaction mixture was stirred for 75 min and then quenched with 10% aqueous NH4Cl solution (20 mL) and EtOAc (20 mL). The bi-phasic mixture was stirred for 1 h and then the layers were separated, and the aqueous phase extracted with EtOAc (2 × 50 mL). The combined organic phases were dried (MgSO4), filtered and concentrated under reduced pressure to afford the crude (±)-17 as a pale yellow oil, which was used immediately in the next step without characterization.
(±)-trans-4-cyclopentyl-3-hydroxypyrrolidine [(±)-25]
Palladium (50 mg, 0.05 mmol, 10 wt% on carbon) was added to a solution of crude (±)-17 in MeOH (20 mL) under argon. The reaction mixture was placed under a hydrogen atmosphere and stirred for 1 h and then further catalyst (50 mg) was added. The reaction mixture was placed under a hydrogen atmosphere for a further 1 h and then filtered through Celite® and concentrated under reduced pressure. Flash chromatography of the residue (5 : 4.6 : 0.4, CH2Cl2 : MeOH : 28% aq. NH4OH) afforded crude (±)-25 (43 mg) which was used without further purification and characterization in the next step.
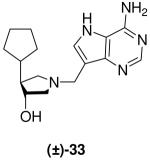
(±)-trans-4-Cyclopentyl-1-[(9-deaza-adenin-9-yl)methyl]-3-hydroxypyrrolidine [(±)-33]
Formaldehyde (35 L, 0.4 mmol, 37 wt% solution in water) followed by 9-deazaadenine (42 mg, 0.3 mmol) were added to a solution of crude (±)-25 (43 mg) in 1,4-dioxane (1 mL) and water (1 mL). The reaction mixture was stirred at room temperature for 94 h, absorbed onto silica and eluted down a silica column with a gradient of 5% to 30% (7 N NH3 in MeOH) in CH2Cl2. The crude product was collected, concentrated and subjected to flash chromatography (5 : 4.95 : 0.05, CH2Cl2 : MeOH : 28% aq. NH4OH) to afford (±)-33 as an off-white solid (28 mg, 10%, over 3 steps). 1H NMR (500 MHz, CD3OD): δ = 8.16 (s, 1H), 7.50 (s, 1H), 3.97 - 3.94 (m, 1H), 3.85 (d, J = 13.4 Hz, 1H), 3.80 (d, J = 13.4 Hz, 1H), 3.06 (dd, J = 9.6, 8.1 Hz, 1H), 2.76 - 2.68 (m, 2H), 2.39 (dd, J = 9.7, 8.1 Hz, 1H), 1.89 - 1.50 (m, 8H), 1.34 -1.30 (m, 1H) and 1.21 - 1.08 ppm (m, 1H). 13C NMR (125 MHz, CD3OD): δ = 152.1, 151.0, 147.0, 130.2, 115.2, 112.4, 76.7, 63.0, 59.1, 54.2, 49.1, 44.7, 32.2, 31.8 and 26.1 (2C) ppm. ESI-HRMS for C16H23N5ONa [MNa]+ calcd, 324.1800; found, 324.1802.
(±)-Benzyl trans-4-(cyclohexylmethyl)-3-hydroxypyrrolidine-1-carboxylate [(±)-18]
A solution of epoxide (meso-11) (243 mg, 1.1 mmol) and CuBr·DMS (50 mg, 0.22 mmol) in THF (10 mL) was cooled to −30 °C (internal temperature). Cyclohexylmethylmagnesium bromide (11 mL, 5.5 mmol, 0.5 M solution in THF) was added drop-wise over 20 min. After complete addition the reaction was allowed to warm to −25 °C over 30 min and then quenched with 10% aqueous NH4Cl solution (20 mL) and EtOAc (20 mL). The mixture was stirred at room temperature for 30 min and then the layers were separated. The aqueous phase was extracted with EtOAc (3 × 30 mL) and the combined organic phases were dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography of the residue (gradient 2 : 8 to 4 : 6, EtOAc : Petrol) afforded (±)-18 as an off-white solid (286 mg, 81%). 1H NMR (500 MHz, CDCl3): δ = 7.40 - 7.30 (m, 5H), 5.13 (s, 2H), 4.01 (t, J = 4.8 Hz, 1H), 3.72 - 3.64 (m, 2H), 3.33 - 3.26 (m, 1H), 3.16 - 3.10 (m, 1H), 2.19 - 2.11 (m, 1H), 1.74 - 1.64 (m, 6H), 1.36 - 1.07 (m, 6H) and 0.95 - 0.81 ppm (m, 2H). 13C NMR (125 MHz, CDCl3): δ = 155.1, 137.0, 128.5, 128.0, 127.9, 75.9, 75.1 (rotamers), 66.8, 52.9, 52.5 (rotamers), 49.8, 49.5 (rotamers), 43.5, 43.0 (rotamers), 39.4, 39.2 (rotamers), 35.8, 35.7 (rotamers), 34.0, 32.9, 26.5, 26.2 and 26.2 ppm. ESI-HRMS for C19H27NO3Na [MNa]+ calcd, 340.1895; found, 340.1895.
(±)-trans-4-(cyclohexylmethyl)-3-hydroxypyrrolidine [(±)-26]
Palladium (20 mg, 0.02 mmol, 10 wt% on carbon) was added to a solution of (±)-18 (286 mg, 0.9 mmol) in MeOH (10 mL) under argon. The reaction mixture was placed under a hydrogen atmosphere and stirred for 1 h and then further catalyst (20 mg, 0.02 mmol, 10 wt% on carbon) was added. The reaction mixture was stirred under a hydrogen atmosphere for a further 1 h then filtered through Celite® and concentrated under reduced pressure. Flash chromatography of the residue (5 : 4.8 : 0.2 then 5 : 4.5 : 0.5, CH2Cl2 : MeOH : 28% aq. NH4OH) afforded (±)-26 as an off-white solid (115 mg, 70%). 1H NMR (500 MHz, CD3OD): δ = 3.88 (q, J = 4.2 Hz, 1H), 3.20 (dd, J = 11.2, 7.6 Hz, 1H), 2.97 (dd, J = 12.0, 5.5 Hz, 1H), 2.76 (dd, J = 12.0, 3.4 Hz, 1H), 2.45 (dd, J = 11.2, 6.4 Hz, 1H), 2.07 - 2.01 (m, 1H), 1.80 - 1.65 (m, 5H), 1.39 - 1.10 (m, 6H) and 0.97 - 0.86 ppm (m, 2H). 13C NMR (125 MHz, CD3OD): δ = 78.8, 54.5, 52.1, 46.2, 41.6, 37.4, 35.1, 34.3, 27.7 and 27.4 (2 × C) ppm. ESI-HRMS for C11H22NO [MH]+ calcd, 184.1701; found, 184.1696.
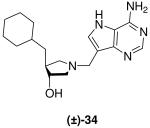
(±)-trans-4-(Cyclohexylmethyl)-1-[(9-deaza-adenin-9-yl)methyl]-3-hydroxypyrrolidine [(±)-34]
Formaldehyde (35 L, 0.4 mmol, 37 wt% solution in water) followed by 9-deazaadenine (42 mg, 0.31 mmol) were added to a solution of (±)-26 (51 mg, 0.28 mmol) in 1,4-dioxane (1.5 mL) and water (1.5 mL). The reaction mixture was stirred at room temperature for 67 h, absorbed onto silica and eluted down a silica column with a gradient of 5% to 30% (7 N NH3 in MeOH) in CH2Cl2. The crude product was collected, concentrated and subjected to flash chromatography (5 : 4.9 : 0.1, CH2Cl2 : MeOH : 28% aq. NH4OH) to afford (±)-34 as a pale yellow solid (42 mg, 46%). 1H NMR (500 MHz, CD3OD): δ = 8.16 (s, 1H), 7.49 (s, 1H), 3.86 - 3.78 (m, 3H), 3.08 - 3.05 (m, 1H), 2.75 (dd, J = 10.4, 6.4 Hz, 1H), 2.70 (dd, J = 10.4, 4.0 Hz, 1H), 2.16 (dd, J = 9.5, 8.1 Hz, 1H), 2.11 - 2.01 (m, 1H), 1.75 - 1.62 (m, 5H), 1.43 - 1.38 (m, 1H), 1.28 - 1.11 (m, 5H) and 0.95 - 0.81 ppm (m, 2H). 13C NMR (125 MHz, CD3OD): δ = 152.1, 151.1, 147.0, 130.1, 115.1, 112.5, 78.1, 62.2, 59.8, 49.1, 45.8, 42.4, 37.4, 35.1, 34.2, 27.7 and 27.4 (2 × C) ppm. ESI-HRMS for C18H28N5O [MH]+ calcd, 330.2294; found, 330.2297.
(±)-Benzyl trans-3-hydroxy-4-phenylpyrrolidine-1-carboxylate [(±)-19]
A solution of epoxide (meso-11) (149 mg, 0.68 mmol) and CuBr·DMS (30 mg, 0.15 mmol) in THF (5.6 mL) was cooled to −30 °C. Phenylmagnesium bromide (1.5 mL, 1.5 mmol, 1 M solution in THF) was added drop-wise over 10 min, keeping the internal temperature below −25 °C. After complete addition the reaction was allowed to warm to −15 °C over 90 min then cooled back to −30 °C and further phenylmagnesium bromide (1.5 mL, 1.5 mmol, 1 M solution in THF) was added. The reaction mixture was allowed to warm to −20 °C over 1 h and then quenched with 10% aqueous NH4Cl solution (20 mL) and EtOAc (20 mL). The mixture was stirred at room temperature for 30 min and then the layers were separated. The aqueous phase was extracted with EtOAc (3 × 20 mL), and the combined organic phases were dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography of the residue (gradient 3 : 7 to 1 : 1, EtOAc : Petrol) afforded a mixture of (±)-19 and unreacted epoxide (meso-11) as a pale yellow oil (162 mg, 2 : 1, (±)-19 : meso-11 which was used without characterization.
(±)-trans-3-Hydroxy-4-phenylpyrrolidine [(±)-27]
Palladium (25 mg, 0.02 mmol, 10 wt% on carbon) was added to a solution of (±)-19 : meso-11 (2 : 1) (160 mg) in MeOH (12 mL) under argon. The reaction mixture was placed under a hydrogen atmosphere and stirred for 75 min and then filtered through Celite® and concentrated under reduced pressure. Flash chromatography of the residue (5 : 4.8 : 0.2 then 5 : 4.6 : 0.4, CH2Cl2 : MeOH : 28% aq. NH4OH) afforded (±)-27 as a yellow oil (39 mg, 35%, over 2 steps). 1H NMR (500 MHz, CD3OD): δ = 7.32 - 7.26 (m, 4H), 7.22 - 7.19 (m, 1H), 4.30 - 4.27 (m, 1H), 3.46 (dd, J = 11.5, 8.2 Hz, 1H), 3.18 (dd, J = 11.9, 6.0 Hz, 1H), 3.12 (td, J = 8.2, 5.4 Hz, 1H) and 2.92 - 2.88 ppm (m, 2H). 13C NMR (125 MHz, CD3OD): δ = 142.7, 129.7, 128.5, 127.8, 80.0, 55.3, 54.9 and 53.6 ppm. ESI-HRMS for C10H14NO [MH]+ calcd. 164.1075; found, 164.1068.
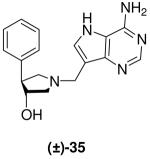
(±)-trans-1-[(9-Deaza-adenin-9-yl)methyl]-3-hydroxy-4-phenylpyrrolidine [(±)-35]
Formaldehyde (35 L, 0.4 mmol, 37 wt% solution in water) followed by 9-deazaadenine (39 mg, 0.24 mmol) were added to a solution of (±)-27 (30 mg, 0.22 mmol) in 1,4-dioxane (0.4 mL) and water (0.8 mL). The reaction mixture was stirred at room temperature for 17 h, absorbed onto silica and eluted down a silica column with a gradient of 10% to 20% (7 N NH3 in MeOH) in CH2Cl2. The crude product was collected, concentrated and subjected to flash chromatography (5 : 4.9 : 0.1 then 5 : 4.8 :0.2, CH2Cl2 : MeOH : 28% aq. NH4OH) to afford (±)-35 as a white solid (38 mg, 55%). 1H NMR (500 MHz, CD3OD): δ = 8.17 (s, 1H), 7.52 (s, 1H), 7.26 (br. s, 2H), 7.25 (br. s, 2H), 7.18 - 7.15 (m, 1H), 4.28 - 4.25 (m, 1H), 3.89 (q, J = 13.4 Hz, 2H), 3.28 - 3.25 (m, 1H), 3.16 (td, J = 8.7, 5.7 Hz, 1H), 3.00 (dd, J = 10.3, 6.8 Hz, 1H), 2.85 (dd, J = 10.3, 4.3 Hz, 1H) and 2.64 ppm (t, J = 9.3 Hz, 1H). 13C NMR (125 MHz, CD3OD): δ = 152.1, 151.0, 147.0, 143.6, 130.1, 129.5, 128.6, 127.5, 115.2, 112.6, 79.3, 62.8, 61.2, 54.9 and 49.1 ppm. ESI-HRMS for C17H19N5ONa [MNa]+ calcd, 332.1487; found, 332.1490.
(±)-tert-Butyl trans-3-hydroxy-4-vinylpyrrolidine-1-carboxylate [(±)-36]23
A solution of epoxide (meso-10) (331 mg, 1.8 mmol) and CuBr·DMS (73 mg, 0.35 mmol) in THF (15 mL) was cooled to −30 °C (internal temperature). Vinylmagnesium bromide (8 mL, 8.0 mmol, 1 M solution in THF) was added drop-wise over 15 min. After complete addition the reaction was allowed to slowly warm to −10 °C over 1 h and then quenched with 10% aqueous NH4Cl solution (20 mL) and EtOAc (50 mL). The mixture was stirred at room temperature for 1 h and then the layers were separated. The aqueous phase was extracted with EtOAc (3 × 50 mL) and the combined organic phases were dried (MgSO4), filtered and concentrated under reduced pressure to afford (±)-36 as a pale yellow oil (286 mg, 75%). No further purification was necessary. 1H NMR (500 MHz, CDCl3): δ = 5.74 - 5.67 (m, 1H), 5.20 (d, J = 17.2 Hz, 1H), 5.15 (d, J = 10.4 Hz, 1H), 4.14 - 4.08 (m, 1H), 3.73 - 3.61 (m, 2H), 3.29 - 3.17 (m, 2H), 2.71 - 2.65 (m, 1H), 2.71 (br. s, 1H) and 1.46 ppm (s, 9H). 13C NMR (125 MHz, CDCl3): δ = 154.5, 136.3, 117.3, 79.5, 74.5, 74.2 (rotamers), 52.2, 51.9 (rotamers), 50.5, 49.9 (rotamers), 48.8, 48.3 (rotamers) and 28.5 ppm. ESI-HRMS for C11H19NO3Na [MNa]+ calcd, 236.1263; found, 236.1263.
(±)-trans-3-Hydroxy-4-vinylpyrrolidine [(±)-38]23
36% aq. HCl (1 mL) was added to a solution of (±)-36 (286 mg, 1.34 mmol) in MeOH (20 mL). The reaction mixture was concentrated under reduced pressure and then azeotroped with MeOH (20 mL) followed by toluene (20 mL). Flash chromatography of the residue (5 : 4.6 : 0.4, CH2Cl2 : MeOH : 28% aq. NH4OH) afforded (±)-38 as a brown oil (95 mg, 63%). 1H NMR (500 MHz, CD3OD): δ = 5.78 (ddd, J = 17.3, 10.4, 7.9 Hz, 1H), 5.12 (dt, J = 17.3, 1.5 Hz, 1H), 5.06 (ddd, J = 10.4, 1.5, 1.0 Hz, 1H), 4.03 (dt, J = 5.6, 4.3 Hz, 1H), 3.21 (dd, J = 11.4, 7.6 Hz, 1H), 3.03 (dd, J = 11.9, 5.7 Hz, 1H), 2.77 (dd, J = 11.8, 4.1 Hz, 1H), 2.68 (dd, J = 11.3, 6.7 Hz, 1H) and 2.61 - 2.56 ppm (m, 1H). 13C NMR (125 MHz, CD3OD): δ = 139.4, 116.1, 78.3, 54.6, 53.5 and 51.3 ppm. ESI-HRMS for C6H12NO [MH]+ calcd, 144.0919; found, 114.0911.
(±)-trans-1-[(9-Deaza-adenin-9-yl)methyl]-3-hydroxy-4-vinylpyrrolidine [(±)-40]
Formaldehyde (45 L, 0.56 mmol, 37 wt% solution in water) followed by 9-deazaadenine (61 mg, 0.46 mmol) were added to a solution of (±)-38 (42 mg, 0.37 mmol) in 1,4-dioxane (1 mL) and water (1 mL). The reaction mixture was stirred at room temperature for 17 h, absorbed onto silica and eluted down a silica column with a gradient of 5% to 20% (7 N NH3 in MeOH) in CH2Cl2. The crude product was collected, concentrated and subjected to flash chromatography (5 : 4.5 : 0.5, CH2Cl2 : MeOH : 28% aq. NH4OH) to afford (±)-40 as an off-white solid (55 mg, 57%). 1H NMR (500 MHz, CD3OD): δ = 8.16 (s, 1H), 7.49 (s, 1H), 5.79 (ddd, J = 17.2, 10.3, 8.0 Hz, 1H), 5.06 (dt, J = 17.2, 1.4 Hz, 1H), 4.99 (dq, J = 10.3, 1.0 Hz, 1H), 4.01 - 3.98 (m, 1H), 3.87 (d, J = 13.5 Hz, 1H), 3.82 (d, J = 13.5 Hz, 1H), 3.05 (dd, J = 9.8, 8.1 Hz, 1H), 2.86 (dd, J = 10.4, 6.7 Hz, 1H), 2.73 (dd, J = 10.4, 4.5 Hz, 1H), 2.65 - 2.59 (m, 1H) and 2.42 ppm (dd, J = 9.9, 8.3 Hz, 1H). 13C NMR (125 MHz, CD3OD): δ = 152.1, 151.0, 147.0, 139.7, 130.3, 116.0, 115.2, 112.1, 77.2, 61.9, 58.7, 52.9 and 49.1 ppm. ESI-HRMS for C13H18N5O [MH]+ calcd, 260.1511; found, 260.1511.
(±)-tert-Butyl trans-4-allyl-3-hydroxypyrrolidine-1-carboxylate [(±)-37]23
A solution of epoxide (meso-10) (227 mg, 1.2 mmol) in diethyl ether (2.6 mL) was added drop-wise to a solution of allylmagnesium chloride (1.4 mL, 2.8 mmol, 2 M solution in THF) in diethyl ether (4.4 mL) at 0 °C (internal temperature). The reaction mixture was stirred at 0 °C for 15 min and then warmed to room temperature. After 90 min the reaction mixture was quenched by the addition of saturated aqueous NH4Cl solution (20 mL) and then extracted with EtOAc (3 × 50 mL). The combined organic phases were washed with brine (2 × 50 mL), dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography of the residue (1 : 9, EtOAc : Petrol) afforded (±)-37 as a pale yellow oil (138 mg, 50%). 1H NMR (500 MHz, CDCl3): δ = 5.82 - 5.75 (m, 1H), 5.09 - 5.04 (m, 2H), 4.04 (dd, J = 10.1, 4.8 Hz, 1H), 3.64 - 3.54 (m, 2H), 3.26 - 3.21 (m, 1H), 3.11 - 3.05 (m, 1H), 2.24 - 2.19 (m, 1H), 2.17 - 2.11 (m, 1H), 2.07 - 1.98 (m, 1H) and 1.45 ppm (s, 9H). 13C NMR (125 MHz, CDCl3): δ = 154.8, 135.8, 116.7, 79.5, 74.7, 74.0 (rotamers), 52.7, 52.4 (rotamers), 49.0, 48.7 (rotamers), 45.5, 45.0 (rotamers), 35.6 and 28.7 ppm. ESI-HRMS for C12H21NO3Na [MNa]+ calcd, 250.1419; found, 250.1416.
(±)-trans-4-Allyl-3-hydroxypyrrolidine [(±)-39]23
36% aq. HCl (1 mL, 33 mmol) was added to a stirred solution of (±)-37 (59 mg, 0.26 mmol) in methanol (5 mL). The reaction mixture was concentrated under reduced pressure and subsequently azeotroped with methanol (10 mL) followed by toluene (10 mL). The residue was absorbed onto silica and eluted down a flash column (5 : 4.5 : 0.5, CH2Cl2 : MeOH : 28% aq. NH4OH) to afford (±)-39 as a yellow oil (40 mg, 95%). 1H NMR (500 MHz, CD3OD): δ = 5.87 - 5.79 (m, 1H), 5.08 - 5.00 (m, 2H), 3.95 - 3.93 (m, 1H), 3.15 (dd, J = 11.5, 7.0 Hz, 1H), 2.95 (dd, J = 12.0, 5.5 Hz, 1H), 2.74 (dd, J = 12.0, 3.4 Hz, 1H), 2.48 (dd, J = 11.6, 5.5 Hz., 1H), 2.24 - 2.19 (m, 1H) and 2.04 - 1.96 ppm (m, 2H). 13C NMR (125 MHz, CD3OD): δ = 136.7, 116.7, 77.4, 54.3, 51.0, 48.3 and 37.3 ppm. ESI-HRMS for C7H14NO [MH]+ calcd, 128.1075; found, 128.1072.
(±)-trans-4-Allyl-1-[(9-deaza-adenin-9-yl)methyl]-3-hydroxypyrrolidine [(±)-41]
Formaldehyde (44 L, 0.54 mmol, 37 wt% solution in water) followed by 9-deazaadenine (44 mg, 0.33 mmol) were added to a solution of (±)-39 (40 mg, 0.32 mmol) in 1,4-dioxane (0.6 mL) and water (1.2 mL). The reaction mixture was stirred at room temperature for 17 h, absorbed onto silica and eluted down a silica column with a gradient of 5% to 40% (7 N NH3 in MeOH) in CH2Cl2. The crude product was collected, concentrated and subjected to flash chromatography (5 : 4.9 : 0.1, CH2Cl2 : MeOH : 28% aq. NH4OH) to afford (±)-41 as an off-white solid (20 mg, 24%). 1H NMR (500 MHz, CD3OD): δ = 8.16 (s, 1H), 7.49 (s, 1H), 5.83 - 5.74 (m, 1H), 5.05 - 4.95 (m, 2H), 3.89 (dt, J = 6.3, 4.1 Hz, 1H), 3.85 (d, J = 13.4 Hz, 1H), 3.80 (d, J = 13.6 Hz, 1H), 3.02 (dd, J = 9.9, 7.6 Hz, 1H), 2.80 (dd, J = 10.4, 6.4 Hz, 1H), 2.68 (dd, J = 10.4, 4.1 Hz, 1H), 2.30 - 2.24 (m, 2H) and 2.10 - 2.02 ppm (m, 2H). 13C NMR (125 MHz, CD3OD): δ = 152.1, 151.0, 147.0, 138.0, 130.1,116.3, 115.1, 112.5, 76.8, 62.2, 59.0, 49.1, 48.0 and 38.2 ppm. ESI-HRMS for C14H20N5O [MH]+ calcd, 274.1668; found, 274.1661.
(±)-tert-Butyl trans-3-hydroxy-4-((trimethylsilyl)ethynyl)pyrrolidine-1-carboxylate [(±)-42]
n-Butyllithium (4.6 mL, 6.0 mmol, 1.3 M solution in hexanes) was added, over 5 min, to a solution of trimethylsilylacetylene (1.1 mL, 7.8 mmol) in THF (8.5 mL) at −78 °C. After 30 min BF3.OEt2 (1.4 mL, 11.4 mmol) was added over 5 min. After stirring for a further 30 min at −78 °C a solution of epoxide (meso-10) (550 mg, 3.0 mmol) in THF (10 mL) was added. The reaction mixture was stirred at −78 °C for 1.5 h and then allowed to warm to room temperature. The reaction was stirred for 16 h and then quenched by the addition of saturated aqueous NH4Cl solution (20 mL), and then partitioned between water (50 mL) and EtOAc (50 mL). The layers were separated and the aqueous phase was extracted with EtOAc (3 × 50 mL). The combined organic phases were washed with brine (2 × 50 mL), dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography of the residue (2 : 8, EtOAc : Petrol) afforded (±)-42 as a yellow gum (478 mg, 57%). 1H NMR (500 MHz, CDCl3): δ = 4.33 (q, J = 4.7, 1H), 3.77 - 3.65 (br. m, 2H), 3.48 - 3.36 (br. m, 1H), 3.32 - 3.22 (br. m, 1H), 2.90 (br. s, 1H), 1.58 (br. s, 1H), 1.46 (s, 9H) and 0.15 ppm (s, 9H). 13C NMR (125 MHz, CDCl3): δ = 154.7, 104.7, 87.8, 79.8, 75.5, 74.7 (rotamers), 52.4, 52.1 (rotamers), 49.9, 49.4 (rotamers), 39.2, 38.7 (rotamers), 28.5 and 0.0 ppm. ESI-HRMS for C14H25NO3NaSi [MNa]+ calcd, 306.1501; found, 306.1505.
(±)-trans-3-Hydroxy-4-((trimethylsilyl)ethynyl)pyrrolidine [(±)-43]
36% aq. HCl (1 mL, 33 mmol) was added to a solution of (±)-42 (478 mg, 1.7 mmol) in methanol (20 mL) and then concentrated under reduced pressure. The residue was subsequently azeotroped with methanol (20 mL) followed by toluene (20 mL) and then absorbed on to silica and eluted down a flash column (5 : 4.8 : 0.2, CH2Cl2 : MeOH : 28% aq. NH4OH) to afford (±)-43 as a yellow solid (276 mg, 89%). 1H NMR (500 MHz, CD3OD): δ = 4.28 (dt, J = 5.0, 2.8 Hz, 1H), 3.35 (dd, J = 11.2, 7.4 Hz, 1H), 3.09 (dd, J = 12.2, 5.0 Hz, 1H), 2.87 - 2.78 (m, 3H) and 0.13 ppm (s, 9H). 13C NMR (125 MHz, CD3OD): δ = 107.6, 87.6, 79.0, 54.8, 52.8, 41.8 and 0.0 ppm. ESI-HRMS for C9H18NOSi [MH]+ calcd, 184.1158; found, 184.1161.
(±)-trans-1-[(9-Deaza-adenin-9-yl)methyl]-3-hydroxy-4-((trimethylsilyl)ethynyl)pyrrolidine [(±)-44]
Formaldehyde (55 L, 0.69 mmol, 37 wt% solution in water) followed by 9-deazaadenine (73 mg, 0.54 mmol) were added to a solution of 45 (81 mg, 0.44 mmol) in 1,4-dioxane (2.5 mL) and water (2.5 mL). The reaction mixture was stirred at room temperature for 66 h and then concentrated under reduced pressure. Crude (±)-44 was used directly in the next step without characterization.
(±)-trans-1-[(9-Deaza-adenin-9-yl)methyl]-4-ethynyl-3-hydroxypyrrolidine [(±)-45]
Sodium methoxide (10 L, 0.05 mmol, 30 wt% solution in methanol) was added to a solution of crude (±)-44 (145 mg, 0.44 mmol) in methanol (5 mL). The reaction mixture was stirred at room temperature for 2.5 h and then further sodium methoxide (10 L, 0.05 mmol, 30 wt% solution in methanol) was added. After stirring for a further 17 h the reaction mixture was absorbed onto silica and eluted down a silica column (5 : 4.95 : 0.05, CH2Cl2 : MeOH : 28% aq. NH4OH) to afford (±)-45 (29 mg, 25%). A sample was purified by preparative HPLC to analytical purity as a TFA salt. 1H NMR (500 MHz, CD3OD): δ = 8.47 (s, 1H), 8.07 (s, 1H), 4.68 (s, 2H), 4.53 (quin, J = 2.2 Hz. 1H), 3.91 (dd, J = 11.8, 7.4 Hz, 1H), 3.74 (dd, J = 12.3, 4.1 Hz, 1H), 3.59 (dd, J = 11.7, 4.1 Hz, 1H), 3.49 (d, J = 12.4 Hz, 1H), 3.22 - 3.19 (m, 1H) and 2.78 ppm (d, J = 2.6 Hz, 1H). 13C NMR (125 MHz, CD3OD): δ = 159.2, 152.2, 145.3, 136.0, 114.1, 104.3, 81.2, 75.9, 75.1, 60.5, 58.1, 50.2 and 39.2 ppm (resonance signals due to CF3COOH have not been quoted). ESI-HRMS for C13H16N5O [MH]+ calcd, 258.1355; found, 258.1355.
(±)-tert-Butyl trans-4-azido-3-hydroxypyrrolidine-1-carboxylate [(±)-46]29
Sodium azide (1.02 g, 15.7 mmol) was added to a solution of epoxide (meso-10) (1.0 g, 5.4 mmol) in 1,4-dioxane (9 mL) and water (1.8 mL). The resulting suspension was heated to 100 °C for 65 h and then cooled to 0 °C and water (20 mL) was added. The mixture was extracted with EtOAc (3 × 50 mL) and the combined organic phases were washed with brine (50 mL), dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography of the residue (3 : 7, then 4 : 6, EtOAc : Petrol) afforded (±)-46 as a pale yellow oil (1.21 g, 98%). 1H NMR (500 MHz, CDCl3): δ = 4.23 (br. s, 1H), 3.93 (br. s, 1H), 3.70 - 3.66 (m, 1H), 3.60 - 3.56 (m, 1H), 3.46 - 3.16 (m, 3H) and 1.46 ppm (s, 9H). 13C NMR (125 MHz, CDCl3): δ = 154.6, 80.3, 74.1, 73.3 (rotamers), 65.4, 64.9 (rotamers), 51.9, 51.6 (rotamers), 48.7, 48.2 (rotamers) and 28.4 ppm. ESI-HRMS for C9H16N4O3Na [MNa]+ calcd, 251.1120; found, 251.1121.
(±)-tert-Butyl trans-3-hydroxy-4-(5-(trimethylsilyl)-1H-1,2,3-triazol-1-yl)pyrrolidine-1-carboxylate [(±)-47]
Trimethylsilylacetylene (2.2 mL, 15.6 mmol) was added to a solution of (±)-46 (730 mg, 3.2 mmol) in toluene (35 mL). The resulting mixture was heated to reflux for 88 h and then allowed to cool and concentrated under reduced pressure to afford a mixture of (±)-47 and (±)-48. This mixture was used directly in the next step without characterization.
(±)-tert-Butyl trans-3-hydroxy-4-(1H-1,2,3-triazol-1-yl)pyrrolidine-1-carboxylate [(±)-48]
The mixture of (±)-47 and (±)-48 was taken up in THF (20 mL) and TBAF (4.8 mL, 4.8 mmol, 1 M solution in THF) was added. The reaction mixture was stirred for 4 h and then further TBAF (1.6 mL, 1.6 mmol, 1 M solution in THF) was added. The reaction was stirred for a further 16 h and then partitioned between EtOAc (30 mL) and water (30 mL). The layers were separated and the aqueous phase was extracted with EtOAc (2 × 50 mL). The combined organic phases were washed with brine (2 × 50 mL), dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography of the residue (100% EtOAc) afforded (±)-48 as a pale yellow gum (510 mg, 63%, over 2 steps) and recovered (±)-47 as a pale yellow oil (140 mg, 14%). (±)-48. 1H NMR (500 MHz, CDCl3): δ = 7.63 (d, J = 6.8 Hz, 1H), 7.58 (s, 1H), 5.44 (br. s, 1H), 4.93 - 4.89 (m, 1H), 4.60 (d, J = 15.9 Hz, 1H), 4.00 (dd, J = 9.5, 7.2 Hz, 1H), 3.77 - 3.67 (m, 2H), 3.36 (dd, J = 11.8, 4.6 Hz, 1H) and 1.38 ppm (s, 9H). 13C NMR (125 MHz, CDCl3): δ = 154.4, 154.2 (rotamers), 133.6, 123.1, 80.4, 74.2, 73.4 (rotamers), 65.4, 64.9 (rotamers), 51.7, 51.1 (rotamers), 48.8, 48.4 (rotamers) and 28.4 ppm. ESI-HRMS for C11H18N4O3Na [MNa]+ calcd, 277.1277; found, 277.1275. (±)-47. 1H NMR (500 MHz, CDCl3): δ = 7.51 (s, 1H), 4.92 - 4.78 (m, 2H), 4.49 (br. s, 1H), 4.10 - 4.03 (m, 1H), 3.89 - 3.79 (m, 2H), 3.44 (br. s, 1H), 1.47 (s, 9H) and 0.29 ppm (s, 9H). 13C NMR (125 MHz, CDCl3): δ = 155.6, 155.4 (rotamers), 147.7, 129.5, 81.5, 75.5, 74.6 (rotamers), 66.5, 66.0 (rotamers), 52.6, 52.1 (rotamers), 50.2, 49.7 (rotamers), 29.6 and 0.0 ppm. ESI-HRMS for C14H26N4O3NaSi [MNa]+ calcd, 349.1672; found, 349.1669.
(±)-trans-3-Hydroxy-4-(1H-1,2,3-triazol-1-yl)pyrrolidine [(±)-49]
36% aq. HCl (1 mL, 33 mmol) was added to a solution of (±)-48 (500 mg, 2.0 mmol) in methanol (25 mL). The reaction mixture was concentrated under reduced pressure and subsequently azeotroped with methanol (2 × 20 mL) followed by toluene (10 mL). Flash chromatography of the residue (5 : 4.6 : 0.4, CH2Cl2 : MeOH : 28% aq. NH4OH) afforded (±)-49 as an off-white foam (185 mg, 61%). 1H NMR (500 MHz, CD3OD): δ = 8.06 (d, J = 0.9 Hz, 1H), 7.75 (d, J = 0.9 Hz, 1H), 4.94 - 4.91 (m, 1H), 4.54 - 4.51 (m, 1H), 3.58 (dd, J = 12.5, 7.3 Hz, 1H), 3.37 - 3.34 (m, 1H), 3.27 (dd, J = 12.6, 4.7 Hz, 1H) and 2.92 ppm (dd, J = 12.2, 3.9 Hz, 1H). 13C NMR (125 MHz, CD3OD): δ = 134.5, 125.4, 78.7, 69.8, 54.7 and 52.3 ppm. ESI-HRMS for C6H11N4O [MH]+ calcd, 155.0933; found, 155.0931.
(±)-trans-1-[(9-Deaza-adenin-9-yl)methyl]-3-hydroxy-4-(1H-1,2,3-triazol-4-yl)pyrrolidine [(±)-50]
Formaldehyde (75 L, 0.9 mmol, 37 wt% solution in water) followed by 9-deazaadenine (100 mg, 0.75 mmol) were added to a solution of (±)-49 (96 mg, 0.62 mmol) in 1,4-dioxane (1.5 mL) and water (1.5 mL). After stirring for 66 h the reaction mixture was absorbed onto silica and eluted down a silica column with a gradient of 10% to 20% (7 N NH3 in MeOH) in CH2Cl2. The crude product was collected, concentrated and subjected to flash chromatography (5 : 4.98 : 0.02, CH2Cl2 : MeOH : 28% aq. NH4OH) to afford (±)-50 (58 mg, 31%). A sample was purified by preparative HPLC to analytical purity as a TFA salt. 1H NMR (500 MHz, CD3OD): δ = 8.45 (s, 1H), 8.14 (d, J = 0.8 Hz, 1H), 8.08 (s, 1H), 7.80 (d, J = 0.7 Hz, 1H), 5.33 (dt, J = 7.3, 2.8 Hz, 1H), 4.82 (s, 2H), 4.66 (quin, J = 2.2 Hz, 1H), 4.33 (dd, J = 13.2, 7.5 Hz, 1H), 4.15 (dd, J = 13.2, 3.6 Hz, 1H), 3.92 (dd, J = 12.4, 4.5 Hz, 1H) and 3.66 ppm (dd, J = 12.3, 1.7 Hz, 1H). 13C NMR (125 MHz, CD3OD): δ = 152.0, 145.1, 139.9, 136.1, 135.1, 126.6, 114.1, 104.6, 75.8, 66.6, 60.3, 57.2 and 49.9 ppm (resonance signals due to CF3COOH have not been quoted). ESI-HRMS for C13H17N8O [MH]+ calcd, 301.1525; found, 301.1530.
(±)-tert-Butyl trans-4-ethynyl-3-hydroxypyrrolidine-1-carboxylate [(±)-51]
Tetrabutylammonium fluoride (3 mL, 3 mmol, 1.0 M solution in THF) was added drop-wise to a stirred solution of (±)-42 (569 mg, 2 mmol) in THF (15 mL). After stirring for 1 h at room temperature the reaction mixture was quenched by the addition of water (100 mL) and then extracted with EtOAc (3 × 75 mL). The combined organic phase was washed with brine (80 mL) and then dried (MgSO4), filtered and concentrated under reduced pressure to afford crude (±)-51 as a yellow oil (420 mg, 99%). No further purification was necessary. 1H NMR (500 MHz, CDCl3): δ = 4.26 (dd, J = 8.4, 3.8 Hz, 1H), 3.63 - 3.56 (m, 2H), 3.39 - 3.32 (m, 1H), 3.22 (t, J = 11.5 Hz, 1H), 2.83 (br. s, 1H), 2.11 (br. s, 1H) and 1.37 ppm (s, 9H). 13C NMR (125 MHz, CDCl3): δ = 154.7, 82.7, 79.9, 75.0, 74.2 (rotamers), 71.3, 52.3, 52.1 (rotamers), 49.7, 49.2 (rotamers), 37.8, 37.2 (rotamers) and 28.4 ppm. ESI-HRMS for C11H17NO3Na [MNa]+ calcd, 234.1106; found, 234.1108.
(±)-tert-Butyl trans-4-(1-benzyl-1H-1,2,3-triazol-4-yl)-3-hydroxypyrrolidine-1-carboxylate [(±)-52]
Sodium ascorbate (14 mg, 0.07 mmol) and then copper(II) sulfate (20 L, 0.02 mmol, 1.0 M aqueous solution) were added to a solution of (±)-51 (122 mg, 0.6 mmol) and benzyl azide (111 mg, 0.8 mmol) in t-BuOH (1 mL) and water (1 mL). After stirring at room temperature for 18.5 h the mixture was partitioned between water (10 mL) and EtOAc (10 mL). The layers were separated and the aqueous phase was extracted with EtOAc (2 × 20 mL). The combined organic phases were washed with 5% aqueous NH4OH solution (2 × 20 mL) and brine (20 mL), dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography of the residue (gradient 50 - 100% EtOAc in Petrol) afforded (±)-52 as a yellow oil (114 mg, 57%). 1H NMR (500 MHz, CDCl3): δ = 7.34 - 7.30 (m, 4H), 7.22 - 7.21 (m, 2H), 5.44 (s, 2H), 4.43 (br. d, J = 38 Hz, 1H), 4.18 (d, J = 11.0 Hz, 1H), 3.83 (dd, J = 11.0, 7.6 Hz, 1H), 3.67 - 3.58 (m, 1H), 3.51 - 3.44 (m, 1H), 3.41 - 3.33 (m, 1H), 3.29 - 3.25 (m, 1H) and 1.40 ppm (s, 9H). 13C NMR (125 MHz, CDCl3): δ = 154.6, 154.5 (rotamers), 147.2, 147.0 (rotamers), 134.5, 129.1, 128.8, 128.1, 120.9, 79.6, 74.9, 74.1 (rotamers), 54.2, 52.2, 51.9 (rotamers), 49.1, 48.6 (rotamers), 43.5, 43.0 (rotamers) and 28.5 ppm. ESI-HRMS for C18H24N4O3Na [MNa]+ calcd, 367.1746; found, 367.1747.
(±)-trans-4-(1-Benzyl-1H-1,2,3-triazol-4-yl)-3-hydroxypyrrolidine [(±)-53]
36% aq. HCl (500 L, 16 mmol) was added to a solution of (±)-52 (114 mg, 0.3 mmol) in methanol (10 mL). The reaction mixture was concentrated under reduced pressure and then azeotroped with methanol (2 × 20 mL) followed by toluene (10 mL). Flash chromatography of the residue (20% (7N NH3 in MeOH) in CH2Cl2) afforded (±)-53 as an off-white solid (60 mg, 74%). 1H NMR (500 MHz, CD3OD): δ = 7.81 (s, 1H), 7.38 - 7.31 (m, 5H), 5.55 (s, 2H), 4.36 (dt, J = 5.7, 3.9 Hz, 1H), 3.43 (dd, J = 11.4, 7.8 Hz, 1H), 3.29 - 3.25 (m, 1H), 3.14 (dd, J = 12.1, 5.7 Hz, 1H), 2.97 (dd, J = 11.4, 6.5 Hz, 1H) and 2.85 ppm (dd, J = 12.0, 3.7 Hz, 1H). 13C NMR (125 MHz, CD3OD): δ = 149.8, 136.8, 130.0, 129.6, 126.2, 123.1, 78.9, 55.1, 54.9, 52.4 and 46.8 ppm. ESI-HRMS for C13H17N4O [MH]+ calcd, 245.1402; found, 245.1401.
(±)-trans-4-(1-Benzyl-1H-1,2,3-triazol-4-yl)-1-[(9-deaza-adenin-9-yl)methyl]-3-hydroxypyrrolidine [(±)-54]
Formaldehyde (35 L, 0.44 mmol, 37 wt% solution in water) followed by 9-deazaadenine (40 mg, 0.30 mmol) were added to a solution of (±)-53 (60 mg, 0.25 mmol) in 1,4-dioxane (0.6 mL) and water (1.2 mL). The reaction mixture was stirred at room temperature for 17 h and then further formaldehyde (20 L, 0.25 mmol, 37 wt% solution in water) was added. After stirring for 60 h the reaction mixture was absorbed onto silica and eluted down a silica column with a gradient of 10% to 30% (7 N NH3 in MeOH) in CH2Cl2. The crude product was collected, concentrated and subjected to flash chromatography (5 : 4.9 : 0.1, CH2Cl2 : MeOH : 28% aq. NH4OH) to afford (±)-54 as an off-white solid (49 mg, 51%). 1H NMR (500 MHz, CD3OD): δ = 8.14 (s, 1H), 7.79 (s, 1H), 7.48 (s, 1H), 7.37 - 7.29 (m, 5H), 5.53 (s, 2H), 4.33 - 4.30 (m, 1H), 3.89 (d, J = 13.4 Hz, 1H), 3.84 (d, J = 13.4 Hz, 1H), 3.25 - 3.21 (m, 1H), 2.96 (dd, J = 10.3, 6.8 Hz, 1H), 2.77 (dd, J = 10.3, 4.0 Hz, 1H) and 2.68 - 2.65 ppm (m, 1H). 13C NMR (125 MHz, CD3OD): δ = 152.1, 151.0, 150.3, 147.0, 136.8, 130.0 (2 C), 129.6, 129.1, 123.1, 115.2, 112.7, 77.8, 62.2, 59.4, 54.9, 48.9 and 46.2 ppm. ESI-HRMS for C20H23N8O [MH]+ calcd, 391.1995; found, 391.1994.
(±)-tert-Butyl trans-4-[3-(benzylthio)propyl]-3-hydroxypyrrolidine-1-carboxylate [(±)-55]
1,1′-Azobis(cyanocyclohexane) (20 mg, 0.08 mmol) was added to a solution of (±)-37 (245 mg, 1.1 mmol) and benzyl mercaptan (1.9 mL, 16 mmol) in 1,4-dioxane (1.9 mL). The reaction mixture was heated to 90 °C for 22 h, with further 1,1′-azobis(cyanocyclohexane) (32 mg, 0.1 mmol) being added at intervals of 3, 5 and 6 h. The reaction was allowed to cool and concentrated under reduced pressure. Flash chromatography of the residue (1 : 9 then 1 : 1, EtOAc : petrol) afforded a 5 : 1 mixture of (±)-55 : (±)-37 as a colorless oil (272 mg). 1H NMR (500 MHz, CDCl3): δ = 7.31 - 7.22 (m, 5H), 3.96 (br. s, 1H), 3.70 (s, 1H), 3.62 - 6.52 (m, 2H), 3.25 - 3.16 (m, 1H), 3.03 - 2.98 (m, 1H), 2.43 - 2.41 (m, 2H), 2.21 - 1.95 (m, 2H), 1.61 - 1.51 (m, 2H), 1.45 (s, 9H) and 1.30 - 1.24 ppm (m, 1H). 13C NMR (125 MHz, CDCl3): δ = 154.6, 138.5, 128.8, 128.5, 127.0, 79.4, 75.5, 74.7 (rotamers), 52.8, 52.5 (rotamers), 49.4, 48.9 (rotamers), 45.9, 45.4 (rotamers), 36.4, 35.7 (rotamers), 31.3, 30.6, 28.5 and 27.3 ppm. ESI-HRMS for C19H29NO3NaS [MNa]+ calcd, 374.1766; found, 374.1761.
(±)-trans-4-[3-(Benzylthio)propyl]-3-hydroxypyrrolidine [(±)-56]
36% aq. HCl (500 L, 16 mmol) was added to a solution of (±)-55 : (±)-37 (272 mg, 5 : 1) in methanol (10 mL). The reaction mixture was concentrated under reduced pressure and then azeotroped with methanol (2 × 20 mL) followed by toluene (10 mL). Flash chromatography of the residue (5 : 4.6 : 0.4, CH2Cl2 : MeOH : 28% aq. NH4OH) afforded (±)-56 as a yellow oil (115 mg, 60% over 2 steps). 1H NMR (500 MHz, CD3OD): δ = 7.33 - 7.28 (m, 4H), 7.23 - 7.20 (m, 1H), 4.14 (s, 1H), 3.72 (s, 2H), 3.53 - 3.49 (m, 1H), 3.38 - 3.35 (m, 1H), 3.14 (d, J = 12.3 Hz, 1H), 2.96 (dd, J = 11.6, 5.5 Hz, 1H), 2.47 - 2.44 (m, 2H), 2.16 (br. s, 1H), 1.64 - 1.50 (m, 3H) and 1.39 - 1.32 ppm (m, 1H). 13C NMR (125 MHz, CD3OD): δ = 140.3, 130.0, 129.5, 128.0, 75.1, 52.5, 49.9, 47.0, 37.1, 32.1, 31.2 and 28.5 ppm. ESI-HRMS for C14H22NOS [MH]+ calcd, 252.1422; found, 252.1417.
(±)-trans-4-[3-(Benzylthio)propyl]-1-[(9-deaza-adenin-9-yl)methyl]-3-hydroxypyrrolidine [(±)-57]
Formaldehyde (40 L, 0.5 mmol, 37 wt% solution in water) followed by 9-deazaadenine (46 mg, 0.3 mmol) were added to a solution of (±)-56 (75 mg, 0.3 mmol) in 1,4-dioxane (0.8 mL) and water (0.8 mL). After stirring for 16 h the reaction mixture was absorbed onto silica and eluted down a silica column with a gradient of 5% to 50% (7 N NH3 in MeOH) in CH2Cl2. The crude product was collected, concentrated and subjected to flash chromatography (5 : 4.95 : 0.05 then 5 : 4.8 : 0.2, CH2Cl2 : MeOH : 28% aq. NH4OH) to afford (±)-57 as an off-white solid (23 mg, 20%). 1H NMR (500 MHz, CD3OD): δ = 8.16 (s, 1H), 7.48 (s, 1H), 7.30 - 7.23 (m, 4H), 7.18 - 7.15 (m, 1H), 3.83 - 3.79 (m, 3H), 3.67 (s, 2H), 3.00 (t, J = 8.3 Hz, 1H), 2.74 (dd, J = 10.4, 6.4 Hz, 1H), 2.67 (dd, J = 10.3, 4.0 Hz, 1H), 2.38 (t, J = 7.0 Hz, 1H), 2.13 (dd, J = 9.6, 8.0 Hz, 1H), 1.92 - 1.86 (m, 1H), 1.97 - 1.48 (m, 3H) and 1.37 - 1.30 ppm (m, 1H). 13C NMR (125 MHz, CD3OD): δ = 152.1, 151.0, 147.0, 140.3, 130.1, 130.0, 129.4, 127.8, 115.2, 112.6, 77.6, 62.4, 59.5, 49.1, 48.2, 36.9, 33.4, 32.2 and 28.9 ppm. ESI-HRMS for C21H28N5OS [MH]+ calcd, 398.2015; found, 398.2013.
(3R,4R)-tert-Butyl 4-(benzoyloxymethyl)-3-hydroxypyrrolidine-1-carboxylate [(3R,4R)-59]40
A solution of (3R,4R)-5837 (4.10 g, 19 mmol) and dibutyltin oxide (5.17 g, 21 mmol) in toluene (60 mL) was refluxed in a Dean-Stark apparatus for 1 h and then cooled to 5 °C. Benzoyl chloride (2.2 mL, 19 mmol) was added drop-wise while the internal temperature was kept below 10 °C. After stirring at room temperature for 17 h the reaction mixture was concentrated under reduced pressure. Flash chromatography of the residue (40% EtOAc in Petrol) afforded (3R,4R)-59 as a yellow oil (2.48 g, 41%). 1H NMR (500 MHz, CDCl3): δ = 8.03 (d, J = 7.6 Hz, 2H), 7.58 (t, J = 7.4 Hz, 1H), 7.47 - 7.44 (m, 2H), 4.41 - 4.37 (m, 1H), 4.35 - 4.26 (m, 2H), 3.76 - 3.67 (m, 2H), 3.36 - 3.25 (m, 2H), 2.60 - 2.52 (m, 1H), 2.10 (br. s, 1H) and 1.46 ppm (s, 9H). 13C NMR (125 MHz, CDCl3): δ = 166.6, 154.6, 133.2, 129.7, 129.6, 128.5, 79.7, 72.1, 71.4 (rotamers), 64.1, 64.0 (rotamers), 52.7, 52.5 (rotamers), 46.9, 46.4 (rotamers), 45.7, 45.2 (rotamers) and 28.5 ppm.
(3R,4R)-tert-Butyl 4-(benzoyloxymethyl)-3-(tert-butyldimethylsilyloxy)pyrrolidine-1-carboxylate [(3R,4R)-60]40
tert-Butylchlorodimethylsilane (2.33 g, 15 mmol) was added to a stirred solution of (3R,4R)-59 (2.48 g, 7.7 mmol) and imidazole (2.1 g, 31 mmol) in DMF (4 mL). After 17 h the reaction mixture was diluted with toluene (50 mL) and water (50 mL). The phases were separated and the aqueous phase was extracted with toluene (2 × 50 mL). The combined organic phases were washed with water (2 × 50 mL) and brine (2 × 50 mL), dried (MgSO4), filtered and concentrated under reduced pressure to afford (3R,4R)-60 as a yellow oil (3.31 g, 99%). No further purification was required. 1H NMR (500 MHz, CDCl3): δ = 8.03 - 8.01 (m, 2H), 7.60 - 7.56 (m, 1H), 7.47 - 7.43 (m, 2H), 4.37 - 4.33 (m, 1H), 4.28 - 4.20 (m, 2H), 3.73 - 3.57 (m, 2H), 3.33 - 3.17 (m, 2H), 2.51 (quin, J = 6.3 Hz, 1H), 1.46 (s, 9H), 0.85 (s, 9H) and 0.05 ppm (s, 6H).
(3R,4R)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-4-(hydroxymethyl)pyrrolidine-1-carboxylate [(3R,4R)-61]40
Sodium methoxide (1.8 mL, 7.9 mmol, 25 wt% in methanol) was added to a solution of (3R,4R)-60 (3.31 g, 7.6 mmol) in methanol (10 mL). The reaction mixture was stirred at room temperature for 3 h and then diluted with chloroform (40 mL). The mixture was washed with water (2 × 20 mL), dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography of the residue (60% EtOAc in Petrol) afforded (3R,4R)-61 as a yellow oil (1.2 g, 47%). 1H NMR (500 MHz, CDCl3): δ = 4.20 - 4.14 (m, 1H), 3.71 - 3.51 (m, 4H), 3.18 - 3.10 (m, 2H), 2.31 - 2.23 (m, 1H), 1.46 (s, 9H), 0.88 (s, 9H), 0.08 (s, 3H) and 0.07 ppm (s, 3H). 13C NMR (125 MHz, CDCl3): δ = 154.6, 79.4, 72.8, 72.1 (rotamers), 62.5, 62.4 (rotamers), 53.2, 52.5 (rotamers), 48.9, 48.3 (rotamers), 46.4, 45.9 (rotamers), 28.5, 25.8 (rotamers), 17.9, −4.7 and −4.9 ppm.
(3R,4S)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-4-formylpyrrolidine-1-carboxylate [(3R,4S)-62]40
Alcohol (3R,4R)-61 (1.1 g, 3.3 mmol) was added to a suspension of Dess-Martin periodinane (1.54 g, 3.6 mmol) in CH2Cl2 (30 mL) and the reaction mixture was stirred at room temperature for 2.5 h. The reaction mixture was diluted with ether (150 mL) and washed with 1 : 1 solution of saturated aqueous sodium hydrogen carbonate solution : 10% aqueous sodium thiosulfate solution (2 × 100 mL) and then dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography of the residue (1 : 9 then 2 : 8, EtOAc : Petrol) afforded (3R,4S)-62 as a pale yellow oil (980 mg, 90%). 1H NMR (500 MHz, CDCl3): δ = 9.67 (s, 1H), 4.54 - 4.52 (m, 1H), 3.68 - 3.53 (m, 3H), 3.19 (br. s, 1H), 2.96 (br. s, 1H), 1.43 (s, 9H), 0.86 (s, 9H) and 0.06 ppm (s, 6H). 13C NMR (125 MHz, CDCl3): δ = 199.7, 154.2, 79.8, 71.5, 70.9 (rotamers), 59.1, 58.4 (rotamers), 53.6, 53.0 (rotamers), 43.5, 28.4, 25.6, 17.9, −4.8 and −4.9 ppm.
(3R,4S)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-4-vinylpyrrolidine-1-carboxylate [(3R,4S)-63]
n-Butyllithium (4.3 mL, 6.8 mmol, 1.6 M solution in hexanes) was added drop-wise to a stirred suspension of methyltriphenylphosphonium bromide (2.44 g, 6.8 mmol) in THF (10 mL) at 0 °C. After 30 min the suspension was added to a solution of (3R,4S)-62 (977 mg, 3.0 mmol) in THF (10 mL) at −78 °C. After stirring at −78 °C for 1 h the reaction mixture was warmed to room temperature, stirred for 2.5 h and then quenched with water (30 mL) and extracted with CH2Cl2 (100 mL). The combined organic phases were washed with saturated aqueous sodium hydrogen carbonate solution (50 mL) and brine (50 mL) and then dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography of the residue (1 : 9, EtOAc : Petrol) afforded (3R,4S)-63 as a colorless oil (867 mg, 89%). 1H NMR (500 MHz, CDCl3): δ = 5.67 (ddd, J = 17.2, 10.4, 7.8 Hz, 1H), 5.13 - 5.07 (m, 2H), 3.99 (q, J = 5.9 Hz, 1H), 3.64 - 3.49 (m, 2H), 3.22 - 3.07 (m, 2H), 2.67 - 2.60 (m, 1H), 1.45 (s, 9H), 0.86 (s, 9H) and 0.04 ppm (s, 6H). 13C NMR (125 MHz, CDCl3): δ = 154.5, 136.6, 116.8, 79.3, 75.8, 74.8 (rotamers), 52.8, 52.3 (rotamers), 50.8, 50.0 (rotamers), 48.5, 48.0 (rotamers), 28.5, 25.7, 18.0 and −4.8 ppm. ESI-HRMS for C17H33NO3NaSi [MNa]+ calcd, 350.2127; found, 350.2128.
(3R,4S)-tert-Butyl 3-(tert-butyldimethylsilyloxy)-4-ethylpyrrolidine-1-carboxylate [(3R,4S)-65]
Palladium (100 mg, 0.9 mmol, 10 wt% on carbon) was added to a solution of (3R,4S)-63 (870 mg, 2.7 mmol) in ethanol (25 mL) under an argon atmosphere. The reaction mixture was placed under a hydrogen atmosphere and stirred for 15 h and then filtered through Celite® and concentrated under reduced pressure to afford (3R,4S)-65 (770 mg, 88%). No further purification was necessary. 1H NMR (500 MHz, CDCl3): δ = 3.90 (d, J = 5.4 Hz, 1H), 3.59 (br. s, 1H), 3.54 - 3.48 (m, 1H), 3.08 (br. s, 1H), 3.00 - 2.94 (m, 1H), 1.90 (br. s, 1H), 1.45 (s, 9H), 1.23 - 1.13 (m, 2H), 0.93 (t, J = 7.5 Hz, 3H), 0.88 (s, 9H), 0.06 (s, 3H) and 0.05 ppm (s, 3H). 13C NMR (125 MHz, CDCl3): δ = 154.7, 79.1, 75.5, 74.8 (rotamers), 53.4, 52.6 (rotamers), 49.0, 48.5 (rotamers), 48.3, 47.6 (rotamers), 28.5, 25.7, 24.3, 18.0, 12.2, −4.7 and −4.8 ppm. ESI-HRMS for C17H35NO3NaSi [MNa]+ calcd, 352.2284; found, 352.2288.
(3R,4S)-4-Ethyl-3-hydroxypyrrolidine [(3R,4S)-67]
A solution of (3R,4S)-65 (770 mg, 2.3 mmol) in trifluoroacetic acid (20 mL, 260 mmol) was stirred at room temperature for 17 h and then concentrated under reduced pressure. The residue was dissolved in water (50 mL) and washed with chloroform (2 × 50 mL). The aqueous phase was absorbed onto silica and eluted down a silica column (5 : 4.5 : 0.5, CH2Cl2 : MeOH : 28% aq. NH4OH) to afford (3R,4S)-67 as a yellow oil (205 mg, 76%). 1H NMR (500 MHz, CD3OD): δ = 3.96 (dt, J = 5.4, 3.5 Hz, 1H), 3.26 (dd, J = 11.4, 7.6 Hz, 1H), 3.01 (dd, J = 12.1, 5.4 Hz, 1H), 2.82 (dd, J = 12.0, 3.4 Hz, 1H), 2.55 (dd, J = 11.4, 6.2 Hz, 1H), 1.91 - 1.84 (m, 1H), 1.55 - 1.46 (m, 1H), 1.33 - 1.24 (m, 1H) and 0.97 ppm (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CD3OD): δ = 77.7, 54.4, 51.3, 50.7, 26.1 and 12.8 ppm. ESI-HRMS for C16H14NO [MH]+ calcd, 116.1075; found, 116.1080. [α]D21 = + 5.04 (c = 1.15, MeOH).
(3R,4S)-1-[(9-Deaza-adenin-9-yl)methyl]-4-ethyl-3-hydroxypyrrolidine [(3R,4S)-69]
Formaldehyde (53 L, 0.7 mmol, 37 wt% solution in water) followed by 9-deazaadenine (54 mg, 0.4 mmol) were added to a solution of (3R,4S)-67 (44 mg, 0.38 mmol) in 1,4-dioxane (0.7 mL) and water (1.4 mL). The reaction mixture was stirred at room temperature for 66 h, absorbed onto silica and eluted down a silica column using a gradient 5 - 30% (7 N NH3 in MeOH) in CH2Cl2. The crude product was collected, concentrated and subjected to flash chromatography (5 : 4.5 : 0.5, CH2Cl2 : MeOH : 28% aq. NH4OH) to afford (3R,4S)-69 as an off-white solid (67 mg, 67%). 1H NMR (500 MHz, CD3OD): δ = 8.15 (s, 1H), 7.49 (s, 1H), 3.86 - 3.77 (m, 3H), 3.06 (dd, J = 9.5, 8.2 Hz, 1H), 2.75 (dd, J = 10.4, 6.3 Hz, 1H), 2.69 (dd, J = 10.4, 4.0 Hz, 1H), 2.18 (dd, J = 9.7, 7.9 Hz, 1H), 1.91 - 1.84 (m, 1H), 1.59 - 1.50 (m, 1H), 1.38 - 1.28 (m, 1H) and 0.92 ppm (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CD3OD): δ = 152.1, 151.0, 147.0, 130.1, 115.1, 112.6, 77.4, 62.4, 59.4, 50.4, 49.2, 27.1 and 12.9 ppm. ESI-HRMS for C13H20N5O [MH]+ calcd, 262.1668; found, 262.1665. [α]D21 = + 3.48 (c = 1.03, MeOH).
(3R,4S)-tert-Butyl-4-(but-1-enyl)-3-(tert-butyldimethylsilyloxy)-pyrrolidine-1-carboxylate [(3R,4S)-64]
n-Butyllithium (2.7 mL, 3.8 mmol, 1.4 M solution in hexanes) was added drop-wise to a stirred suspension of n-propyltriphenylphosphonium bromide (1.743 g, 4.52 mmol) in THF (20 mL) at 0 °C. After 20 minutes the suspension was cooled to −40 °C and a solution of (3R,4S)-62 (497 mg, 1.5 mmol) in THF (10 mL) was added and the resulting mixture was allowed to warm to −10 °C and kept at that temperature for 30 min. The reaction mixture was then quenched with water (50 mL) and extracted with ethyl acetate (50 mL). The organic layer was separated and washed with water (50 mL) and brine (50 mL) and then dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography of the resulting residue (1 : 19, EtOAc : Petrol) afforded (3R,4S)-64 as an oil (300 mg, 56 %). 1H NMR (500 MHz, CDCl3): δ = 5.51 (dt, J = 10.8, 7.3 Hz, 1H), 5.10 (br. t, J = 10.1 Hz, 1H), 3.99 - 3.91 (m, 1H), 3.67 - 3.58 (m, 1H), 3.56 - 3.49 (m, 1H), 3.17 - 2.89 (m, 3H), 2.17 - 2.00 (m, 2H), 1.45 (s, 9H), 0.97 (t, J = 7.6 Hz, 3H), 0.87 (s, 9H) and 0.04 ppm (s, 6H). 13C NMR (125 MHz, CDCl3): δ = 154.6, 135.0, 127.6, 79.3, 76.3, 75.6 (rotamers), 53.0, 52.5 (rotamers), 49.6, 49.2 (rotamers), 45.2, 44.5 (rotamers), 28.6, 25.8, 21.0, 18.1, 14.4 and −4.8 ppm. ESI-HRMS for C19H37NNaO3Si [MNa]+ calcd, 378.2440; found, 378.2438.
(3R,4S)-4-Butyl-3-hydroxypyrrolidine [(3R,4S)-68]
A suspension of (3R,4S)-64 (260 mg, 0.73 mmol) and Pearlman’s catalyst (50 mg, cat., 20% b/w) in ethanol (5 mL) was stirred under an atmosphere of hydrogen at room temperature for 18 h. The reaction mixture was then filtered through Celite® and concentrated under reduced pressure to afford, presumably, (3R,4S)-tert-butyl-4-(butyl)-3-(tert-butyldimethylsilyloxy)-pyrrolidine-1-carboxylate [(3R,4S)-66] as a colorless oil. 1H NMR confirmed the absence of any olefinic protons and compound (3R,4S)-66 was committed to the next step without further characterization or purification. 36% aq. HCl (1 mL, 12 mmol) was added to a solution of (3R,4S)-66 (270 mg, 0.76 mmol) in methanol (2 mL) and the resulting solution concentrated under reduced pressure. The resulting residue was dissolved in 36% aq. HCl (1 mL, 12 mmol) and concentrated under reduced pressure and the resulting residue partitioned between water (10 mL) and CHCl3 (5 mL). The water layer was washed again with CHCl3 (5 mL) and concentrated under reduced pressure to afford the hydrochloride salt of (3R,4S)-68 as a white foam (136 mg, 100%). 1H NMR (500 MHz, D2O): δ = 4.18 - 4.14 (m, 1H), 3.47 (dd, J = 11.9, 7.4 Hz, 1H), 3.34 (dd, J = 12.7, 5.3 Hz, 1H), 3.11 (dd, J = 12.7, 2.9 Hz, 1H), 2.93 (dd, J = 11.9, 6.1 Hz, 1H), 2.16 - 2.08 (m, 1H), 1.42 - 1.31 (m, 1H), 1.25 - 1.14 (m, 5H) and 0.75 ppm (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, D2O): δ = 73.9, 51.1, 48.9, 45.3, 30.1, 29.1, 21.9 and 13.3 ppm. ESI-HRMS for C8H18NO [MH]+ calcd, 144.1388; found, 144.1383.
(3R,4S)-4-Butyl-1-[(9-deaza-adenin-9-yl)methyl]-3-hydroxypyrrolidine [(3R,4S)-70]
Formaldehyde (86 L, 1.1 mmol, 37 wt% solution in water) followed by 9-deazaadenine (112 mg, 0.84 mmol) were added to a solution of (3R,4S)-68 (100 mg, 0.56 mmol) in 1,4-dioxane (1 mL) and water (2 mL). The reaction mixture was warmed to 85 °C and after 1 h the crude reaction mixture was absorbed onto silica and eluted down a silica column using a gradient 5 - 30% (7 N NH3 in MeOH) in CH2Cl2. The crude product was collected, concentrated and subjected to flash chromatography (5 : 4.5 : 0.5, CH2Cl2 : MeOH : 28% aq. NH4OH) to afford (3R,4S)-70 as an off-white solid (90 mg, 56%). 1H NMR (500 MHz, CD3OD): δ = 8.17 (s, 1H), 7.49 (s, 1H), 3.86 - 3.83 (m, 1H), 3.81 (q, J = 13.2 Hz, 2H), 3.05 (dd, J = 9.6, 8.0 Hz, 1H), 2.74 (dd, J = 10.4, 6.3 Hz, 1H), 2.69 (dd, J = 10.4, 4.0 Hz, 1H), 2.17 (dd, J = 9.7, 8.0 Hz, 1H), 1.98 - 1.90 (m, 1H), 1.57 - 1.47 (m, 1H), 1.34 - 1.24 (m, 5H) and 0.89 ppm (t, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CD3OD): δ = 152.1, 151.0, 147.0, 130.1, 115.1, 112.6, 77.8, 62.4, 59.7, 49.1, 48.6, 34.0, 31.5, 23.8 and 14.4 ppm. ESI-HRMS for C15H24N5O [MH]+ calcd, 290.1981; found, 290.1988
(±)-Benzyl cis-3-(benzoyloxy)-4-ethylpyrrolidine-1-carboxylate [(±)-71]
Benzoic acid (430 mg, 3.5 mmol) and triphenylphosphine (909 mg, 3.5 mmol) were added to a stirred solution of (±)-12 (719 mg, 2.8 mmol) in THF (24 mL). The reaction mixture was cooled to −10 °C and DIAD (680 L, 3.5 mmol) was added drop-wise over 10 min. After stirring at −10 °C for 45 min the reaction mixture was warmed to room temperature and stirred for 22 h and then concentrated under reduced pressure. Flash chromatography of the residue (1 : 9 then 2 : 8, EtOAc : Petrol) afforded (±)-71 as a pale colorless oil (995 mg, 98%). 1H NMR (500 MHz, CDCl3): δ = 8.12 - 8.00 (m, 2H), 7.60 - 7.56 (m, 1H), 7.49 - 7.28 (m, 7H), 5.57 - 5.53 (m, 1H), 5.20 - 5.09 (m, 2H), 3.89 - 3.67 (m, 3H), 3.29 (dt, J = 19.1, 10.7 Hz, 1H), 2.35 - 2.24 (m, 1H), 1.68 - 1.46 (m, 2H) and 0.98 - 0.94 ppm (m, 3H). 13C NMR (125 MHz, CDCl3): δ = 166.0, 165.9, 154.8, 136.9, 136.7, 133.5, 133.3, 133.2, 130.2, 130.0, 129.9, 129.7, 128.5, 128.4, 127.9, 74.7, 73.8 (rotamers), 67.0, 66.9 (rotamers), 53.3, 53.0 (rotamers), 49.8, 49.5 (rotamers), 45.1, 44.3 (rotamers), 20.1 and 12.4 ppm. ESR-HRMS for C21H23NO4Na [MNa]+ calcd, 376.1525; found, 376.1521.
(±)-Benzyl cis-4-ethyl-3-hydroxypyrrolidine-1-carboxylate [(±)-72]
A solution of K2CO3 (583 mg, 4.2 mmol) in water (20 mL) was added to a solution of (±)-71 (995 mg, 2.8 mmol) in ethanol (40 mL). The resulting mixture was heated to reflux for 90 min and then allowed to cool and concentrated under reduced pressure. The residue was partitioned between DCM (50 mL) and water (50 mL) and the layers were separated and the aqueous phase was extracted with EtOAc (3 × 50 mL). The combined organic phases were washed with brine (2 × 50 mL), dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography of the residue (3 : 7, EtOAc : Petrol) afforded (±)-72 as a yellow oil (476 mg, 68%). 1H NMR (500 MHz, CDCl3): δ = 7.35 - 7.29 (m, 5H), 5.15 - 5.08 (m, 2H), 4.23 (br. s, 1H), 3.67 - 3.47 (m, 3H), 3.14 (dd, J = 10.8, 3.6 Hz, 1H), 2.24 (br. d, J = 45 Hz, 1H), 2.01 - 1.94 (m, 1H), 1.61 - 1.53 (m, 1H), 1.51 - 1.43 (m, 1H) and 0.98 - 0.94 ppm (m, 3H). 13C NMR (125 MHz, CDCl3): δ = 155.2, 155.0, 136.9, 128.4, 127.9, 127.8, 71.7, 70.7 (rotamers), 66.8, 66.7 (rotamers), 55.5, 55.0 (rotamers), 49.0, 48.8 (rotamers), 46.0, 45.3 (rotamers), 19.6 and 12.4 ppm. ESI-HRMS for C14H19NO3Na [MNa]+ calcd, 272.1263; found, 272.1268.
(±)-cis-4-Ethyl-3-hydroxypyrrolidine [(±)-73]
Palladium (10 mg, 0.01 mmol, 10 wt% on carbon) was added to a solution of (±)-72 (146 mg, 0.5 mmol) in MeOH (10 mL) under argon. The reaction mixture was placed under a hydrogen atmosphere and stirred for 1 h and then filtered through Celite® and concentrated under reduced pressure. Flash chromatography of the residue (gradient 10 to 40% (7N NH3 in MeOH) in DCM) afforded (±)-73 as a yellow oil (42 mg, 62%). 1H NMR (500 MHz, CD3OD): δ = 4.17 (td, J = 4.5, 1.4 Hz, 1H), 3.04 (dd, J = 12.2, 4.4 Hz, 1H), 2.99 (dd, J = 10.5, 7.9 Hz, 1H), 2.84 (dd, J = 12.2, 1.3 Hz, 1H), 2.61 (t, J = 10.6 Hz, 1H), 1.86 - 1.79 (m, 1H), 1.65 - 1.56 (m, 1H), 1.46 - 1.37 (m, 1H) and 0.98 ppm (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CD3OD): δ = 73.2, 56.0, 50.6, 48.6, 21.0 and 13.3 ppm. ESI-HRMS for C6H14NO [MH]+ calcd, 116.1075; found, 116.1077.
(±)-cis-1-[(9-Deaza-adenin-9-yl)methyl]-4-ethyl-3-hydroxypyrrolidine [(±)-74]
Formaldehyde (35 L, 0.4 mmol, 37 wt% solution in water) followed by 9-deazaadenine (52 mg, 0.4 mmol) were added to a solution of (±)-73 (32 mg, 0.3 mmol) in 1,4-dioxane (1 mL) and water (1 mL). The reaction mixture was stirred at room temperature for 68 h, absorbed onto silica and eluted down a silica column using a gradient 10 - 50% (7 N NH3 in MeOH) in CH2Cl2. The crude product was collected, concentrated and subjected to flash chromatography (5 : 4.9 : 0.1 then 5 : 4.8 : 0.2, CH2Cl2 : MeOH : 28% aq. NH4OH) to afford (±)-74 as an off-white solid (45 mg, 62%). 1H NMR (500 MHz, CD3OD): δ = 8.16 (s, 1H), 7.49 (s, 1H), 4.20 (td, J = 5.8, 3.3 Hz, 1H), 3.89 (s, 2H), 3.17 (dd, J = 10.9, 5.5 Hz, 1H), 2.95 (dd, J = 9.4, 7.5 Hz, 1H), 2.57 (dd, J = 10.9, 3.3 Hz, 1H), 2.41 (t, J = 9.9 Hz, 1H), 2.00 - 1.92 (m, 1H), 1.61 - 1.53 (m, 1H), 1.38 - 1.28 (m, 1H) and 0.92 ppm (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, CD3OD): δ = 152.1, 151.0, 147.0, 130.1, 115.1, 112.7, 72.5, 63.0, 58.3, 49.4, 46.6, 21.4 and 13.2 ppm. ESI-HRMS for C13H20N5O [MH]+ calcd, 262.1668; found, 262.1663.
Protein Expression and Inhibition Assays
E. coli MTAN and human MTAP used in the biological evaluation of the compounds were expressed as IPTG-inducible His-tagged recombinant enzymes purified using Ni-NTA chromatography. Enzyme inhibition assays were carried out using a xanthine oxidase-coupling enzyme that converts the adenine product of the MTAN and MTAP reactions to 2,8-dihydroxyadenine monitored at 293nm (ε293 = 15.2 mM−1cm−1). The 1 mL reaction mixture consisted of 100 mM HEPES, pH 7.4, 50 mM KCl, 250 μM MTA, 100 mM potassium phosphate (only for the MTAP reaction), and varying concentrations of inhibitor, in the presence of xanthine oxidase (Sigma). The reaction was initiated by addition of 4 nM E. coli MTAN or 10 nM human MTAP and monitored for 60 - 90 minutes at 25°C. Controls without inhibitor and without enzyme were included in the experiment. Ki values were determined using the following equation for competitive inhibition:
where v0’ and v0 are initial rates in the presence and absence of inhibitor, respectively; Km and [S] are the Michaelis constant and concentration of MTA, respectively; and Ki and [I] are the inhibition constant and inhibitor concentration, respectively. If [I] is less than ten times the enzyme concentration, the following correction is applied:
where I’ is the effective inhibitor concentration to be used in the equation for competitive inhibition above; and Et is the final enzyme concentration. In some instances, a second slower rate phase is observed in progress curves of tight binding inhibitors. These steady state rates are taken instead (vs’/vs) and used in the inhibition equation above to determine the overall inhibition constant Ki*, which reflects the slow onset inhibition of the enzyme-inhibitor complex.
Acknowledgment
This work was supported by the New Zealand Foundation for Research, Science and Technology and the US National Institute of Health Grant GM41916.
Footnotes
Abbreviations: AI, autoinducer; 9-DAA, 9-deazaadenine; DADMe, 5′-deaza-1′-aza-2′-deoxy-1′-(9-methylene); DIAD, diisopropyl azodicarboxylate, ImmA, immucillin-A; MT, methylthio; MTA, methylthioadenosine; MTAN, 5′-Methylthioadenosine/S-adenosylhomocysteine nucleosidase; MTAP, 5′-Methylthioadenosine phosphorylase; PNP, purine nucleoside phosphorylase; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine; SRH, S-ribosylhomocysteine
for clarity only the data for (±)-15 is quoted.
References
- 1.Schramm VL. Enzymatic Transition State Theory and Transition State Analogue Design. J. Biol. Chem. 2007;282:28297–28300. doi: 10.1074/jbc.R700018200. [DOI] [PubMed] [Google Scholar]
- 2.Lewandowicz A, Schramm VL. Transition State Analysis for Human and Plasmodium falciparum Purine Nucleoside Phosphorylases. Biochemistry. 2004;43:1458–1468. doi: 10.1021/bi0359123. [DOI] [PubMed] [Google Scholar]
- 3.Singh V, Schramm VL. Transition state structure of human 5′-Methylthioadenosine phosphorylase. J. Am. Chem. Soc. 2006;128:14691–14696. doi: 10.1021/ja065419p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh V, Lee JE, Nunez S, Howell PL, Schramm VL. Transition state structure of 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase from Escherichia Coli and its similarity to transition state analogues. Biochemistry. 2005;44:11647–11659. doi: 10.1021/bi050863a. [DOI] [PubMed] [Google Scholar]
- 5.Singh V, Luo M, Brown RL;, Norris GE;, Schramm VL. Transition state structure of Neisseria meningitides 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase. J. Am. Chem. 2007;129:13831–13833. doi: 10.1021/ja0754204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh V, Schramm VL. Transition state analysis of S. pneumoniae 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase. J. Am. Chem. Soc. 2007;129:2783–2795. doi: 10.1021/ja065082r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans GB. The Synthesis of N-Ribosyl Transferase Inhibitors Based on a Transition State Blueprint. Aus. J. Chem. 2004;57:837–854. [Google Scholar]
- 8.Basu I, Cordovano G, Das I, Belbin TJ, Guha C, Schramm VL. A Transition State Analogue of 5′-Methylthioadenosine Phosphorylase Induces Apoptosis in Head and Neck Cancers. J. Biol. Chem. 2007;282:21477–21486. doi: 10.1074/jbc.M702287200. [DOI] [PubMed] [Google Scholar]
- 9.Gutierrez JA, Luo M, Singh V, Li L, Brown RL, Norris GE, Evans GB, Furneaux RH, Tyler PC, Painter GF, Lenz DH, Schramm VL. Picomolar Inhibitors as Transition-State Probes of 5′-Methylthioadenosine nucleosidases. ACS Chem. Biol. 2007;2:725–734. doi: 10.1021/cb700166z. [DOI] [PubMed] [Google Scholar]
- 10.Stroeher UH, Paton AW, Ogunniyi AD, Paton JC. Mutation of luxS of Streptococcus pneumoniae affects virulence in a mouse model. Infect. Immun. 2003;71:3206–3212. doi: 10.1128/IAI.71.6.3206-3212.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winzer K, Sun YH, Green A, Delory M, Blackley D, Hardie KR, Baldwin TJ, Tang CM. Role of Neisseria meningitidis luxS in cell-to-cell signaling and bacteremic infection. Infect. Immun. 2002;70:2245–2248. doi: 10.1128/IAI.70.4.2245-2248.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parsek MR, Val DL, Hanzelka BL, Cronan JE, Jr, Greenberg EP. Acyl homoserine-lactone quorum-sensing signal generation. Proc. Natl. Acad. Sci. U.S.A. 1999;96:4360–4365. doi: 10.1073/pnas.96.8.4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schauder S, Shokat K, Surette MG, Bassler BL. The LuxS family of bacterial autoinducers: Biosynthesis of a novel quorum-sensing signal molecule. Mol. Microbiol. 2001;41:463–476. doi: 10.1046/j.1365-2958.2001.02532.x. [DOI] [PubMed] [Google Scholar]
- 14.Thomas T, Thomas TJ. Polyamines in cell growth and cell death: molecular mechanisms and therapeutic applications. Cell. Mol. Life Sci. 2001;58:244–258. doi: 10.1007/PL00000852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marton LJ;, Pegg AE. Polyamines as targets for therapeutic intervention. Annu. Rev. Pharmacol. Toxicol. 1995;35:55–91. doi: 10.1146/annurev.pa.35.040195.000415. [DOI] [PubMed] [Google Scholar]
- 16.Seiler N, Atanassov CL, Raul F. Polyamine metabolism as target for cancer chemoprevention (review) Int. J. Oncol. 1998;13:993–1006. doi: 10.3892/ijo.13.5.993. [DOI] [PubMed] [Google Scholar]
- 17.Chattopadhyay S, Zhao R, Tsai E, Schramm VL, Goldman ID. The effect of a novel transition state inhibitor of methylthioadenosine phosphorylase on pemetrexed activity. Mol. Cancer Ther. 2006;5:2549–2555. doi: 10.1158/1535-7163.MCT-06-0313. [DOI] [PubMed] [Google Scholar]
- 18.Singh V, Evans GB, Lenz DH, Mason JM, Clinch K, Mee S, Painter GF, Tyler PC, Furneaux RH, Lee JE, Howell PL, Schramm VL. Femtomolar Transition Analogue Inhibitors of 5′-Methylthioadenosine/S-Adenosylhomocysteine Nucleosidase from Escherichia coli. J. Biol. Chem. 2005;280:18265–18273. doi: 10.1074/jbc.M414472200. [DOI] [PubMed] [Google Scholar]
- 19.Evans GB, Furneaux RH, Lenz DH, Painter GF, Schramm VL, Singh V, Tyler PC. Second Generation Transition State Analogue Inhibitors of Human 5′-Methylthioadenosine Phosphorylase. J. Med. Chem. 2005;48:4679–4689. doi: 10.1021/jm050269z. [DOI] [PubMed] [Google Scholar]
- 20.Gutierrez JA, Crowder T, Rinaldo-Matthis A, Ho M-C, Almo SC, Schramm VL. Transition state analogs of 5′-Methylthioadenosine nucleosidase disrupt quorum sensing. Nat. Chem. Biol. 2009;5:251–257. doi: 10.1038/nchembio.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schramm VL. Unpublished results. [Google Scholar]
- 22.Evans GB, Furneaux RH, Tyler PC, Schramm VL. Synthesis of a Transition State Analogue Inhibitor of Purine Nucleoside Phosphorylase via the Mannich Reaction. Org. Lett. 2003;5:3639–3640. doi: 10.1021/ol035293q. [DOI] [PubMed] [Google Scholar]
- 23.Hansen SU, Bols M. 1-Azaribofuranoside Analogues as Designed Inhibitors of Purine Nucleoside Phosphorylase. Synthesis and Biological Evaluation. Acta Chem. Scand. 1998;52:1214–1222. doi: 10.3891/acta.chem.scand.52-1214. [DOI] [PubMed] [Google Scholar]
- 24.Rives A, Génisson Y, Faugeroux V, Zedde C, Lepetit C, Chauvin R, Andrieu-Abadie N, Colié S, Levade T, Baltas M. Highly Regioselective Oxirane Ring-Opening of a Versatile Epoxypyrrolidine Precursor of New Imino-Sugar-Based Sphingolid Mimics. Eur. J. Org. Chem. 2009:2474–2489. [Google Scholar]
- 25.Cren S, Gurcha SS, Blake AJ, Besra GS, Thomas NR. Synthesis and biological evaluation of new inhibitors of UDP-Galf transferase-a key enzyme in M. tuberculosis cell wall biosynthesis. Org. Biomol. Chem. 2004;2:2418–2420. doi: 10.1039/B411554F. [DOI] [PubMed] [Google Scholar]
- 26.Kamal A, Shaik AA, Sandbhor M, Malik MS, Azeeza S. Chemoenzymatic synthesis of (3R,4S)- and (3S,4R)-3-methoxy-4-methylaminopyrrolidine. Tetrahedron: Asymmetry. 2006;17:2876–2883. [Google Scholar]
- 27.Yamaguchi M, Hirao I. An efficient Method for the Alkynylation of oxiranes using alkynyl Boranes. Tetrahedron Lett. 1983;24:391–394. [Google Scholar]
- 28.Tron GC, Pirali T, Billington RA, Canonico PL, Sorba G, Genazzani AA. Click Chemistry Reactions in Medicinal Chemistry: Applications of the 1,3-dipolar Cycloaddtion Between Azides and Alkynes. Medicinal Research Reviews. 2008;28:278–308. doi: 10.1002/med.20107. [DOI] [PubMed] [Google Scholar]
- 29.Tsuzuki Y, Chiba K, Mizuno K, Tomita K, Suzuki K. Practical Synthesis of (3S, 4S)-3-methoxy-4-methylpyrolidine. Tetrahedron: Asymmetry. 2002;12:2989–2997. [Google Scholar]
- 30.Tornoe CW, Christensen C, Meldal M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 31.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 32.Kalisiak J, Sharpless KB, Fokin VV. Efficient Synthesis of 2-Substituted-1,2,3-triazoles. Org. Lett. 2008;10:3171–3174. doi: 10.1021/ol8006748. [DOI] [PubMed] [Google Scholar]
- 33.Loren JC, Krasiński, Fokin VV, Sharpless KB. NH-1,2,3-Triazoles from Azidomethyl Pivalate and Carbamates: Base-Labile N-Protecting Groups. Synlett. 2005:2847–2850. [Google Scholar]
- 34.Becer CR, Hoogenboom R, Schubert US. Click Chemistry beyond Metal-Catalyzed Cycloaddition. Angew. Chem. Int. Ed. 2009;48:4900–4908. doi: 10.1002/anie.200900755. [DOI] [PubMed] [Google Scholar]
- 35.Heidecke CD, Lindhorst TK. Iterative Synthesis of Spacered Glycodendrons as Oligomannoside Mimetics and Evaluation of Their Antiadhesive Properties. Chem. Eur. J. 2007;13:9056–9067. doi: 10.1002/chem.200700787. [DOI] [PubMed] [Google Scholar]
- 36.Henin F, Muzart J. Cis- and Trans-Opening of Oxirane by Vinyl Grignard Reagents. Synth. Commun. 1984:1355–1358. [Google Scholar]
- 37.Clinch K, Evans GB, Furneaux RH, Lenz D, Mason JM, Mee SP, Tyler PC, Wilcox SJ. A Practical Synthesis of (3R, 4R)-N-tert-butoxycarbonyl-4-hydroxymethylpyrrolidin-3-ol. Org. Biomol. Chem. 2007;5:2800–28002. doi: 10.1039/b708796a. [DOI] [PubMed] [Google Scholar]
- 38.Mitsunobu O. The Use of Diethyl Azodicarboxylate and Triphenylphosphine in Synthesis and Transformation of Natural Products. Synthesis. 1981:1–28. [Google Scholar]
- 39.Clinch K, Evans GB, Fleet GW, Furneaux RH, Johnson SW, Lenz D, Mee SP, Rands PR, Schramm VL, Taylor Ringia EA;, Tyler PC. Syntheses and bio-activities of the L-enantiomers of two potent transition state analogue inhibitors of purine nucleoside phosphorylases. Org. Biomol. Chem. 2006;4:1131–1139. doi: 10.1039/b517883e. [DOI] [PubMed] [Google Scholar]
- 40.Murkin AS, Tyler PC, Schramm VL. Transition State Interactions Revealed in Purine Nucleoside Phosphorylase by Binding Isotope effects. J. Am. Chem. Soc. 2008;130:2166–2167. doi: 10.1021/ja7104398. [DOI] [PubMed] [Google Scholar]