

Abstract
Post-transcriptional chemical covalent modification of adenosine, guanosine, uridine and cytidine occurs frequently in all types of ribonucleic acids (RNAs). In ribosomal RNA (rRNA) and transfer RNA (tRNA) these modifications make important contributions to RNA structure and stability and to the accuracy and efficiency of protein translation. The functional dynamics, synergistic nature and regulatory roles of these posttranscriptional nucleoside modifications within the cell are not well characterized. These modifications are present at very low levels and isolation of individual nucleosides for analysis requires a complex multi-step approach. The focus of this study is to characterize the reproducibility of a liquid chromatography method used to isolate and quantitatively characterize modified nucleosides in tRNA and rRNA when nucleoside detection is performed using ultraviolet and mass spectrometric detection (UV and MS, respectively). Despite the analytical challenges of sample isolation and dynamic range, quantitative profiling of modified nucleosides obtained from bacterial tRNAs and rRNAs is feasible at relative standard deviations of 5% RSD or less.
Keywords: tRNA, rRNA, HPLC, Posttranscriptional modification, Metabolites
1. Introduction
Chemical modification of RNA occurs post-transcriptionally in all types of RNA. Currently there are 109 known structurally unique RNA nucleoside modifications, which include base or ribose methylation, isomerization of uridine to pseudouridine, thiolation, alkylation, acetylation and complex hypermodifications [1]. In tRNA, post-transcriptional modifications in the anticodon region directly influence decoding [2,3] and reading frame maintenance[4–6]. Modifications have also been shown to contribute to recognition of the correct aminoacyl synthetase for each tRNA [7]. In tRNA, modification status varies depending on cellular conditions such as temperature, nutrient availability, growth phase, growth rate and cell cycle progression [8–14]. Some studies suggest definite modification networks exist for the control or adaptation to changing cellular conditions and tRNA degradation [8–10]. The synergistic nature of these modifications and the mechanisms of dynamic control of RNA modification have not been explored, primarily due to lack of methods to quantitatively evaluate the global degree of modification in cellular RNA.
Post-transcriptional RNA modifications are located in functionally important regions such as the peptidyl transferase center of the ribosome and the anticodon of transfer RNA [11,12]. Several studies have shown ribosome function can also be attributed to specific rRNA modifications [13–20]. RNA modifications outside the functional centers are important in maintaining tertiary structure, for example, by contributing rigidity through enhanced base stacking or increasing flexibility [21] in the RNA biopolymer. The effects of the absence or presence of most individual modifications, especially outside of the functional center, is not easily evaluated, rarely results in any observable phenotype and often does not appear to affect function. However, combinations of modification deficiencies in rRNA will negatively impact cell growth, ribosome production and translation [15,19,22–24]. Characterization of modification status of large RNA molecules requires enzymatic hydrolysis of tRNA or rRNA to yield individual nucleosides. The extent of variation in the identity and relative quantities of modified nucleosides present in this complete profile under different cellular conditions is a valuable measure of global modification status that can be used to assess the dynamic nature of RNA modification.
For the analysis of modified nucleosides a separation technique such as HPLC, UPLC, GC or CE is usually combined with UV or mass spectrometry (MS)-based detection [25,26]. Reversed phase HPLC with UV detection is the most commonly used method for the quantitation of modified nucleosides during modification profiling [27]. The aromatic nature of nucleosides yields characteristic UV spectra, which along with relative retention time determined by suitable reference standards can be used for positive identification. A more powerful approach for nucleoside identification is to couple reversed phase HPLC to electrospray ionization mass spectrometry (ESI-MS). During ESI-MS, the glycosidic bond between the nucleobase and ribose of the nucleoside fragments easily yielding the characteristic protonated molecular ion of the nucleoside and a protonated nucleobase ion. Further tandem mass spectrometry (MS/MS) via collision-induced dissociation (CID) of the nucleobase ion yields ring opening of the base and produces fragments containing even greater structural detail. Accurate mass measurements of molecular ions, base ions and fragments made with high resolution mass spectrometers can also be used to confirm nucleoside identity. The MS data along with retention time and UV absorbance characteristics further confirm nucleoside structure [28].
Increasingly, LC/MS is being used for the quantitative analysis of modified nucleosides in urine [29,30]. Recently, quantitative studies of urinary nucleosides have used MS/MS techniques based on the known fragmentation behavior of nucleosides to monitor multiple molecular ion to nucleobase ion transitions (multiple reaction monitoring, MRM) of several nucleosides known to be present [30]. This type of MS/MS scan is both selective and sensitive, albeit some what limited in the number of nucleosides that can be characterized in one experiment.
The reproducibility of LC/UV and various LC/MS techniques for the quantitative analysis of urinary nucleosides has been reported[29–34]. Those methods were characterized using a set of urinary nucleoside standards and yielded reproducibilities in the range of 1–15% RSD using either UV or MS as the detector. When quantifying biologically isolated urinary nucleosides with UV detection, reproducibility ranges from a low of 1% RSD for well-behaved urinary nucleosides to as high as 30–35% RSD for poorly behaved urinary nucleosides. Among the various MS/MS techniques used for quantification, Wang et al. characterized nine urinary nucleosides with a reproducibility ranging from 5 to 16% RSD using selected ion monitoring MS [33]. More recently, Teichert et al. used constant neutral loss scans to quantify 35 urinary nucleosides with reproducibilities ranging from 6 to 50% RSD [34].
The significant differences between quantifying urinary nucleosides and modified nucleosides present within cellular RNAs arise from the multi-step procedures required for rRNA and tRNA isolation from cells and the presence of large amounts of unmodified (major) nucleosides generated after enzymatic digestion of those isolated RNAs. Despite these challenges, similar LC/MS methods have been used extensively in the identification and structural characterization of post-transcriptional modifications in tRNA and rRNA. The total census of modifications present in tRNA and rRNA of several different organisms have been determined using LC/MS, often with on-line UV detection as well [35–39]. Recently, accurate relative and absolute quantification of modified nucleosides has been demonstrated through the use of stable-isotope labeling [40–42]. Although the use of stable-isotope labeling can yield accurate and precise measurements on the number of modified nucleosides present in the sample, it cannot be used in a general strategy for total nucleoside analysis unless the specific modifications are limited to those with stable-isotope analogs [40,41] or the organism can easily be cultured in labeled media [42].
Additional analytical challenges faced during the quantitative analysis of modified nucleosides in tRNAs and rRNAs are dynamic range and specificity of analysis. For example, in the bacterial ribosome of Escherichia coli, only 11 nucleotides are modified of the total 1542 nucleotides present in 16S RNA of the small subunit; similarly, only 23 nucleotides are modified of 2904 nucleotides in the large subunit 23S RNA. Because modification occurs at very low levels, 0.03–0.06% of total nucleotides, an analytical method must be capable of detecting such low levels of modified nucleosides in a background of the significantly more abundant major (unmodified) nucleosides. While such a dynamic range is typically not too challenging for modern LC/MS approaches, it does place significant strains on the upfront chromatographic separation of modified nucleosides.
In this work, we have explored the overall utility of reversed phase HPLC separation of modified nucleosides in an analytical method using LC/MS and LC/UV for both total nucleoside identification and quantitation. One particular interest of this study was to characterize the reproducibility of these methods when attempting to quantify modified nucleosides from RNA isolated from replicate biological samples. To date there have been no reports describing the reproducibility of results one would obtain from isolating tRNAs and/or rRNAs and quantifying their constituent modified nucleosides generated by total nucleoside digestion across multiple samples. Large variability in method precision diminishes the significance of the results, and if the variability is too great important differences could be missed. By establishing the overall method and sample variability for measuring modified nucleosides from tRNA and rRNA without resorting to stable isotope labeling methods, we are now able to define the statistical significance of changes in modification status when comparing biological systems of varying environmental or physiological cellular conditions, which should enable future studies aimed at measuring relative changes in modification profiles under these experimental conditions.
2. Experimental
2.1. Materials
Culture media used was Bacto tryptone and Bacto yeast extract from Becton, Dickinson & Co (Sparks, MD). Lysozyme (from chicken egg), urea, ethanol, isopropanol, sucrose, Tri-Reagent, Nuclease P1, nucleoside test mix, ammonium chloride, ammonium acetate, acetic acid, Tris–HCl, potassium chloride, magnesium acetate, and sodium citrate used in buffers were purchased from Sigma (St. Louis, MO). Sodium chloride, ammonium bicarbonate and EDTA were obtained from Fisher Scientific (Fairlawn, NJ). RNase free DNase was purchased from Promega (Madison, WI). Phenol/chloroform/isoamylalcohol (25:24:1) solution was obtained from Ambion (Austin, TX). Nucleobond AXR-80 gravity flow columns were purchased from Machery-Nagel (Bethlehem, PA). Snake venom phosphodiesterase I was purchased from Worthington Biochemicals (Lakewood, NJ) and antartic phosphatase from New England Biolabs (Ipswich, MA). Nanopure water (18 Mohms) from a Barnstead (Dubuque, IA) filtering system was autoclaved and used in all buffers and solutions.
2.2. Escherichia coli culturing
An Innova 4000 incubator from New Brunswick Scientific (Edison, NJ) was used for culturing. RNA concentration and cell density measurements were made on a Shimadzu Biospec 1601 UV/Vis spectrometer (Columbia, MD) at 260 nm and 600 nm, respectively. A Sorvall RC5C preparative centrifuge was used to pellet bacterial cells. A Beckman Coulter Optima L-XP ultracentrifuge (Fullerton, CA) was used for ribosome subunit separation and pelleting.
Bacterial cultures were prepared by inoculating 100 mL of Luria Broth with a small aliquot of E. coli MRE 600 stock culture and incubating at 37 °C for 16 h with agitation. This culture was then added to 900 mL of fresh Luria Broth and incubated at 30 °C until mid-log phase (OD 0.5–0.7 at 600 nm). Cells were harvested by centrifugation at 10,000 × g for 15 min at 4 °C and washed with buffer (20 mM Tris–HCl, 10.5 mM magnesium acetate, 0.5 mM EDTA, pH 7.5).
2.3. Isolation and purification of RNA
Procedures used for isolation of ribosomes and ribosomal subunits were adapted from previously described standard protocols [43]. The bacterial cells were physically lysed in a French press at 12,000 psi (American Instrument Co., Silver Spring, MD) and cell debris removed via centrifugation. The cell lysate was treated with RNase-free DNase (2.5 units DNase/g of cell). Crude ribosomes were isolated via centrifugation through a 1.1 M sucrose cushion. The resulting 70S pellet was resuspended in a low magnesium buffer (20 mM Tris–HCl at pH 7.6, 10.5 mM magnesium acetate, and 30 mM ammonium chloride) and allowed to dissociate into 50S and 30S subunits overnight at 4 °C. The subunits were separated through a 0–45% sucrose gradient by centrifugation on a Beckman L-XP ultracentrifuge at 19,000 rpm for 17 h in a SW28 rotor. The 30S and 50S subunit fractions were collected, pooled and pelleted via centrifugation at 48,000 rpm for 18 h. Subunits were resuspended in 20 mM Tris–HCl, 10.5 magnesium acetate, 30 mM ammonium chloride, pH 7.6 and stored at −80 °C.
Two methods were used to isolate 16S rRNA. In the first, ribosomal subunits were deproteinized using phenol/chloroform extraction followed by ethanol precipitation. Alternatively, ribosomal proteins were denatured with 6 M urea and the 16S RNA was purified using a Nucleobond (Machery-Nagel) ion exchange column. Denatured subunits were loaded onto the column in a solution of 500 mM potassium chloride, 100 mM tris acetate (pH 6.3) and 15% ethanol then washed with the same solution. A solution of 800 mM potassium chloride, 100 mM tris acetate (pH 6.3) and 15% ethanol was used for a second wash followed by elution of rRNA with 1.5 M potassium chloride, 100 mM tris acetate (pH6.3) 15% ethanol. Isopropanol was used to precipitate the rRNA and the pelleted rRNA was resuspended in autoclaved water.
For tRNA isolation, lysozyme (1 mg/mL) was used for cell lysis. Total tRNA was isolated by guanidinium thiocyanate–phenol– bromochloropropane extraction. rRNA was separated from tRNA in a solution of high salt concentration and stepwise precipitation with isopropanol. Salts and phenol were removed using the Nucleobond ion exchange column as described above. Buffer concentrations were lower for tRNA purification: loading buffer, wash buffer and elution buffers were 200 mM, 400 mM and 750 mM potassium chloride, respectively.
2.4. Enzymatic digestion of RNA
Prior to enzymatic digestion of the RNA to nucleosides, the RNA was denatured at 100 °C for 3 min then chilled in an ice water bath. To lower the pH, 1/10 volume 0.1 M ammonium acetate (pH 5.3) was added. For each 0.5 AU of RNA, 2 units Nuclease P1 was added and incubated at 45 °C for 2 h. The pH was readjusted by adding 1/10 volume of 1.0 M ammonium bicarbonate, then 0.002 units of snake venom phosphodiesterase was added and incubated at 37 °C for 2 h. Finally, 0.5 units of antartic phosphatase was added and incubated at 37 °C for 60 min. The nucleoside digests were stored at −80 °C [44].
2.5. LC–UV and LC–MS conditions
Analysis of the nucleoside test mix and nucleoside digests of RNA by analytical HPLC were done using a Hitachi D-7000 HPLC equipped with a diode array detector. A 4.6 mm × 250 mm Supelcosil LC-18-S 5 μm particle reversed phase column with a 4.0 mm × 20 mm Supelguard LC-18-S guard column was used at a flow rate of 2.0 mL/min and temperature controlled at 30 °C. Mobile phases used were 250 mM ammonium acetate, pH 6.0 (Buffer A) and 40% aqueous acetonitrile (Buffer B). A multilinear gradient was used with only minor modification from that described previously [28].
Analysis of the nucleoside test mix and nucleoside digests of RNA by narrow bore HPLC were done using a Hitachi D-7000 HPLC system. A 2.1 mm × 250 mm Supelcosil LC-18-S (5 μm particle) reversed phase column was used at a flow rate of 300 μL/min and either a temperature controlled 30 °C or a room temperature of approximately 20 °C. Mobile phases used were 5 mM ammonium acetate, pH 5.3 (Buffer A) and 40% aqueous acetonitrile (Buffer B). The gradient used was the same as for analytical HPLC, with slight adjustments for column size differences. The column eluent was split immediately post column, 1/3 to a Thermo LTQ-XL ion trap mass spectrometer and 2/3 to a Hitachi D-7400 UV detector set at 260 nm.
A Thermo LTQ-XL ion trap mass spectrometer equipped with an ion max electrospray source was used for the LC/MS identification of nucleosides. Mass spectra were recorded in the positive ion mode over an m/z range of 105–510 with a capillary temperature of 275 °C, spray voltage of 3.7–4.0 kV and sheath gas, auxiliary gas and sweep gas of 45, 25 and 10 arbitrary units, respectively. Data dependent MS/MS of each of the two most intense ions were recorded throughout the LC/MS run.
3. Results and discussion
3.1. Nucleoside quantitation: technical reproducibility
The LC/MS methods used here are based on several well-established methods for the identification of post-transcriptional modifications in enzymatic digests of RNA [27,28]. In this work the instrumental and biological reproducibility were determined for the quantitative analysis of modified nucleosides in enzymatic digests of tRNA and rRNA. The reproducibility was evaluated using reversed phase analytical (4.6 mm × 250 mm) and narrow bore columns (2.1 mm × 250 mm), both frequently used for the analysis of nucleosides in RNA [10,24,37,39]. The analytical column has greater loading capacity, does not require a low volume detector flow cell and needs less equilibration time between runs than the narrow bore column. The analytical column is useful in the analysis of nucleoside digests of rRNA where it is necessary to load a larger amount due to the dynamic range requirements, for example, in 16S RNA where only 11 of the 1542 nucleotides are modified. The advantages of the narrow bore column are reduced solvent consumption, increased sensitivity and greater compatibility with electrospray ionization due to the lower flow rate and smaller column volume. The narrow bore column is most often used when MS-based nucleoside identification is required. The analysis conditions were the same for both columns except different HPLC instruments were used and the mobile phase flow rate and concentration of ammonium acetate were reduced as required for compatibility with the electrospray process.
Before examining multiple biological samples, the technical precision of the basic HPLC method under several common instrumental configurations was evaluated by performing replicate analyses of a commercially available nucleoside test mix over multiple days. Such information is important to identify whether differences in instrumental configurations would affect subsequent measurements of biological samples and to identify the reproducibility baselines prior to analyzing biological samples. The test mix contained 12 nucleosides found frequently in RNA: pseudouridine (ψ), cytidine (C), uridine (U), 2-thiocytidine (s2C), 3-methylcytidine (m3C), 5-methylcytidine (m5C), 2′-O-methylcytidine (Cm), 1-methyladenosine (m1A), inosine (I), 5-methyluridine (m5U), guanosine (G), and 7-methylguanosine (m7G). The identification of nucleosides in the test mix was based on relative retention times and either UV absorbance characteristics (ratio of absorbance at 260/280 nm) for the analytical column or ESI-MS spectral characteristics for the narrow bore column.
A representative UV chromatogram of the test mix separated on the analytical column is shown in Fig. 1. The instrumental analysis reproducibility, determined as the variability in peak areas and retention times, is summarized in Table 1. Replicate injections (n = 4) yielded relative standard deviations (expressed as a percentage, % RSD) for peak areas with a range of 2.9–4.3% RSD and an average of 3.4% RSD, regardless of the amount of any individual nucleoside injected. Reproducibility of nucleoside retention times was between 1.5% and 3.2% with an average of 2.1% RSD for the analytical column.
Fig. 1.
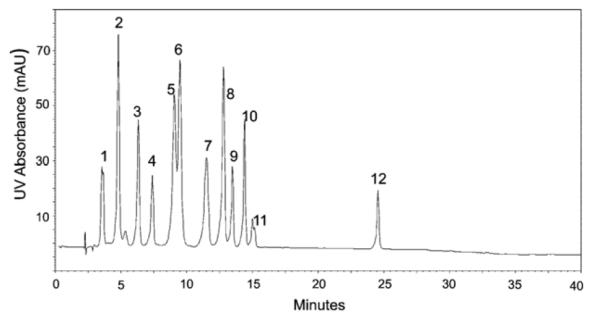
UV chromatogram of nucleoside test mix, 4.6 mm × 250 mm/LC-18S column, 30 °C, 250 mM ammonium acetate pH 6.0 (A) 40% acetonitrile in water (B), 2.0 mL/min. UV detection @ 260 nm.
Table 1.
Reproducibility of UV peak area and retention time for analysis of modified nucleosides in standard test mix, 4.6 mm × 250 mm column used (n = 4).
| Peak | Nucleoside | % RSD peak area | Avg. tr (min) | % RSD tr |
|---|---|---|---|---|
| 1 | Pseudouridine | 2.9 | 3.4 | 2.8 |
| 2 | Cytidine | 3.6 | 4.6 | 1.7 |
| 3 | Uridine | 3.4 | 6.2 | 2.0 |
| 4 | 2-Thiocytidine | 3.4 | 7.3 | 2.0 |
| 5 | 3-Methylcytidine | 3.1 | 9.1 | 3.2 |
| 6 | 5-Methylcytidine | 4.3 | 9.5 | 2.7 |
| 7 | 2′-O-methylcytidine | 3.2 | 11.5 | 2.3 |
| 8 | 5-Methyluridine | 3.1 | 12.8 | 1.8 |
| 9 | Inosine | 3.6 | 13.5 | 1.7 |
| 10 | Guanosine | 3.3 | 14.4 | 1.5 |
| 11 | 7-Methylguanosine | 3.1 | 15.0 | 1.8 |
| 12 | N6-methyladenosine | 3.5 | 24.6 | 1.6 |
A similar evaluation of the reproducibility of the narrow bore column was performed. Two different temperatures, 20 °C and 30 °C, were used for the analysis of nucleosides in the RNA hydrolysates to enable separate optimization for tRNA and rRNA nucleosides, respectively. Fig. 2 contains representative UV chromatograms for the nucleoside test mix, with variability in peak areas and retention times summarized in Table 2 (analysis at 20 °C) and Table 3 (analysis at 30 °C). Peak area RSDs were statistically no different for the narrow bore column at these two temperatures (0.8–5.5% RSD at 20 °C and 1.0–5.8% RSD at 30 °C, F-test <19.6). Variability in retention time was greater (0.4–3.8% RSD) for replicate analyses performed at 20 °C than analyses run at 30 °C (0.2–1.1% RSD), likely due to better thermostatting at 30 °C. Retention time differences between the analytical and narrow bore columns can be attributed to the difference in dead volumes and gradient delays in the two different instruments used and the lower flow rate used (300 μL/min). The method precision reported as percent relative standard deviation in peak areas of the standard test mix here is similar to the method precision reported in studies of modified nucleosides in urine [30,33,34,45].
Fig. 2.

UV chromatogram of nucleoside test mix, 2.1 mm × 250 mm/LC-18S column, 30 °C, 5 mM ammonium acetate pH 5.3 (A) 40% acetonitrile in water (B), 0.3 mL/min. UV detection @ 260 nm. (a) 20 °C and (b) 30 °C.
Table 2.
Reproducibility of UV peak areas and retention times for modified nucleoside in standard test mix, 2.1 mm × 250 mm column used, 20 °C.
| Peak | Nucleoside | % RSD UV peak area | % RSD tr |
|---|---|---|---|
| 1 | Pseudouridine | 0.8 | 1.5 |
| 2 | Cytidine | 2.7 | 3.2 |
| 3 | Uridine | 4.3 | 2.9 |
| 4 | 2-Thiocytidine | 3.1 | 3.1 |
| 5 | 5-Methylcytidine | 1.0 | 3.8 |
| 6 | 3-Methylcytidine | 5.5 | 3.6 |
| 7 | 2′-O-methylcytidine | 4.1 | 2.7 |
| 8 | 1-Methyladenosine | 4.1 | 1.4 |
| 9 | Inosine | 1.9 | 1.3 |
| 10 | 5-Methyluridine | 3.2 | 1.3 |
| 11 | Guanosine | 2.0 | 1.2 |
| 12 | 7-Methylguanosine | 3.0 | 1.0 |
| 13 | 3-Methyluridine | 1.0 | 0.8 |
| 14 | N6-methyladenosine | 0.9 | 0.4 |
Table 3.
Reproducibility of UV and MS peak areas and retention times for modified nucleosides (both molecular ion [MH+] and base ion [BH2 +]) in standard test mix, 2.1 mm × 250 mm column used, 30 °C.
| Peak | Nucleoside/base | (m/z) | % RSD UV peak area |
% RSD tr |
% RSD MS peak area |
|---|---|---|---|---|---|
| 1 | Pseudouridine | (245) | 3.0 | 0.2 | 5.3 |
| (209) | 3.7 | ||||
| 2 | Cytidine | (244) | 3.0 | 0.2 | 5.8 |
| (112) | 6.3 | ||||
| 3 | Uridine | (245) | 3.4 | 0.2 | 11.6 |
| (113) | 1.9 | ||||
| 4 | 2-Thiocytidine | (260) | 4.3 | 0.6 | 1.3 |
| (128) | 1.2 | ||||
| 5 | 5-Methylcytidine | (258) | 3.0 | 0.8 | 4.8 |
| (126) | 8.5 | ||||
| 6 | 3-Methylcytidine | (258) | 3.6 | 0.9 | 1.3 |
| (126) | 4.8 | ||||
| 7 | 2′-O-methylcytidine | (258) | 1.0 | 1.1 | 7.7 |
| (112) | 4.4 | ||||
| 8 | 1-Methyladenosine | (282) | 2.0 | 0.7 | 1.2 |
| (150) | 6.0 | ||||
| 9 | Inosine | (269) | 1.9 | 0.7 | 3.6 |
| (137) | 10.7 | ||||
| 10 | 5-Methyluridine | (259) | * | 1.4 | 7.4 |
| (127) | 7.8 | ||||
| 11 | Guanosine | (284) | 3.2 | 0.3 | 5.6 |
| (152) | 5.9 | ||||
| 12 | 7-Methylguanosine | (298) | 5.8 | 0.2 | 4.7 |
| (166) | 8.2 | ||||
| 13 | 3-Methyluridine | (259) | 5.8 | 0.3 | 5.2 |
| (127) | 12.4 | ||||
| 14 | N6-methyladenosine | (282) | 5.1 | 0.8 | 2.2 |
| (150) | 1.0 |
denotes peaks not well resolved.
Analytically useful differences in modified nucleoside retention and separation were observed during the temperature studies. The major difference in separation of tRNA vs. rRNA nucleosides arises from the similar retention times for 7-methylguanosine and guanosine. 7-Methylguanosine has been detected in E. coli rRNA, although it is a rare modification, whereas this modification is much more common in tRNAs. The large dynamic range between these two nucleosides in rRNA digests requires chromatography at the higher temperature (30 °C) to improve their separation. Improved separation of 3-methyl from 5-methylcytidine as well as 5-methyluridine from inosine was noted when the chromatography was performed at 20 °C. Because tRNAs contain these modified nucleosides, subsequent experiments probing the biological reproducibility of tRNA analyses was performed at 20 °C.
To compare the precision of quantitative measurements of nucleosides using UV detection with mass spectrometry detection, MS peak areas were also determined for each of the nucleosides in the test mix by extracted ion chromatograms (XICs) for each nucleoside molecular ion (MH+) and base ion (BH2+). Because nucleosides containing an N—C glycosidic bond can fragment easily during ESI-MS, leading to the formation of the BH2+ ion, peak areas for both ions were measured. Pseudouridine contains a more stable C—C bond rather than the C—N glycosidic bond and does not fragment to yield a base ion. However, pseudouridine fragments easily forming a characteristic electrospray spectra with an abundant ion at m/z 209, which results from the elimination of 2 water molecules [46] and this ion was measured.
For MS detection, the average percent RSD in peak areas for the ions measured was 5.9% RSD (Table 3). Variability in MS peak areas in this study ranged from 1.0 to 12.4%, which is comparable that reported for similar MS studies of modified nucleosides present in urine [30,33,34,45]. The relative standard deviations are slightly higher for the MS peak areas of most nucleoside ions when compared to the UV peak areas. In addition, differences between molecular ion and base ion precisions are noted for several of the test mix components (e.g., uridine, inosine). Because the experimental conditions utilized source-induced fragmentation to generate the base ions, there is no optimization of fragmentation during the chromatographic run to generate both molecular and base ion signals. For some nucleosides, the glycosidic bond fragments easily leading to higher base ion abundances than molecular ion abundances, whereas in other nucleosides the ratio skews toward molecular ions over base ions. Method optimization, including using only MS/MS responses [35], may yield improved precision for these measurements.
As mass spectra were collected in full scan mode over the m/z range 103–510 in the ion trap, interfering ions may be present that could be contributing to variability in MS peak area measurements. Smaller variations in peak areas may be obtained in more selective analyses such as selected reaction monitoring (SRM), if the interfering ion has a similar mass to the nucleoside molecular or base ion [35]. The SRM approach has been used previously for the analysis of urinary nucleosides resulting in reported precisions of less than 15% RSDs [30].
Two nucleosides not listed as components of the test mix were identified by ESI-MS and found to be N6-methyladenosine (m6A) and 3-methyluridine (m3U). 1-methyladenosine undergoes a Dimroth rearrangement to form N6-methyladenosine that is 90% complete at pH 11 and 50% complete at pH 7 [47]. The presence of N6-methyladenosine is most likely due to this rearrangement because 1-methyladenosine was not found in the older nucleoside test mix used in analyses using the analytical column. The formation of 3-methyluridine is likely an unintended synthesis product of 5-methyluridine.
3.2. Biological reproducibility
Having established the technical reproducibility for LC/UV and LC/MS detection of modified nucleosides, we next sought to determine the biological variability associated with measuring modified nucleosides present in tRNAs and rRNAs. To conduct these measurements, separate cultures of E. coli were grown and the appropriate RNAs were isolated. Except where noted, identical growth and isolation protocols were followed, and any variability associated with the isolation steps will be reflected in the overall variability of the measured nucleoside abundances.
3.2.1. Transfer RNA
Determining the amounts of the various modified nucleosides to be expected in unfractionated tRNA is not a straightforward task. E. coli contains 26 different modified nucleosides (names and abbreviations found in Fig. 3) distributed unevenly among the 46 different tRNAs [48,49]. Some modifications such as pseudouridine and 5-methyluridine are common to all the tRNAs. Other modifications are present in the majority but not all tRNAs, for example D, s4U, m7G and Gm. Several modifications, mnm5U, Cm, mnm5s2U, I, ac4C, m6t6A, m6A, i6A occur in only one or a few specific tRNAs. Most modifications are specific to one location within the tRNA sequence with the exception of dihydrouridine and pseudouridine, which are located at several different positions within the same tRNA. All tRNAs contain pseudouridine at position 55, but several tRNAs contain additional pseudouridines. The D loop and stem domain of tRNA often contain more than one dihyrouridine.
Fig. 3.
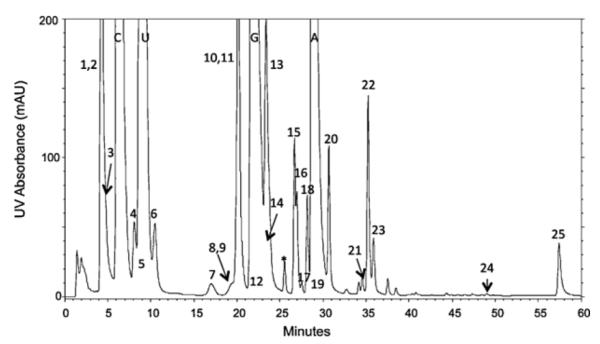
Modified nucleosides in Escherichia coli total tRNA. 1, dihydrouridine (D). 2, pseudouridine (ψ). 3,5-carboxymethoxyuridine (cmo5U). 4,3-(3-amino-3-carboxypropyl)uridine (acp3U). 5,5-methylaminomethyluridine (mnm5U). 6,2-thiocytidine (s2C). 7,2′-O-methylcytidine (Cm). 8,5-carboxylmethylaminomethyl-2′ -O-methyluridine (cmnm5Um). 9,5-methylamino-methyl-2-thiouridine (mnm5s2U). 10,5-methyluridine (m5U). 11, inosine (I). 12,4-thiouridine (s4U). 13,7-methylguanosine (m7G). 14,2′-O-methyluridine (Um). 15,2′-O-methylguanosine (Gm). 16,1-methylguanosine (m1G). 17, N4-acetylcytidine (ac4C). 18, queuosine (Q). 19, lysidine (k2C). 20, N6-threonylcarbamoyladenosine (t6A). 21, N6-methyl-N6-threonylcarbamoyladenosine (m6t6A). 22,2-methyladenosine (m2A). 23, N6-methyladenosine (m6A). 24, N6-isopentenyladenosine (i6A). 25,2-methylthio-N6-isopentenyladenosine (ms2i6A) (*unknown).
Not surprisingly, the different tRNA species are not present in equal abundance within the cell. The relative abundance of the individual tRNAs is thought to be correlated to the frequency with which the cognate codon occurs on the mRNA. Several different codons can specify the same amino acid, and these synonymous codons do not occur with equal frequency in the mRNA pool. In a set of synonymous codons, one dominates in frequency and this codon is read by the most abundant tRNA isoacceptor. Codon bias varies among species [50] varies with cellular conditions such as amino acid availability [51] and is correlated with growth rate[52–54]. Thus the complexity and large dynamic range in the analysis of modified nucleosides present in total tRNA nucleoside digests result from both the differences of frequency in occurrence of specific modifications in the 46 tRNAs and the effects of codon bias which is reflected in the differential expression of individual tRNAs.
Data obtained from the analysis of modified nucleosides present in E. coli tRNAs is presented in Table 4 with a corresponding representative chromatogram shown in Fig. 3. These data were obtained using the narrow bore column following the tRNA isolation and enzymatic digestion protocols described in the Experimental section. Three or four replicate tRNA isolations from three separate cell cultures were analyzed. For each analysis, 25 μg of total unfractionated tRNA was enzymatically digested to nucleosides and injected on column. All of the 25 known modified nucleosides present in E. coli tRNA have been identified in this analysis using the combination of retention time, UV and MS characteristics (Fig. 3). However, only a subset of modifications is adequately chromatographically resolved to obtain UV peak area measurements. The relative amounts of each nucleoside present were calculated based on peak area measurements using N6-threonylcarbamoyladenosine as an internal reference peak. This was used to eliminate differences in injection volume and slight variations in tRNA concentration. The variability of these relative amounts calculated as percent relative standard deviation is reported for this subset of modifications in Table 4 and ranged from 2.6 to 9.2% RSD. UV peak area measurements were not obtained for dihydrouridine because it does not possess a chromophore and thus will not be detected by UV absorbance. Other nucleosides, such as mnm5U, cmnm5Um and k2C, are found only in low abundance tRNAs and thus were present in very small amounts and only detected by MS.
Table 4.
Reproducibility of UV and MS peak areas for modified nucleosides (both molecular ion [MH+] and base ion [BH2 +]) in E. coli tRNA. Pooled percent relative standard deviations are for 3 or 4 replicate analyses within three separate cultures. For MS peak area % RSD 4 replicate analyses were performed on tRNA isolated from a single culture.
| Peak | Nucleoside | tr (avg)(min) | tr% RSD | UV peak area% RSD pooled | [MH+]/[BH2+]m/z | MS peak area% RSD (n = 4) |
|---|---|---|---|---|---|---|
| 1 | D | 4.1 | 3.8% | 247/115 | 4.7%/4.5% | |
| 2 | ψ | 4.2 | 2.4% | 4.4% | 245/209 | 6.0%/5.1% |
| 3 | cmo5U | 4.5 | 3.4% | 319/187 | 4.4%/5.6% | |
| 4 | acp3U | 7.6 | 4.7% | 346/214 | 2.4%/1.0% | |
| 5 | mnm5U | 8.1 | 7.3% | 288/156 | 8.7%/nd | |
| 6 | s2C | 9.7 | 6.7% | 6.5% | 260/128 | 3.0%/3.1% |
| 7 | Cm | 15.7 | 7.1% | 9.2% | 258/112 | 7.2%/9.2% |
| 8 | cmnm5Um | 17.9 | 7.4% | 346/200 | 3.1%/nd | |
| 9 | mnm5s2U | 18.6 | 7.2% | 304/172 | 5.6%/nd | |
| 10 | m5U | 19.3 | 5.0% | 2.6% | 259/127 | 5.6%/9.9% |
| 11 | I | 19.9 | 6.5% | 269/137 | 9.3%/7.9% | |
| 12 | s4U | 22.0 | 5.2% | 261/129 | 1.0%/2.7% | |
| 13 | m7G | 22.6 | 3.5% | 6.1% | 298/166 | 3.5%/4.6% |
| 14 | Um | 23.1 | 4.1% | 259/113 | 5.2%/10.6% | |
| 15 | Gm | 26.1 | 1.9% | 298/152 | */12.0% | |
| 16 | m1G | 26.4 | 1.8% | 5.8% | 298/166 | */1.4% |
| 17 | ac4C | 27.0 | 1.8% | 8.4% | 286/154 | 10.9%/5.5% |
| 18 | Q | 27.8 | 1.6% | 4.6% | 410/295 | 5.9%/5.7% |
| 19 | k2C | 28.9 | 2.0% | 372/240 | 7.8%/nd | |
| 20 | t6A | 30.3 | 1.7% | 413/281 | 5.2%/1.3% | |
| 21 | m6t6A | 34.2 | 1.5% | 4.4% | 427/295 | 5.9%/3.6% |
| 22 | m2A | 34.9 | 1.5% | 5.0% | 282/150 | 4.6%/7.5% |
| 23 | m6A | 35.5 | 1.4% | 3.7% | 282/150 | 6.3%/8.1% |
| 24 | i6A | 49.1 | 0.3% | 5.7% | 336/204 | 6.7%/nd |
| 25 | ms2i6A | 57.6 | 0.3% | 4.4% | 382/250 | 6.6%/5.0% |
denotes isobaric molecular ion and “nd” denotes base ion not detected in MS analysis.
The variability in MS peak areas measured for both the molecular [MH+] and base ions [BH2+] are also reported and ranged from 1 to 12% RSD. The MS peak area data is obtained from four separate tRNA isolations from one culture. For pseudouridine, the molecular ion and the characteristic ion of m/z 209 formed from water loss were measured. For a few nucleosides, such as cmnm5Um, mnm5s2U, k2C and i6A, base ions arising from in-source fragmentation were not detected in the XICs. However, the base ions were detected in the MS/MS analysis. For the isobaric nucleosides Gm and m1G, only the MS peak area of the base ions were measured. The variability shown here for the analysis of modified nucleosides in tRNA is similar to the variability reported for modified nucleosides present in urine for both UV and MS detection [30,33,34,45].
3.2.2. Ribosomal RNA
Quantitative measurements of modified nucleosides in rRNAs present an analytical challenge due to the dynamic range of nucleoside quantities, especially for bacterial rRNAs. The biological variability was determined by analyzing multiple enzymatic digests of 16S rRNA isolated from four different cell cultures using the same conditions as for analysis of the nucleoside test mix. Dimethyladenosine was chosen as an internal reference peak for the calculation of relative amounts of modified nucleosides present since it was baseline resolved, similar in size to other modified nucleosides and present in all 16S digests. An internal reference peak for calculation of relative amounts of modified nucleosides present was used to diminish the effect of variations in the initial amount of digested RNA loaded onto the column for each replicate analysis. Although we sought to load 100 μg of digested RNA, the precision and accuracy of this measurement may be limited due to the presence of varying amounts of phenol and large serial dilutions used for UV measurements.
Variability was first examined by loading 100 μg of total 16S rRNA nucleoside digest on the analytical column with UV detection (n = 3). Peak area and retention time variability are summarized in Table 5 with a representative chromatogram shown in Fig. 4a. The identities of the major and modified nucleosides present are based on UV spectra and comparison of retention times of nucleosides in the test mix and retention time data [28]. Pseudouridine was not well resolved from a large solvent front in many of the digests analyzed on the analytical column, thus it was not included in the culture to culture reproducibility data. The large peak present at 25 min in the chromatogram originates from the phenol used to extract ribosomal proteins, which is incompletely removed in the subsequent chloroform extraction and ethanol precipitation of the RNA. This interference is often reported in other HPLC analyses of 16S rRNA [24], and while not an issue in the detection of modifications in E. coli rRNA this peak elutes where many modified adenosines elute and may prevent their detection in RNA from other organisms.
Table 5.
Biological Reproducibility for analysis of modified nucleosides in E. coli 16S rRNA, 4.6 mm × 250 mm column used. Amounts of modified nucleosides present are based on UV peak areas relative to m6 2A. Percent relative standard deviations in parentheses are for three replicate analyses within each separate culture.
| Nucleoside | Culture 1 | Culture 2 | Culture 3 | Average | % RSD (pooled) |
|---|---|---|---|---|---|
| ψ | NA | ||||
| m5C | 0.457 (8.9%) | 0.465 (4.1%) | 0.424 (4.2%) | 0.449 | 6.2% |
| m7G | 0.319 (8.9%) | 0.339 (7.4%) | 0.349 (8.5%) | 0.336 | 7.5% |
| dG | 0.415 (6.5%) | 0.444 (3.6%) | * | 0.430 | 5.2% |
| m3U | 0.443 (3.6%) | 0.484 (3.4%) | * | 0.464 | 3.5% |
| m4Cm | 0.293 (12.6%) | 0.250 (9.4%) | 0.247 (5.2%) | 0.245 | 10.7% |
| m2G | 1.699 (3.0%) | 1.71 (3.2%) | 1.633 (2.6%) | 1.681 | 3.0% |
| m62A | 1.00 | 1.00 | 1.00 | 1.00 |
denotes peaks not well resolved.
Fig. 4.

(a) UV chromatogram of E. coli 16S RNA nucleoside digest, 4.6 mm × 250 mm/LC-18S column, 30 °C, 250 mM ammonium acetate pH 6.0 (A), 40% acetonitrile in water (B), 2.0 mL/min, UV detection @ 260 nm. (b) UV chromatogram of E. coli 16S RNA nucleoside digest, 2.1 mm × 250 mm/LC-18S column, 30 °C, 5 mM ammonium acetate pH 5.3 (A), 40% acetonitrile in water (B), 0.3 mL/min, UV detection @ 260 nm.
Because of the potential interference of the phenol-associated peak and due to the success of the Nucleobond-based purification that was used for tRNA isolations, subsequent studies avoided phenol–chloroform purification of 16S rRNA. The effect of this change in protocol is easily illustrated in Fig. 4b, which is a representative UV chromatogram of the 16S RNA enzymatic digest analyzed using the narrow bore column. Not only is the phenol related peak absent, but the ion exchange cartridge separated the DNA more effectively than the phenol extraction thus dG and dA are not present, and the decrease in the solvent front improves resolution of pseudouridine enabling higher quality measurements of its abundance in the sample.
For both the analytical column and the narrow bore column the relative amount present of each nucleoside and corresponding % RSD was calculated for replicate analyses within each of the different cell cultures and is shown in parentheses (Tables 5 and 6). The overall biological variability for measurement of each nucleoside was then calculated using the calculated pooled standard deviations for the nucleoside measurements in each of the different cell cultures. The relative standard deviation in peak areas of the modified nucleosides present in 16S RNA ranged from 3.0 to 10.7% (Table 5) when analyzed using the analytical column. Retention time variability was less than 5% for all modified nucleoside peaks. The reproducibility of peak areas of modified nucleosides in 16S RNA digests as determined on the narrow bore column ranged from 1.8% to 15.6% as shown in Table 6.
Table 6.
Biological reproducibility for analysis of modified nucleosides in E. coli 16S RNA, 2.1 mm × 250 mm column used. Amounts of modified nucleosides present are based on UV peak areas relative to m6 2A. Percent relative standard deviations in parentheses are for four replicate analyses within each separate culture.
| Nucleoside | Culture 4 | Culture 5 | Culture 6 | Culture 7 | Average | % RSD (pooled) |
|---|---|---|---|---|---|---|
| ψ | 0.497 (3.9%) | 0.458 (2.9%) | 0.474 (6.1%) | 0.466 (2.6%) | 0.474 | 4.1% |
| m5C | 0.545 (2.6%) | 0.542 (1.0%) | 0.533 (1.9%) | 0.544 (1.6%) | 0.541 | 1.8% |
| m7G | 0.762 (6.5%) | 0.731 (6.9%) | 0.800 (21%) | 0.816 (16.8%) | 0.777 | 14.9% |
| m3U | 0.554 (4.0%) | 0.555 (4.7%) | 0.559 (5.7%) | 0.573 (3.6%) | 0.560 | 4.6% |
| m4Cm | 0.510 (3.7%) | 0.519 (4.0%) | 0.514 (6.9%) | 0.515 (6.7%) | 0.515 | 5.4% |
| m2G | 1.929 (8.7%) | 1.918 (9.1%) | 1.761 (17.5%) | 1.877 (13.5%) | 1.871 | 15.6% |
| m62A | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
The differences in measured amounts of nucleosides (normalized to N6,N6-dimethyladenosine) reported in Tables 5 and 6 likely arise due to both instrumental considerations as well as differences in sample preparation. The rRNA digest analyzed on the 2.1 mm column and prepared using the same sample purification protocol used for tRNAs was free of the interferences from phenol and DNA nucleosides. Additionally, this sample purification protocol maintains the pH at 6.3, whereas the more conventional phenol/chloroform sample purification protocol is performed under more acidic conditions. These results illustrate that the use of solid phase sample purification and narrow bore chromatography should be preferred for comparative studies on modified nucleosides in rRNA based on both the improved chromatography and the ability to measure more modified nucleosides as interferences such as phenol are eliminated.
Although the goal of this work was not to obtain absolute quantitative measurements of levels of modified nucleosides in rRNA, the data in Table 6 agree well with anticipated amounts of these modified nucleosides. A 16S rRNA contains one residue each of pseudouridine, 7-methylguanosine, 3-methyluridine and N4,2′-O-dimethylcytidine. Two residues of 5-methylcytidine and N6,N6-dimethyladenosine and three residues of N2-methylguanosine are present in 16S rRNA. The obtained data, normalized against N6,N6-dimethyladenosine, is consistent with these expected ratios: pseudouridine, 3-methyluridine and N4,2′-O-dimethylcytidine yield peak areas approximately half that for N6,N6-dimethyladenosine. N2-methylguanosine and 7-methylguanosine are slightly higher than their expected ratios, while 5-methylcytidine is less than its expected ratio.
As these ratios were obtained from UV data, these particular ratios must also incorporate differences in molar absorptivities at 260 nm to more accurately report the absolute quantities of each modified nucleoside in the sample. Table 7 incorporates previously reported molar absorptivities [55] and demonstrates that the data obtained here are in good agreement even when differences in UV absorbance among the various modified nucleosides are taken into account. Most importantly, the relative amounts of each modification are reasonable given the assumption that the 16S rRNA analyzed here is modified stoichiometrically, and the combination of the reasonable experimental precision with realistic relative amounts of modifications allows this method to be used for future quantitative comparisons of changes in nucleoside composition.
Table 7.
Estimation of relative amounts of modified nucleosides in E. coli 16S RNA from data in Table 6 where molar absorptivity differences are considered. Molar absorptivities from Ref. [55].
| Nucleoside | Average peak areas (Table 6) |
Molar absorptivities (ε × 10–3) |
Relative peak areas based on ε |
|---|---|---|---|
| ψ | 0.474 | 8.3 | 0.35 |
| m5C | 0.541 | 6 | 0.50 |
| m7G | 0.777 | 11 | 0.46 |
| m3U | 0.560 | 8.5 | 0.35 |
| m4Cm | 0.515 | 10 | 0.42 |
| m2G | 1.871 | 10 | 1.80 |
| m62A | 1.00 | 12 | 1.00 |
4. Conclusions
LC/MS methods for the quantitative analysis of biomolecules enable a wide range of applications in a variety of fields. Surprisingly, although LC/UV and LC/MS analyses of modified nucleosides obtained from tRNAs and rRNAs are well represented in the literature, before now there have been no investigations that establish whether the reproducibility of LC/MS would be sufficient for its use in characterizing changes to modified nucleoside abundance as a function of environmental, physiological or other factors. The results obtained here when measuring modified nucleosides from cellular RNAs are comparable to those obtained via similar LC/UV and LC/MS analyses of urinary nucleosides [30,33,34,45]. Although the dynamic range is not as large in the analysis of urinary nucleosides and fewer steps are involved in the isolation procedures, both analyses seek to compare the total profile and determine statistically significant differences in amounts of nucleosides present in different biological conditions.
Modified nucleosides in tRNAs and, especially, rRNAs may also prove useful when characterizing the extent of drug resistance for certain bacteria/antibiotic combinations. For example, it is known that 16S rRNA methylation is a common mechanism by which pathogenic bacteria develop resistance to common aminoglycosides [56–58]. Analytical methods such as LC/MS that can detect changes in methylation status, including the specific type of methylation, would be attractive options for examining the progression of antibacterial resistance within rRNA. In addition, the ability to monitor changes in tRNA modified nucleoside abundance provide new and unique insights into the regulatory pathways involved in controlling protein translation under a variety of environmental conditions [35]. Based on the findings presented here, quantitative profiling of modified nucleosides from tRNAs and/or rRNAs is feasible, and with reproducibilities of 5% RSD or less for well-behaved modified nucleosides, this method can expand the range of biological investigations that one wishes to conduct into RNA modification.
Acknowledgment
Financial support of this work was provided by the National Institutes of Health (GM58843).
References
- [1].Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang X, Vendeix FA, Fabris D, Agris PF. Nucleic Acids Res. 2010;39:D195. doi: 10.1093/nar/gkq1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Agris PF, Vendeix FA, Graham WD. J. Mol. Biol. 2007;366:1. doi: 10.1016/j.jmb.2006.11.046. [DOI] [PubMed] [Google Scholar]
- [3].Gustilo EM, Vendeix FA, Agris PF. Curr. Opin. Microbiol. 2008;11:134. doi: 10.1016/j.mib.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bjork GR, Durand JMB, Hagervall TG, Leipuviene R, Lundgren HK, Nilson K, Chen P, Qian Q, Urbonavicius J. FEBS Lett. 1999;452:47. doi: 10.1016/s0014-5793(99)00528-1. [DOI] [PubMed] [Google Scholar]
- [5].Namy O, Lecointe F, Grosjean H, Rousset JP. In: Fine-Tuning of RNA Functions by Modification and Editing. Grosjean H, editor. Springer; New York: 2005. [Google Scholar]
- [6].Urbonavicius J, Qian Q, Durand JMB, Hagervall TG, Bjork GR. EMBO J. 2001;20:4863. doi: 10.1093/emboj/20.17.4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Giege R, Sissler MFC. Nucleic Acids Res. 1998;26:5017. doi: 10.1093/nar/26.22.5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Alexandrov A, Chernyakov I, Gu W, Hiley SL, Hughes TR, Grayhack EJ, Phizicky EM. Mol. Cell. 2006;21:87. doi: 10.1016/j.molcel.2005.10.036. [DOI] [PubMed] [Google Scholar]
- [9].Ishida K, Kunibayashi T, Tomikawa C, Ochi A, Kanai T, Hirata A, Iwashita C, Hori H. Nucleic Acids Res. 2010;39:2304. doi: 10.1093/nar/gkq1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tomikawa C, Yokogawa T, Kanai T, Hori H. Nucleic Acids Res. 2010;38:942. doi: 10.1093/nar/gkp1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Brimacombe R, Mitchell P, Osswald M, Stade K, Bochkariov D. FASEB J. 1993;7:161. doi: 10.1096/fasebj.7.1.8422963. [DOI] [PubMed] [Google Scholar]
- [12].Decatur WA, Fournier MJ. TIBS. 2002;27:344. doi: 10.1016/s0968-0004(02)02109-6. [DOI] [PubMed] [Google Scholar]
- [13].Baxter-Roshek JL, Petrov AN, Dinman JD. PLoS ONE. 2007;2:E174. doi: 10.1371/journal.pone.0000174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chow CS, Lamichhane TN, Mahto SK. ACS Chem. Biol. 2007;2:610. doi: 10.1021/cb7001494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Das G, Thotala DK, Kapoor S, Karunanithi S, Thakur SS, Singh NSVU. EMBO J. 2008;27:840. doi: 10.1038/emboj.2008.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ejby M, Sorensen MA, Pedersen S. Proc. Natl. Acad. Sci. U.S.A. 2007;104:19410. doi: 10.1073/pnas.0706558104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Esguerra J, Warringer J, Blomberg A. RNA. 2008;14:649. doi: 10.1261/rna.845808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].King TH, Liu B, McCully RR, Fournier MJ. Mol. Cell. 2003;11:425. doi: 10.1016/s1097-2765(03)00040-6. [DOI] [PubMed] [Google Scholar]
- [19].Liang XH, Lui Q, Fournier MJ. Mol. Cell. 2007;28:965. doi: 10.1016/j.molcel.2007.10.012. [DOI] [PubMed] [Google Scholar]
- [20].Saraiya AA, Lamichhane TN, Chow CS, SantaLucia JCPR., Jr. J. Mol. Biol. 2008;376:645. doi: 10.1016/j.jmb.2007.11.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dalluge JJ, Hashizume T, Sopchik A, McCloskey JA, Davis D. Nucleic Acids Res. 1996;24:1073. doi: 10.1093/nar/24.6.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Baudin-Baillieu A, Fabret C, Liang X, Piekna-Przybylska D, Fournier MJ, Roussel JP. Nucleic Acids Res. 2009;37:7665. doi: 10.1093/nar/gkp816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gutgsell NS, Deutscher MP, Ofengand J. RNA. 2005;11:1141. doi: 10.1261/rna.2550105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Siibak T, Remme J. RNA. 2010;16:2023. doi: 10.1261/rna.2160010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dudley E. In: Mass Spectrometry of Nucleosides and Nucleic Acids. Banoub J, (Eds.) P.A. Limbach, editors. CRC Press; Boca Raton, FL: 2009. [Google Scholar]
- [26].Struck W, Waszczuk W, Jankowska M, Kaliszan R, Markuszewki MJ. Anal. Bioanal. Chem. 2011;401:2039. doi: 10.1007/s00216-011-4789-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gehrke CW, Kuo K. J. Chromatogr. 1989;471:3. doi: 10.1016/s0021-9673(00)94152-9. [DOI] [PubMed] [Google Scholar]
- [28].Pomerantz SC, McCloskey JA. Methods Enzymol. 1990;193:796. doi: 10.1016/0076-6879(90)93452-q. [DOI] [PubMed] [Google Scholar]
- [29].Hsu WY, Lin WD, Tsai YT, Lin CT, Wang HC, Jeng LB, Lee CC, Lin YC, Lai CC, Tsai FJ. Clin. Chim. Acta. 2011;412:1861. doi: 10.1016/j.cca.2011.06.027. [DOI] [PubMed] [Google Scholar]
- [30].Lee HL, Jung BH, Kim SY, Chung BC. Rapid Commun. Mass Spectrom. 2004;18:973. doi: 10.1002/rcm.1400. [DOI] [PubMed] [Google Scholar]
- [31].Liebich HM, Muller-Hagedorn S, Klaus F, Meziane K, Kim KR, Fricken-schmidt A, Kammerer B. J. Chromatogr. A. 2005;1071:271. doi: 10.1016/j.chroma.2004.12.055. [DOI] [PubMed] [Google Scholar]
- [32].Liebich HM, Muller-Hagedorn S, Bacher M, Scheel-Walter HG, Lu X, Frick-enschmidt A, Kammerer B, Kim KR, Gerard H. J. Chromatogr. B: Analyt. Technol. Biomed. Life Sci. 2005;814:275. doi: 10.1016/j.jchromb.2004.10.051. [DOI] [PubMed] [Google Scholar]
- [33].Wang S, Zhao X, Mao Y, Cheng Y. J. Chromatogr. A. 2007;1147:254. doi: 10.1016/j.chroma.2007.02.049. [DOI] [PubMed] [Google Scholar]
- [34].Teichert F, Winkler S, Keun HC, Steward WP, Gescher AJ, Farmer PB, Singh R. Rapid Commun. Mass Spectrom. 2011;25:2071. doi: 10.1002/rcm.5086. [DOI] [PubMed] [Google Scholar]
- [35].Chan CTY, Dyavaiah M, DeMott MS, Taghizadeh K, Dedon PC, Begley TJ. PLoS Genet. 2010;6:e1001247. doi: 10.1371/journal.pgen.1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Emmerechts G, Barbe S, Herdewijn P, Anne J, Rosenski J. Nucleic Acids Res. 2007;35:3494. doi: 10.1093/nar/gkm248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Guymon R, Pomerantz SC, Crain PF, McCloskey JA. Biochemistry. 2006;45:4888. doi: 10.1021/bi052579p. [DOI] [PubMed] [Google Scholar]
- [38].McCloskey JA, Graham DE, Zhou S, Crain PF, Ibba M, Konisky J, Soll D, Olsen GJ. Nucleic Acids Res. 2001;29:4699. doi: 10.1093/nar/29.22.4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Noon KR, Bruenger E, McCloskey JA. Bacteriol. 1998;180:2883. doi: 10.1128/jb.180.11.2883-2888.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Brückl T, Globisch D, Wagner M, Müller M, Carell T. Angew. Chem. Int. Ed. 2009;48:7932. doi: 10.1002/anie.200902740. [DOI] [PubMed] [Google Scholar]
- [41].Globisch D, Pearson D, Hienzsch A, Brückl T, Wagner M, Thoma I, Thumbs P, Reiter V, Kneuttinger AC, Müller M, Sieber SA, Carell T. Angew. Chem. Int. Ed. 2011;50:9739. doi: 10.1002/anie.201103229. [DOI] [PubMed] [Google Scholar]
- [42].Waghmare SP, Dickman MJ. Anal. Chem. 2011;83:4894. doi: 10.1021/ac200547y. [DOI] [PubMed] [Google Scholar]
- [43].Spedding G. Ribosomes and Protein Synthesis: A Practical Approach. Oxford University Press; 1990. [Google Scholar]
- [44].Crain PF. Methods Enzymol. 1990;193:782. doi: 10.1016/0076-6879(90)93450-y. [DOI] [PubMed] [Google Scholar]
- [45].Hsu WY, Chen WTL, Lin WD, Tsai FJ, Tsai Y, Lin CT, Lo WY, Jeng LB, Lai CC. Clin. Chim. Acta. 2009;402:31. doi: 10.1016/j.cca.2008.12.009. [DOI] [PubMed] [Google Scholar]
- [46].Dudley E, Tuytten R, Bond A, Lemiere F, Brenton AG, Esmans EL, Newton RP. Rapid Commun. Mass Spectrom. 2005;19:3075. doi: 10.1002/rcm.2151. [DOI] [PubMed] [Google Scholar]
- [47].Macon JB, Wolfenden R. Biochemistry. 1968;7:3453. doi: 10.1021/bi00850a021. [DOI] [PubMed] [Google Scholar]
- [48].Juhling F, Morl M, Hartmann RK, Sprinzl M, Stadler PF, Putz J. Nucleic Acids Res. 2009;37:D159. doi: 10.1093/nar/gkn772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Czerwoniec A, Dunin-Horkawicz S, Purta E, Kaminska KH, Kasprzak JM, Bujnicki JM, Grosjean H, Rother K. Nucleic Acids Res. 2009;37:D118. doi: 10.1093/nar/gkn710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sharp PM, Cowe E, Higgins DG, Shields DC, Wolfe KH, Wright F. Nucleic Acids Res. 1988;16:8207. doi: 10.1093/nar/16.17.8207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Elf J, Nilsson D, Tenson T, Ehrenberg M. Science. 2003;300:1718. doi: 10.1126/science.1083811. [DOI] [PubMed] [Google Scholar]
- [52].Dong H, Nilsson L, Kurland CG. J. Mol. Biol. 1996;260:649. doi: 10.1006/jmbi.1996.0428. [DOI] [PubMed] [Google Scholar]
- [53].Ikemura T. J. Mol. Biol. 1981;146:1. doi: 10.1016/0022-2836(81)90363-6. [DOI] [PubMed] [Google Scholar]
- [54].Ikemura T. Mol. Biol. Evol. 1985;2:13. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- [55].The Japanese Biochemical Society, editor. Handbook of Biochemistry. vol. I. Tokya Kagaku Dozin, Tokyo; Japan: 1979. [Google Scholar]
- [56].Doi Y, Arakawa Y. Clin. Infect. Dis. 2007;45:88. doi: 10.1086/518605. [DOI] [PubMed] [Google Scholar]
- [57].Shakil S, Khan R, Zarrilli R, Khan AU. J. Biomed. Sci. 2008;15:5. doi: 10.1007/s11373-007-9194-y. [DOI] [PubMed] [Google Scholar]
- [58].Moric I, Savic M, Ilic-Tomic T, Vojnovic S, Bajkic S, Vasiljevic B. J. Med. Biochem. 2010;29:165. [Google Scholar]