

Abstract
There is a current need in biomedical sciences to perform live imaging with higher spatial and temporal resolution. To achieve this goal in a comprehensive manner, three-dimensional acquisitions are necessary. Ideal features of a modern microscope system should include high imaging speed, high contrast ratio, low photo-bleaching and photo-toxicity, good resolution in a 3D context and mosaic acquisition for large samples. Considering the importance collecting data in live sample increases the technical challenges required to solve these issues.
This work presents a practical version of a microscopy method, Selective Plane Illumination Microscopy reintroduced by Huisken (2004). This method is gaining importance in the biomedical field, with its load of technical solutions but has been limited by difficulties associated with unconventional microscope construction using two objectives and sample preparation. Based on the selective plane illumination principle but with a design similar to the Total Internal Reflection Fluorescence microscope, Dunsby (2008) demonstrated the Oblique Plane Microscope (OPM) using a single objective which uses conventional sample preparation protocols. However, the Dunsby instrument was not intended to be part of a commercial microscope.
In this work we describe a system with the advantages of OPM and that can be used as an add-on to commonly used microscopes, such as IX-71 Olympus, simplifying the construction of the OPM and increasing performance of a conventional microscope. We named our design inclined Selective Plane Illumination Microscope (iSPIM).
Introduction
The need for fast 3D microscopy has increased over the years in bio-imaging facilities around the world. In response, the confocal microscope principle has been pushed to faster speed such as the spinning disk technology (Nipkow disk optimizations) and fast laser scanning microscopes. Recently, another technique has been proposed, Selective Plane Illumination Microscopy (SPIM), a method based on illumination of the sample from the side with a sheet of light. The original idea was described in 1903 (Siedentopf and Zsigmondy, 1903) and pioneered in 1993 (Voie et al., 1993) with the Orthogonal Plane Fluorescence Optical Sectioning (OPFOS). It was subsequently re-described in a modern incarnation by Huisken et al (2004) who proposed a practical basic setup for SPIM systems.
Techniques like SPIM, generally called light-sheet microscopy techniques, are gaining popularity because of the high efficiency in accessing volumetric information of the specimen while minimizing photo-bleaching and energy load. For this reason multiple labs have focused on the development of light-sheet microscopy with higher efficiency and have introduced specific advantages. Working on the OPFOS idea, Buytaert (Buytaert and Dirckx, 2007) developed High-resolution orthogonal-plane fluorescence optical sectioning (HROPFOS) with the main advantage of a translating thinner light-sheet. Holekamp et al (2008) with their Objective Coupled Planar Illumination microscopy (OCPI) develop a single-sided light-sheet illumination mounted on the detection lens. The same principle of lateral sectioning through light-sheet was then optimized for large samples with Ultramicroscopy by Dodt et al (2007) where dual sided light-sheet illumination is implemented and Multidirectional SPIM (mSPIM) by Huisken (Huisken and Stainier, 2007). These developments separate the use of SPIM for large and small specimens establishing the use of cylindrical lens-shaped beam for large samples while objective-coupled excitation beam as in Digital Scanned Laser Light-Sheet fluorescence microscopy (DSLM) by Keller et al (2008) was used for small samples.
A further development of the SPIM method was proposed by the application of Bessel beams to light sheet microscopy by Planchon et al (2011). Their system was not based on cylindrical lenses but rather on a swept-light-sheet obtained by rapidly shifting the Bessel beam on the plane of interest. Despite the innovations in the excitation modality and pattern in these designs, the perpendicularity between excitation and collection objectives is maintained, following the main SPIM original design.
A different path was proposed by Tokunaga et al (2008) with the Highly Inclined and Laminated Optical sheet microscopy (HILO). Using the same single objective lens the system excites the sample with an oblique light sheet and collects the emitted fluorescence. The most important differences of this method with respect to the SPIM classical formulation reside in the geometry of illumination/detection on the sample. The 90 degrees angle between excitation and collected beams is substituted by a misalignment of the emitting plane and focal plane of the objective. The main drawback of HILO resides in the narrow field of view (FOV) due to severe defocusing affecting the regions of the sample depending on the distance from the central focusing line.
Following this idea Dunsby (2008) developed the Oblique Plane Microscope (OPM) that overcomes the limitation of the narrow FOV of HILO. The inspiration comes from the work of Botcherby et al (2008) that solves the issue of aberration of images collected at positions out of the focal plane using a refocusing method before the detector. The application of this optical arrangement to HILO overcomes the limitation of the defocusing problem related to the inclined illumination.
Advantages of Inclined Light Sheet Microscopy
The developments in light sheet microscopy can also be summarized in terms of advantages. Here we first compare laser scanning microscopes (LSM) and light sheet based microscopes, followed by a comparison with SPIM microscopes and OPM.
In laser sheet based microscopes the entire laser intensity is focused on a diffraction limited flat beam, which excites the area of interest of the sample. Considering this principle in terms of illumination efficiency, the maximum total loss along mirrors and lenses will not exceed 5%, thus achieving a 95% efficiency (Keller and Stelzer, 2008). Furthermore, given the illumination efficiency, the necessary exposure time for achieving a specific amount of signal is reduced compared to LSM. In fact LSM sectioning is performed through discrimination of the out-of-focus light using the detection pinhole. In other words, the whole sample is illuminated and the out-of-focus signal is discarded, yielding a decrease in efficiency as the depth within the sample increases.
Two-photon excitation microscopes (TPE) efficiency is higher than LSM due to the selection criterion induced by the probability density function of the two-photon excitation process: only the fluorophores close to the focal plane are excited. However there is a surface contribution that appears at high laser power when the depth within the specimen increases.
Based on the efficiency of these systems and their respective basic theory, we can discuss the respective energy load, photo-bleaching and photo-toxicity advantages. Since SPIM systems perform a one-dimensional scan, while the LSM and TPE use a 2-dimensional scan, the total scanning speed is higher in the SPIM systems, where scanning is only limited by the acquisition speed of the camera (Keller and Stelzer, 2008).
Considering a frame size of 256×256 pixels, at a reasonably fast scanning speed of CFM of 400Hz, the theoretical time for covering the full frame is approximately 640 ms. Instead, for the camera the average acquisition time for a 256×256 pixel frame can be as high as 60 fps using conventionally cooled CCD cameras and even faster when using the new cMOS scientific camera which provides a 16ms frame acquisition time. Therefore the exposure of the sample to the laser is reduced by a factor of 40 or higher. Furthermore because of the excitation efficiency of SPIM systems compared to LSM, the energy load of a laser sheet microscope is reduced by a factor n, which is defined as the thickness of the sampled acquisition divided by the width of the laser sheet.
If the illumination efficiency is taken into consideration, photo-bleaching, photo-damage and energy load are reduced by a factor n in SPIM systems compared to LSM, where n can reach a value of 100 or higher (Keller and Stelzer, 2008). The photo-toxicity comparison between SPIM and TPE is complicated by the difference of illumination wavelengths used in the processes (e.g. 488nm and 930nm respectively for GFP). Nevertheless TPE still illuminates the whole sample when acquiring a single plane, thus the limitations due to LSM still apply. In addition TPE necessitates short high energy pulses for excitation process to yield a small sampling cross section. From recent studies (Keller et al., 2008) the energy load of TPE is approximately six orders of magnitude higher compared to SPIM systems.
We next compare spatial resolution, in particular lateral and axial resolution. In terms of lateral resolution, the performance of a SPIM system is similar to that of LSM. The latter will perform better, and TPE is worse in regard to lateral resolution (Stelzer et al., 1994). Furthermore, considering that lateral and axial resolutions depend on excitation wavelengths, NA (Numerical Apertures) and Signal to Noise Ratio (SNR), assuming the maximum average SNR of LSM is 60:1 (Keller et al., 2008), confocal microscopes will not be able to take advantage of this improved lateral resolution. Things are different when considering axial resolution. The SPIM illumination method is advantageous when compared to the planar optical sectioning of LSM. Data reported in literature show an increase of 50% in axial extent, when considering the strong dependence of axial resolution from the thickness of the illumination profile (Engelbrecht and Stelzer, 2006). Moreover, the high SNR yielded by SPIM systems permits a better use of image deconvolution providing further increase in resolution.
Comparing the Signal to Noise Ratio is the next point. The relevant parameter for this comparison is the detection system performance of SPIM systems. The use of EMCCD cameras provides an advantage over detection in confocal systems (PMTs, APDs). This advantage derives from the parallel acquisition in contrast with serial single point detection. Considering for example for a frame acquired in a specific time T, then each camera pixels will be exposed in parallel for the entire time T. With confocal detection, given the serial acquisition, the exposure per pixel will be T divided by the number of pixels composing the frame, leading to a lower physical amount of photons collected. The high dynamic range of SPIM has the advantage of allowing image deconvolution, thus increasing resolution (Verveer et al., 2007). The high SNR also increases precision of any type of signal quantification based analysis (e.g. embryonic development). An important point of comparison is also imaging speed. As discussed previously SPIM systems achieve a theoretical 33.7 frames per second at full frame (512×512) resolution (or 60 fps at 256×256), limited by the acquisition speed of the camera in use. It is possible to generalize the concept to pixels per second. Common camera used in SPIM setup performs at approximately 10 Mega pixel per second while commercial confocal microscopes achieve about 1 Mega pixel per second. This means not only a tenth of the speed but also a lower sensitivity.
In the confocal microscopes family we can identify a different approach based on the Spinning Disk confocal systems (SD). These systems use a Nipkow Disk, a disk with arrays of pinholes distributed along an Archimedean spiral, to insert multiple beams on the sample in parallel. This system achieves consistently higher speeds. Considering a rotational speed of 1600rpm and 1/12 of revolution necessary to cover a full frame, in theory the system has capability to achieve 26.6 rounds per second, which in principle covers 320 times the frame every second. Thus the system is, like SPIM systems, limited by the acquisition speed of the camera. On the other hand, the same issues related to confocal systems apply. In particular with a fixed pinhole size, a consistent deterioration in SNR is visible proportionally to the depth of the focal plane in the specimen.
At this point the advantages of SPIM systems over LSM and TPE has been discussed. In the following we show advantages of the Oblique Light Sheet Microscopes (OLSM) over the regular SPIM system. The SPIM setup, as previously described, consists in a double objective system L-shaped, with excitation objective introducing the laser sheet laterally and detection objective collecting the emission perpendicularly. OLSM instead use a single objective to excite and collect the light. The advantage of OLSM therefore is the decreased mechanical complexity. In fact, SPIM necessitates moving elements for both objectives in order to change the excited plane and to change the focusing plane, thus requiring complex coordination of moving optical paths. Alternatively, the sample can be moved, but with increasing acquisition time. These issues are resolved in the OLSM design. The single objective system requires only stage movement for scanning through the sample and no modification to the optical path or complex mechanical moving parts are necessary. Furthermore the nature of the OLSM setup is similar to that of commercially available microscopes. This is important since through custom designed parts, OLSM can be applied to any microscope body. Decreasing the complexity of the setup has also the beneficial effect of increasing the interest and dissemination of this novel SPIM system from a scientific point of view.
The other important advantage of OLSM is the sample preparation. The SPIM double objective design in fact requires a special sample preparation in which the sample is cylindrically shaped. This is accomplished by embedding the specimen in an Agarose gel filled cylinder, but on the other hand changes the condition of the experiment, thus complicating the interpretation of the results and comparison with datasets acquired with different microscopy systems. This particular issue, in addition of a dedicated new type of microscope, has limited the widespread use of SPIM systems. OLSM, taking full advantage of the single objective setup, has no particular sample preparation requirement. Regular culture dishes can be used, decreasing the preparation complexity compared to SPIM. The design of iSPIM maintains the same advantages of the OLSM with ease of adaptability to commercial body microscopes.
Material and Methods
The schematic setup of iSPIM is shown in figure 1. The excitation light is provided by a laser launcher (ISS, Champaign IL) at 488nm and 436nm. The laser beam is expanded (x10) using a custom beam expander built with matching achromatic doublet lenses (Achromatic Doublet Lens 5mm Dia. × 10mm FL, MgF2 Coating and Achromatic Doublet Lens 25mm Dia. × 100mm FL, MgF2 Coating NT32-327, Edmund Optics, Karlsruhe, Germany). This component, with two mirrors, is connected to a metal base mounted on a vertical manual stage.
Figure 1.

Setup of iSPIM. Panel A shows the overview of the system. Laser beam is inserted into a 10x beam expander and reflected by a mirror (M) both mounted on a vertically moving component (panel B). Cylindrical lens (CL) and achromatic doublet (L1) are mounted on the “injection arm” base (panel C). The dichroic mirror (D) reflects the beam vertically into the back aperture of the objective (Obj1) (panel D). The emission light collected through Obj1 is reflected out of the body microscope into a tube containing two achromatic doublet lenses (L2). The virtual image is formed after the second objective (Obj2) and its light is collected perpendicularly with Objective three (Obj3), mounted on an objective wheel (panel E). The resulting image is focused on the camera through tube lens (L3).
The “injection arm”. It includes a cylindrical lens and a focusing achromatic lens (Edmund Optics, Lens Cyl 10×20×75 mm FL VIS-NIR, Lens Ach 25×75 mm MgF2 TS) on to the same metal base. This ensures the pre-aligned optics to maintain the precise distance of the 150mm determined by the lenses focuses. Furthermore it also serves as optical adaptor for inserting the light sheet in a commercial body microscope, in this case an IX-71 Olympus. The apochromatic lens in fact refocuses the light on to the back aperture of the objective. The dichroic mirror used is a beam splitter 20/80 (21008 20/80 Beamsplitter, Chroma). The “injection arm” and the assembly containing the vertically moving beam expander are mounted on a linear manual stage base that allows the horizontal movement of the two components.
The emission light is refocused using a back-to-back microscope system, composed by the first objective (UPlanApo 60x/1.35 oil immersion, Olympus) and lens L2, the latter lens L3 and the second objective (UPlanApo 40x/0.85 air objective, Olympus). The objectives are chosen according to the aberration-free rule from Botcherby (2008), thus the chosen magnification must equal the ratio of n1/n2, where n1 is the refraction index of the immersion medium of the specimen and n2 is the refraction index of the second objective. Since the first is an oil objective the magnification is chosen to be 1.5x.
The tilted component consists of a lens turret with a 40x (LucplFln 40x/.60 air objective, Olympus) and a 10x (UPlanApo 10/.40 air objective, Olympus) followed by an achromatic doublet lens (Edmund Optics, Lens Ach 50 X 350 MgF2 TS) that focuses the image on the sensor of an EMCCD camera (Evolve 512, Photometrics, Tucson, AZ). This allows the user to choose among two magnifications, depending on typology of specimen.
The microscope stage in use is programmable (Applied Scientific Instruments, MS-2000 Eugene, OR). Since the light sheet position and angle are fixed, the only moving component is the stage. This is used for scanning through the sample.
The stage has been programmed to perform an array scan. Size, distance and array cell dwell time are customizable. Upon reaching a new position the stage sends a TTL pulse to the trigger port of the camera, initializing acquisition. The camera, set to image in triggered mode, outputs a TTL pulse when reaching ‘frame readout’. In a feedback loop this pulse is used to activate stage motion to the next position.
The acquisition algorithm, written in Matlab with C++, optimize acquisition times considering exposure time, read out, dwell time and time to reach next position on the stage. The acquisition protocol has been simplified by programming a user friendly Graphical User Interface that guides the user along the acquisition steps. Algorithms for mosaic/tiled acquisition have been implemented.
The resulting FWHM of the light sheet on the sample results to be 2.6μm whereas the confocal parameter is 80μm. A geometrical model for excitation angle, acceptance angle and emission (Dunsby, 2008) is presented in figure 2, where θ is the acceptance angle of the first objective, Φex the half angle of the excitation sheet, Φem the half angle of the emission. Considering illumination and collection separated by π/2 it is possible to obtain the equation (Dunsby, 2008):
Figure 2.
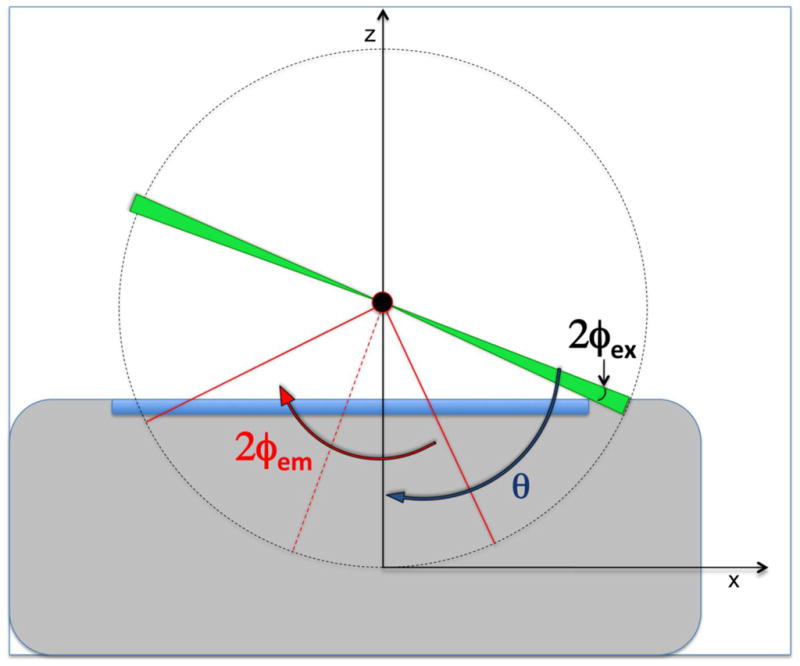
geometrical model for excitation angle. The scheme represents an objective lens with acceptance angle θ. The light sheet, in green, has an half angle of the excitation Φex. The sample in the center (black circle), has a half angle of emission Φem.
From the definition of Numerical Aperture (NA), with a non-fluorescent oil refractive index of n=1.479 (Cargille Oil) the value of θobj1=73.7deg. Consequently, being Φex=3deg the equation above gives Φem=54.4deg. The potential NA of the system, considering only the first objective is NApotential=1.20.
One issue also outlined by Dunsby is that the limiting factor in the NA of the system is located in the refocusing part with the third objective. In this setup the real NA obtained are 0.88 for the LucplFln 40x/.60 and 0.59 for the UPlanApo 10/.40. This value considers the fact that the last objective image is the already magnified sample from the second objective.
The angle subtended by the laser sheet with the first objective, α, is set to be 30 degrees whereas the FWHM of the focal spot is 344nm.
Results
Samples of MMT cell line (American Type Culture Collection, CCL-51) transfected with H2B-EGFP are prepared on standard culture dishes (MatTek, Ashland, MA) coated with fibronectin. Cells are imaged live after overnight seeding and further fixed. Fixation is done with PFA (Paraformaldehyde) and mounting media (polyvinyl alcohol mounting medium with DABCO, antifading- 10981) for further sampling. Imaging is performed using 488nm laser and shifting the sample with the stage movement. The dataset is then corrected for the acquisition geometry, shearing the frames accordingly. Rendering is performed using Amira, (San Diego, CA).
A comparison with confocal images of the same sample collected with Olympus FV1000 using comparable optical settings to iSPIM is shown in figure 4. It is possible to distinguish structures inside the nucleus on the intensity based volumetric representation. For proper understanding, one of the sections, in gray scale, is shown in the rendering. This section is a 30 degrees angle section of the sample.
Figure 4.
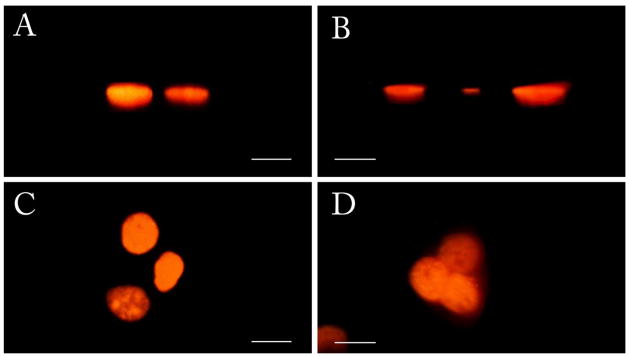
comparison of images of live MMT H2B-EGFP acquired with confocal (A, C) and iSPIM (B, D). Scale bars are 20 μm. A and B represent the x-z view. The lateral resolution observed is comparable to confocal. C and D show the view of x-y plane. In these two panels the advantage of gray levels camera-based acquisition is noticeable. The confocal acquisition is performed using settings comparable to those used for the iSPIM system and considering that the zooming function of a confocal microscope is not applicable to a SPIM camera based system.
Furthermore, to show the mosaic capability of the system a tiled acquisition of 9012 frames full frame resolution (512 by 512 pixels) is performed. Images are then stitched, shifted and rendered. The area covered is 842μm by 442μm. The resulting image is shown in figure 5.
Figure 5.
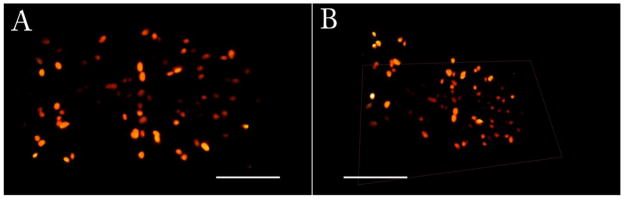
Mosaic of 9012 frames 512 × 512 pixels acquired with iSPIM. Scale bar is 100μm. Due to size issues, the dataset has been downsampled a factor of 2 to allow visualization. 4a) vertical view; 4b) inclined view with reference plane resulting from stitched frames. The fluorescence image corresponds to single cell nuclei
Given the acquisition speed the quality of high resulting resolution of the images demonstrate the potential of this imaging method. The mosaic size of 9012 frames results in a manageable raw file of 4.6GB which after stitching and shifting, considering 30% overlap and conversion to TIFF format, weights 4GB, yielding 2061×1273×751 volumetric pixel size.
Discussion
As shown by the images presented in this article, iSPIM has the capability of sectioning the sample with a simple linear translation of the specimen over the light sheet. The only moving component in the setup is in fact the stage.
This setup still shows extensive optics as in Dunsby’s prototype. Nevertheless the system has improved stability and simplified alignment. Once aligned the components do not require any further movement. Critical alignment points are the laser sheet insertion into the microscope and the tilting component between the second and third objective. To simplify the insertion of the excitation light, the block containing vertically the moving mirror, beam expander and secondary mirror, together with the block of the “injection arm” have been pre-aligned and mounted on a single axis manual translation stage. It is critical that the light sheet has the correct angle subtended with the first objective lens. This angle depends on the entrance angle of the beam on the back aperture of the objective.
A simple rule arises from geometrical considerations. Moving the light sheet on the x-y plane of the microscope system will change the exit angle of the beam from the objective. Altering the z-position will shift the exit position on the objective lens. Therefore moving the single axis manual translation stage will alter the exit angle. The mirror, along with beam expander and secondary mirror in this block is installed on a kinematic mirror mount, thus it is possible to move the beam along the z-axis and consequently change the exit position.
The tilting component, the interface between the re-imaged light sheet and the camera acquisition, is another critical alignment point of the system. The addition of the objective wheel and thus the possibility to change the final magnification increases the demand for a simplified and quick repositioning of the third objective. The solution adopted in this setup is to uncouple the block with a third objective from the camera arm. The filter wheel is mounted on an x-y-z manual translation stage. As a consequence switching between objectives and adjusting for a different working distance is performed with reduced time expense.
Another relevant point is regarding the three dimensional data acquired. The camera device acquires continuously data in a single sequenced not spaced vector. As a consequence when the raw data is rendered the resulting volume is cubical. Considering the acquisition setup, the physical volume has a parallelogram shape in cross-section. It is then necessary to post-process the data shifting each frame accordingly to render the correct physical volume (figure 5).
Mosaic acquisition
Through programming of the MS-2000 ASI stage controller an array can be acquired with custom chosen overlap. Serpentine mode has been used to optimize acquisition time. The frames collected are then stitched using the fast algorithm developed by Preibisch et al (2009) implemented in FIJI (ImageJ). The final composite image is then processed for volumetric rendering. This process, due to the number of frames collected, increases the size of the final data file considerably. One common problem in the field is related to huge dataset volumetric representation. A possible solution is the increase of Random Access Memory of the computer in use. On the other hand with the increasing size of camera chipsets and the upgraded acquisition speed, a constant increase in datasets can be expected.
Even though iSPIM data acquired did not exceed 16GB, thus in the RAM limits of the workstation, an algorithm is being implemented for subsampling the image resolution to different level of zoom.
Conclusions
This paper shows the design of inclined Selective Plane Illumination device as an adaptor for commercial body microscopes. As for the original Oblique Plane Microscope (Dunsby, 2008) it carries the same advantages of SPIM systems but with simplified mechanical component and through-the-objective TIRF-like operation. It furthermore allows the user to maintain standard sample preparation and standard horizontal positioning of the sample.
The results presented demonstrate the potential of the system when applied to cellular imaging. The capability to acquire volumetric information by simply translating the sample in a direction and the iteration of this to form of mosaic is shown in the images.
The competing limiting factors are camera acquisition speed, camera pixel size and stage movement speed. Using Sci-CMOS cameras could allow higher acquisition speed with reduced pixel dimension, increasing the resolution but with the trade off of a lower sensitivity. Optimization of stage movement, considering still time during exposure and acceleration value during camera readout will also decrease acquisition time.
The optomechanical design is enhanced to provide simplified alignment among the critical points of the system, defining an increased stability of the system. The optical components are chosen with the perspective of multicolor imaging, which yields the possibility to image multiple channels.
Furthermore our inclined design is fully compatible with 2-Photon excitation which could increase the system illumination efficiency with an augmented penetration in the sample.
Figure 3.

images of live MMT H2B-EGFP acquired with iSPIM with a step size of 0.5μm. The fluorescence signal derives from the nucleus. Renderings are in color, whereas reference section slices are in grayscale. Scale bars are 10μm. A–B) 98 frames stack; C) 280 frames stack; D) 361 frames stack.
Figure 6.

outline of volumetric acquisition through iSPIM. The image represents an objective (in black) with inclined light sheet excitation (in green) exiting the lens. The specimen (in red) placed over the objective is thus sectioned by the excitation light yielding a real volume subtended by the inclined light sheet, that is a 3D parallelogram.
Acknowledgments
The authors would like to thank Alexander Dvornikov for the mechanical help, Christoph Gohlke for programming, Milka Stakic and Chi-Li Chiu for sample preparation. This work was supported by the National Center for Research Resources (5P41RR003155), the National Institute of General Medical Sciences (8P41GM103540 and 5P50 GM076516) divisions of the National Institutes of Health.
References
- Botcherby EJ, Juskaitis R, Booth MJ, Wilson T. An optical technique for remote focusing in microscopy. Optics Communications. 2008;281(4):880–887. [Google Scholar]
- Buytaert JA, Dirckx JJ. Design and quantitative resolution measurements of an optical virtual sectioning three-dimensional imaging technique for biomedical specimens, featuring two-micrometer slicing resolution. J Biomed Opt. 2007;12(1):014039. doi: 10.1117/1.2671712. [DOI] [PubMed] [Google Scholar]
- Dodt HU, Leischner U, Schierloh A, Jahrling N, Mauch CP, Deininger K, Deussing JM, Eder M, Zieglgansberger W, Becker K. Ultramicroscopy: three-dimensional visualization of neuronal networks in the whole mouse brain. Nat Methods. 2007;4(4):331–6. doi: 10.1038/nmeth1036. [DOI] [PubMed] [Google Scholar]
- Dunsby C. Optically sectioned imaging by oblique plane microscopy. Opt Express. 2008;16(25):20306–16. doi: 10.1364/oe.16.020306. [DOI] [PubMed] [Google Scholar]
- Engelbrecht CJ, Stelzer EH. Resolution enhancement in a light-sheet-based microscope (SPIM) Opt Lett. 2006;31(10):1477–9. doi: 10.1364/ol.31.001477. [DOI] [PubMed] [Google Scholar]
- Holekamp TF, Turaga D, Holy TE. Fast three-dimensional fluorescence imaging of activity in neural populations by objective-coupled planar illumination microscopy. Neuron. 2008;57(5):661–72. doi: 10.1016/j.neuron.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Huisken J, Stainier DYR. Even fluorescence excitation by multidirectional selective plane illumination microscopy (mSPIM) Optics Letters. 2007;32(17):2608–2610. doi: 10.1364/ol.32.002608. [DOI] [PubMed] [Google Scholar]
- Huisken J, Swoger J, Del Bene F, Wittbrodt J, Stelzer EH. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science. 2004;305(5686):1007–9. doi: 10.1126/science.1100035. [DOI] [PubMed] [Google Scholar]
- Keller PJ, Schmidt AD, Wittbrodt J, Stelzer EH. Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy. Science. 2008;322(5904):1065–9. doi: 10.1126/science.1162493. [DOI] [PubMed] [Google Scholar]
- Keller PJ, Stelzer EH. Quantitative in vivo imaging of entire embryos with Digital Scanned Laser Light Sheet Fluorescence Microscopy. Curr Opin Neurobiol. 2008;18(6):624–32. doi: 10.1016/j.conb.2009.03.008. [DOI] [PubMed] [Google Scholar]
- Planchon TA, Gao L, Milkie DE, Davidson MW, Galbraith JA, Galbraith CG, Betzig E. Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination. Nat Methods. 2011;8(5):417–23. doi: 10.1038/nmeth.1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preibisch S, Saalfeld S, Tomancak P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics. 2009;25(11):1463–5. doi: 10.1093/bioinformatics/btp184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siedentopf H, Zsigmondy R. Über Sichtbarmachung und Grössenbestimmung ultramikroskopischer Teilchen, mit besonderer Anwendung auf Goldrubingläser. Annalen der Physik. 1903;(10):1–39. [Google Scholar]
- Stelzer EHK, Hell S, Lindek S, Stricker R, Pick R, Storz C, Ritter G, Salmon N. Nonlinear absorption extends confocal fluorescence microscopy into the ultra-violet regime and confines the illumination volume. Optics Communications. 1994;104(4–6):223–228. [Google Scholar]
- Tokunaga M, Imamoto N, Sakata-Sogawa K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat Methods. 2008;5(2):159–61. doi: 10.1038/nmeth1171. [DOI] [PubMed] [Google Scholar]
- Verveer PJ, Swoger J, Pampaloni F, Greger K, Marcello M, Stelzer EH. High-resolution three-dimensional imaging of large specimens with light sheet-based microscopy. Nat Methods. 2007;4(4):311–3. doi: 10.1038/nmeth1017. [DOI] [PubMed] [Google Scholar]
- Voie AH, Burns DH, Spelman FA. Orthogonal-plane fluorescence optical sectioning: three-dimensional imaging of macroscopic biological specimens. J Microsc. 1993;170(Pt 3):229–36. doi: 10.1111/j.1365-2818.1993.tb03346.x. [DOI] [PubMed] [Google Scholar]