

Abstract
Epigenetics is believed to play a role in Alzheimer's disease (AD). DNA methylation, the most investigated epigenetic hallmark, is a reversible mechanism that modifies genome function and chromosomal stability through the addition of methyl groups to cytosine located in CpG dinucleotides to form 5 methylcytosine (5mC). Methylation status of repetitive elements (i.e. Alu, LINE-1 and SAT- α) is a major contributor of global DNA methylation patterns and has been investigated in relation to a variety of human diseases. However, the role of methylation of repetitive elements in blood of AD patients has never been investigated so far. In the present study, a quantitative bisulfite-PCR pyrosequencing method was used to evaluate methylation of Alu, LINE-1 and SAT- α sequences in 43 AD patients and 38 healthy donors. In multivariate analysis adjusting for age and gender, LINE-1 was increased in AD patients compared with healthy volunteers (ADs: 83.6 %5mC, Volunteers: 83.1 %5mC, p-value: 0.05). The group with best performances in mini mental state examination (MMSE) showed higher levels of LINE-1 methylation compared to the group with worst performances (MMSE>22: 83.9 %5mC; MMSE<=22: 83.2 %5mC; p=0.05). Our data suggest that LINE-1 methylation may lead to a better understanding of AD pathogenesis and course, and may contribute to identify novel markers useful to assess risk stratification. Further prospective investigations are warranted to evaluate the dynamics of DNA methylation from early-stage AD to advanced phases of the disease.
Keywords: Alzheimer's Disease, Epigenetics, DNA methylation, Repetitive elements, Peripheral Blood Leukocytes
Introduction
Alzheimer disease (AD) is a neurodegenerative disorder clinically characterized by a progressive impairment in memory and other cognitive abilities, and accounts for the vast majority of dementia cases (Blennow et al., 2006). AD pathology is characterized by senile plaques and neurofibrillary tangles, combined with massive neuronal loss, mainly in the hippocampus and association regions of the neocortex. The majority of AD cases occur in the elderly. Owing to changing demographics, and in the current absence of a successful cure or preventive strategy, the number of dementia patients worldwide is projected to increase from 35.6 million in 2010 to 115.4 million in 2050 (Alzheimer's Disease International, 2009).
Mutations in genes encoding for Amyloid Precursors Protein or presenilin 1 and presenilin 2 genes (APP, PSEN1 and PSEN2, respectively) account for about 5% of cases, characterized by an early onset (before 65 years of age). The majority of cases of AD are however sporadic, and likely several genetic and environmental factors contribute to their development. It is known that the presence of the ε4 allele of the Apolipoprotein E gene is a susceptibility factor, increasing the risk of about 4 fold. A number of additional genetic factors, including cytokines, chemokines, Nitric Oxide Synthases, contribute to the susceptibility for the disease.
Some studies have suggested a possible role for epigenetic changes in AD etiology. Epigenetics relate to stable and heritable patterns of gene expression and genomic functions that do not involve changes in DNA sequence (Feinberg, 2007). In particular, DNA methylation, the most investigated epigenetic hallmark, is a reversible mechanism that modifies genome function and chromosomal stability through the addition of methyl groups to cytosine located in CpG dinucleotides to form 5 methylcytosine (5mC). AD patients display high homocysteine and low B12 vitamin and folate in blood, suggesting a dysregulation in the S-adenosylmethionine cycle that contributes methyl donors for DNA methylation (Scarpa et al., 2006). Moreover, an age-specific epigenetic drifts associated with unusual methylation patterns in late onset AD was identified, supporting a potential role for epigenetic effects in the development of the disease (Wang et al., 2008).
Repetitive elements comprise ∼45% of the human genome. Those include one million Alu sequences occupying ∼10% of the genome, and LINE-1 elements also representing a large genomic portion (Ehrlich, 2002). These interspersed repeated DNA sequences, as well as tandem repeats including DNA satellites (i.e. SAT-α) generally found in centromeres or centromere-adjacent heterochromatin (Lee et al., 1997), contain numerous CpG dinucleotides. Therefore, the methylation status of these sequences is a major contributor of global DNA methylation patterns (Yang et al., 2004) and have been investigated in relation to a variety of human diseases (Pogribny and Beland, 2009).
In the past few years, prefrontal cortex and lymphocytes from AD patients have been used to analyze DNA methylation patterns in genes with a potential role in AD etiology (Zawia et al., 2009). However, the role of methylation of repetitive elements in peripheral blood leukocytes of AD patients has never been investigated so far.
The purpose of the present study was to evaluate Alu, LINE-1 and SAT-α methylation changes in AD by means of a quantitative approach. In particular, we estimated global DNA methylation in LINE-1 and Alu elements and centromeric SAT-α sequences in a population of 43 AD patients and 38 non-demented donors. Global DNA methylation was evaluated and correlated with Mini Mental Scale Examination score, APOE status, Cerebrospinal fluid (CSF) levels of β-amyloid (Ab), total Tau and Tau phosphorylated at position 181 (Ptau).
Materials and Methods
Subjects
The study included 43 patients with AD consecutively recruited between 2009 and 2010 at the Alzheimer Unit, Fondazione Ca' Granda, IRCCS Ospedale Maggiore Policlinico, Milan, Italy. In this patients, the mean duration of the disease is 3 years (MIN-MAX: 1-10 years). All patients underwent a standard battery of examinations, including medical history, physical and neurological examination, screening laboratory tests, neurocognitive evaluation, brain magnetic resonance imaging (MRI) or computed tomography (CT) and, if indicated, positron emission computed tomography (PET). Dementia severity was assessed by the clinical dementia rating (CDR) and the mini mental state examination (MMSE). As a control group we enrolled 38 healthy volunteers, matched for age, region, and smoking habits. The mean age at the time of enrollment was 75 years (MIN-MAX: 55-87; SD 8.0) for AD cases and 67 years (MIN-MAX: 56-87; SD 8.6) for healthy controls. Twenty patients were males (46.5%) and 23 females (53.5 %) while in healthy control group, 25 subjects were males (64.9%) and 13 subjects were females (35.1%). In accordance with our institutional guidelines, all of the patients gave their informed consent. Diagnosis of AD was done in accordance to McKhann et al criteria (McKhann et al., 1984).
For all AD patients and healthy volunteers, we collected an EDTA blood sample. CSF samples were obtained from AD patients through diagnostic lumbar punctures. Blood and CSF samples were stored at -80°C until analysis. The characteristics of the AD patients are reported in table 1 and include APOE polymorphisms, MMSE, CSF β-amyloid, CSF Tau total and CSF Tau phosphorylated.
Table 1.
Characteristics of the Alzheimer Disease subjects (n=43)
| Variable | |
|---|---|
| Age | |
| Mean (SD) | 75 (8.0) |
| Min-Max | 55-87 |
| Gender | |
| Male n (%) | 20 (46.5) |
| Female n (%) | 23 (46.5) |
| APOE Polymorphisms | |
| ε3- ε3 n (%) | 25 (58.1%) |
| ε3- ε4 n (%) | 15 (34.9%) |
| ε2- ε4 n (%) | 2 (4.7%) |
| ε4- ε4 n (%) | 1 (2.3%) |
| Mini Mental State Examination (MMSE) | |
| Mean (SD) | 20.8 (4.4) |
| Min-Max | 8.0-28 |
| β-amyloid 1-42 | |
| Mean (SD) | 462.8 (208.2) |
| Min-Max | 152-1044 |
| Tau (total) | |
| Mean (SD) | 879.5 (553.0) |
| Min-Max | 119-2121 |
| Tau (phosphorylated-181) | |
| Mean (SD) | 73.6 (35.7) |
| Min-Max | 17-166 |
DNA extraction and bisulfite treatment of the DNA
High-molecular weight DNA was isolated from whole blood using a Flexigene Kit (Qiagen, Hildren, Gemany), as described by the manufacturer. The amount of DNA for each sample was determined by measuring the optical density at 260 nm wavelength using a spectrophotometer (Eppendorf AG, Germany). DNA samples were aliquoted and stored at −20°C.1μg DNA (concentration 50 ng/μl) was treated using EZ DNA Methylation-Gold™ Kit (Zymo Research, Orange, CA, USA) according to the manufacturer's protocol. Final elution was performed with 30 μl of M-Elution Buffer. Bisulfite-treated DNA was stored at –20°C and used shortly after treatment.
PCR and pyrosequencing
DNA methylation was quantitated using bisulfite-PCR and Pyrosequencing (Yang et al., 2004). In brief, the samples were bisulfite treated and PCR amplified. Repetitive elements primers were designed towards a consensus Alu or LINE-1 sequence and allowed the amplification of a representative pool of repetitive elements to serve as a surrogate for global DNA methylation changes (Yang et al., 2004). Analysis of DNA methylation in Alu, LINE-1 and SAT-α repetitive element was performed using previously published methods (Bollati et al., 2007; Bollati et al., 2009a; Yang et al., 2004). For each reaction, a 50 μl PCR was carried out in 50 μl of GoTaq Green Master mix (Promega, Madison, WI, USA), 1 pmol of the forward primer 1 pmol of the reverse primer, 50 ng of bisulfite-treated genomic DNA and water. Bisulfite-modified DNA was amplified and genotyped with primers as follows: forward 5′-biotin-TTTTTATTAAAAATATAAAAATT-3′, reverse 5′-CCCAAACTAAAATACAATAA-3′ and sequencing primer 5′-AATAACTAAAATTACAAAC-3′; forward 5′- TTTTGAGTTAGGTGTGGGATATA-3′, reverse 5′- biotin-AAAATCAAAAAATTCCCTTTC -3′ and sequencing primer 5′- AGTTAGGTGTGGGATATAGT-3′; forward 5′- biotin-TGTAAGTGGATATTTGGATTATTGG-3′, reverse 5′- TTTCCAAAAAAATCTTCAAAAAAAT-3′ and sequencing primer 5′-CTCAAAAATTTCTAAAAATACTTCTC-3 for Alu, LINE-1 and SAT-α, respectively. PCR conditions consisted of 96°C for 90 s, followed by 43°C for 60 s 72°C for 120 s (45 cycles) for Alu; 95°C for 30 s, followed by 50°C for 30 s 72°C for 30 (45 cycles) for LINE-1 and 95°C for 60 s, followed by 55°C for 60 s 72°C for 60 s (45 cycles) for SAT-α. The size of bisulfite PCR products was 168 bp for LINE-1, 148 bp for Alu, and 223 bp for SAT-α.
One of the primers was biotin-labelled and used to purify the final PCR product using Sepharose beads. The PCR product was bound to Streptavidin Sepharose HP (Amersham Biosciences, Uppsala, Sweden) and the Sepharose beads containing the immobilized PCR product were purified, washed, denatured using a 0.2 M NaOH solution, and washed again using the Pyrosequencing Vacuum Prep Tool (Pyrosequencing, Inc., Westborough, MA), as recommended by the manufacturer. Then, 0.3 μM pyrosequencing primer was annealed to the purified single-stranded PCR product and pyrosequencing was performed using the Pyromark MD (Biotage, Sweden) System (Pyrosequencing, Inc.). Methylation quantification was performed using the provided software. The degree of methylation was expressed as %5-methylated cytosines (%5mC) over the sum of methylated and unmethylated cytosines. Every sample was tested three times for each marker to confirm reproducibility of our results. The average of the triplicates was used in the statistical analysis.
Apolipoprotein E gene analysis
High-molecular-weight DNA was amplified by polymerase chain reaction (thermal cycler by Perkin Elmer). The primers (5′TCC AAG GAG CTG CAG GCG GCG CA3′ and 5′ ACA GAA TTC GCC CCG GCC TGG TAC ACT GCC A3′) amplify a 227 bp region of DNA as described (Wenham et al., 1991). The 10-fold PCR buffer contained 100 mM Tris-HCl (pH 8.2), 15 mM MgCl2, 500 mM KCl and 0.1% gelatin. Each reaction mixture contained 500 ng of genomic DNA, 30 pmol each primer, dNTPs 200 mM, dimethyl sulphoxide 10%, 0.2 U Taq DNA polymerase (Amplitaq, Perkin Elmer), 1 × PCR buffer and H2O to a final volume of 25 ml. Cycles parameters were: 5 min at 94°C for the first cycle, denaturation at 94°C for 30 s, annealing at 65°C for 30 s, extension at 72°C for 60 s for the subsequent 35 cycles, and a final extension at 72°C for 10 min. PCR products were digested with 3 units of HhaI at 37°C for 3 h, electrophoresed on a 4% agarose gel (1% agarose, 3% NuSieve) and photographed under ultraviolet light after staining with ethidium bromide: the pattern of obtained genomic fragments was similar to previously reported findings (Hixson and Vernier, 1990).
CSF β-amyloid, Tau total and Tau phosphorylated assessment
CSF samples were obtained in polypropylene tubes by LP at the L4/L5 or L3/L4 interspace, centrifuged at 4 °C and stored at ≤ –30 °C until analysis. CSF cell counts, glucose and proteins were determined. Albumin was measured by rate nephelometry. To evaluate the integrity of the brain blood barrier (BBB) and the intrathecal IgG production, the albumin quotient (CSF albumin/serum albumin) X 103 and the IgG index (CSF albumin/serum albumin)/(CSF IgG/serum IgG) were calculated (Sellebjerg et al., 2000) Aβ42, total tau (tau) and tau phosphorylated at position 181 (P-tau) CSF levels were determined with human specific ELISA kits (Innogenetics), as previously reported (Andreasen and Blennow, 2005).
Statistical analysis
For each of the 81 subjects (43 cases and 38 healthy volunteers) we collected one blood sample and obtained three replications of Pyrosequencing-based analysis of DNA methylation for LINE-1, Alu and SAT-α. In order to evaluate differences we performed a T-test on the means of the replications for unadjusted analysis and implemented simple linear regression model to correct for the effect of age and gender. Partial unadjusted tests were conducted in AD patients on MMSE results and gender and in healthy volunteers on gender. To assess the correlation between our outcomes and covariates we used the Pearson correlation coefficient (ρ) as well as for correlation with MMSE and age in AD patients subgroup. Chi-Square exact (Fisher) test was used to verify presence of association between APOE status (at least one ε4 polymorphism versus no-ε4 polymorphism) and class of methylation in AD patients. All analyses were performed in SAS 9.2 (SAS Institute Inc., Cary, NC).
Results
Methylation levels in AD and healthy subjects
Alu methylation was significantly decreased in patients with AD compared with healthy volunteers (ADs: 25.5 %5mC versus 25.8 %5mC, P=0.01), as well as a significant increase of LINE-1 methylation (ADs: 83.6 %5mC, Healthy volunteers: 83.1 %5mC, p-value: 0.02) in unadjusted analysis (Fig.1). A trend towards a decreased SAT-α DNA methylation was observed in patients with AD as compared with healthy volunteers (70.6 %5mC versus 71.7 %5mC), although the significance threshold was not reached (P=0.29). In AD patients, gender differences were observed in Alu (M: 25.7 %5mC; F: 25.4 %5mC; p=0.05), LINE-1 (M: 84.0 %5mC; F: 83.3 %5mC; p=0.04) and SAT- α (M: 72.2 %5mC; F: 69.2 %5mC; p=0.04) while in healthy volunteers just SAT- α was higher in males than in females (M: 73.2 %5mC; F: 68.9 %5mC; p=0.002) (Fig.1). In multivariate analysis adjusting for age and gender, LINE-1 was increased in AD patients compared with healthy volunteers (P=0.04), whereas the difference in Alu and SAT-α methylation between AD patients and control was not statistically significant (P>0.1).
Figure 1.

Median methylation levels of Alu (1A), LINE-1 (1B) and SAT-α (1C) repetitive genomic sequences in healthy volunteers and Alzheimer Disease Subjects. The medians for males, females and for the entire population are reported.
Correlation between age and DNA methylation
Considering all subjects (AD patients and healthy volunteers), a significant associations between age and Alu elements methylation was observed (ρ=-0.3 %5mC/year, P=0.006), but not between age and LINE-1 and SAT- α methylation (ρ =0.18 %5mC/year, P=0.10 and ρ =0.06 %5mC/year, P=0.6, respectively) (Fig. 2). In AD patients, however, DNA methylation did not show significant associations with age in Alu elements (ρ =-0.22 %5mC/year, P=0.16), LINE-1 (ρ =0.16 %5mC/year, P=0.32) and SAT- α methylation (ρ =0.8 %5mC/year, P=0.62).
Figure 2.
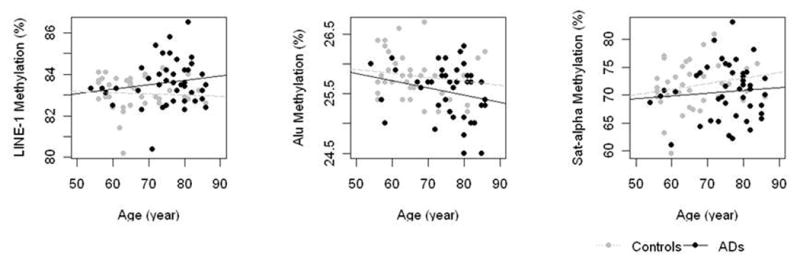
Association between DNA methylation in Alu (2A), LINE-1 (2B) and SAT-α (2C) repetitive genomic sequences and age at time of sampling.
Correlation between CSF markers and DNA methylation
We evaluated the correlations among CSF levels of Aβ, tau and Ptau. As expected, Tau was positively correlated with Ptau (ρ=0.77 %5mC/unit, P<0.0001) (Figure 3).
Figure 3.
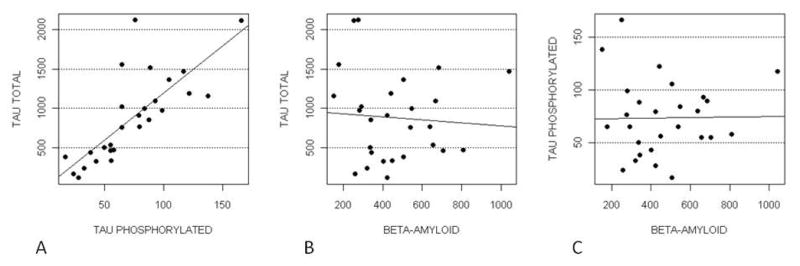
PTau and Tau (Panel A), Beta-amyloid and Tau (Panel B) Beta-amyloid and PTau (Panel C).
We did not observe any correlation between any these clinical markers and DNA methylation of LINE-1, Alu, or SAT-α (Table 2). Also we did not observe any association between APOE status and LINE-1, Alu and SAT-α methylation (data not shown).
Table 2.
Correlation between clinical markers and DNA methylation in Alzheimer Disease subjects (n=43).
| ALU | LINE-1 | SAT-α | ||
|---|---|---|---|---|
| CSF β-amyloid | Beta* | 0.000 | -0.002 | 0.004 |
| SE | 0.000 | 0.001 | 0.847 | |
| p-value | 0.948 | 0.07 | 0.406 | |
| CSF TAU-total | Beta* | 0.000 | 0.000 | 0.000 |
| SE | 0.000 | 0.000 | 0.002 | |
| p-value | 0.579 | 0.370 | 0.879 | |
| CSF TAU-phosporilated | Beta* | 0.000 | 0.000 | 0.002 |
| SE | 0.003 | 0.007 | 0.029 | |
| p-value | 0.939 | 0.996 | 0.936 |
Beta are regression coefficients from linear regression, expressing the size of methylation change associated with the independent variable.
Correlation between MMSE and DNA methylation (Figure 1S)
MMSE was administered to AD patients at time of sampling. MMSE average score was 20.9 point and ranged from 7.99 to 28 points. MMSE did not correlate with Alu (ρ=-0.04 %5mC/point, P=0.79), LINE-1 (ρ=0.16 %5mC/point, P=0.34), and SAT-α methylation (ρ=0.11 %5mC/point, P=0.5). When we divided patients in two groups according to the MMSE median (22/30 score), the group with best performances in MMSE showed border-line higher levels of LINE-1 methylation compared to the group with worst performances (MMSE>22: 83.9 %5mC; MMSE<=22: 83.2 %5mC; p=0.05). AD patients above and below the median MMSE did not show differences in Alu and SAT-α methylation (data not shown).
Discussion
In the present study, we analyzed methylation in Alu, LINE-1 and SAT-α repetitive element in blood of 43 patients with AD and 38 healthy volunteers and found a an increase in LINE1 methylation after adjusting for age and gender. Studies investigating altered DNA methylation in AD have been mainly conducted in human postmortem brain tissues, showing hyper or hypomethylation of several candidate genes (reviewed in (Chouliaras et al., 2010)). A few studies that have investigated DNA methylation in peripheral blood leukocytes in AD patients have obtained heterogeneous findings. To our knowledge, methylation in Alu, LINE-1 and SAT- α repetitive elements has yet never been evaluated.
A major issue for AD molecular studies is the lack of reliable samples in the different stages of the disease. Postmortem brain tissues samples, even if useful to understand disease mechanisms, can be used only to observe the end results of AD processes, which do not necessarily reflect the mechanisms responsible for disease development. Even though methylation patterns in leukocytes and brain are likely to be different, the potential use of blood-based gene expression profiling in the diagnosis of brain disorders have been shown in several independent research (Burczynski and Dorner, 2006; Gladkevich et al., 2004; Lonneborg, 2008; Sharp et al., 2006). In the present study, however, blood DNA is derived from a mixed cell population and we cannot exclude that differences in the number of subpopulations might have contributed to determine the results.
In the present study we also observed hypomethylation of Alu repetitive elements in AD patients compared to healthy volunteers. However, the difference in Alu methylation between AD and healthy volunteers did not reach statistical significance in multivariate models adjusting for age and gender. This may be partly explained by the small size of our population, which provided us with limited statistical power to perform multivariate modeling. In a cohort of elderly individuals in Eastern Massachusetts, we have previously demonstrated that Alu methylation declines individuals age, whereas LINE-1 methylation did not change over time (Bollati et al., 2009b). Our previous findings indicate that genome-wide losses of methylation through aging may account, at least in part, for the increased disease rates through aging (Bjornsson et al., 2004; Jiang et al., 2004; Petronis, 2001). The alterations in Alu methylation observed in the present study, although not statistically significant, might be determined more by the aging process itself, characterized by oxidative stress, disturbed calcium homeostasis, chromosomal instability, impaired DNA repair and accumulation of nuclear and mitochondrial DNA damage (Chouliaras et al., 2010), than by AD development.
Aging-related DNA methylation may represent one of the mechanisms by which environmental and dietary factors can promote AD (Fuso et al., 2009). Aging, in fact, is the most significant risk factor for AD and is associated with multiple epigenetic alterations (Ferri et al., 2005). Fraga found that monozygous twins are epigenetically indistinguishable during the early years of life while older twins exhibited remarkable differences in their overall content and genomic distribution of 5-methylcytosine (Fraga et al., 2005).
LINE-1 methylation was increased in AD patients in our study. Conversely, previous findings showed that LINE-1 hypomethylation could be related to different disease conditions, such as cancer and cardiovascular disease (Jones, 1986), which however have a different pathogenesis than AD.
Sporadic AD is thought to be caused by environmental and/or endogenous factors that may be associated with mitochondrial dysfunction and increased Reactive Oxygen Species (ROS) production followed by DNA damage (James et al., 2003). During DNA damage, DNA methyl-transferases (DNMTs) are up-regulated and bind to the DNA lesions with higher affinity (James et al., 2003). Up-regulation of DNMTs secondary to DNA damage may lead to increased LINE-1 methylation.
The changes we observed are small in size. However, The methylation difference in repetitive elements, apparently small when expressed in percentage over the total number of cytosine in the considered position, involve a large number of molecules distributed over the genome.
Considering MMSE as continuous variable, no association was observed between Alu, LINE1 and SAT-α methylation and MMSE score. However, a borderline significant difference was found comparing 2 groups characterized by a MMSE greater or lower than the median value. Unexpectedly, patients with a MMSE greater than the median value showed higher LINE-1 methylation than patients with a MMSE lower than the median value. In addition, beside MMSE, other cognitive tests (i.e. ADAS-cog) are currently available and should be evaluated in future investigations. Based on our data, we might speculate that Alzheimer disease might have two different phases with respect to LINE-1 methylation. At first, LINE-1 methylation might increase in AD patients, whereas in advanced-stage AD, a compromised methyl cycle may result in a decrease in DNA methylation. However, these results are far from being conclusive and deserve further investigation.
In conclusion, our data suggest that LINE-1 methylation may lead to a better understanding of AD pathogenesis and course, and may contribute to identify novel markers useful to assess risk stratification. However, because of the small sample size and the number of statistical tests performed, we cannot exclude that some of our findings might be due to chance. Further prospective investigations are warranted to evaluate the dynamics of DNA methylation from early-stage AD to advanced disease.
Supplementary Material
Acknowledgments
This study was supported in part by New Investigator funding from the HSPH-NIEHS Center for Environmental Health (ES000002), PS39 from Italian Ministry of Health, Monzino Foundation and Ing. Cesare Cusan.
References
- Prince M, Jackson J. Alzheimer's Disease International. World Alzheimer Report 2009. 2009. [Google Scholar]
- Andreasen N, Blennow K. CSF biomarkers for mild cognitive impairment and early Alzheimer's disease. Clin Neurol Neurosurg. 2005;107:165–173. doi: 10.1016/j.clineuro.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Bjornsson HT, Fallin MD, Feinberg AP. An integrated epigenetic and genetic approach to common human disease. Trends Genet. 2004;20:350–358. doi: 10.1016/j.tig.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- Bollati V, Baccarelli A, Hou L, Bonzini M, Fustinoni S, Cavallo D, Byun HM, Jiang J, Marinelli B, Pesatori AC, Bertazzi PA, Yang AS. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res. 2007;67:876–880. doi: 10.1158/0008-5472.CAN-06-2995. [DOI] [PubMed] [Google Scholar]
- Bollati V, Fabris S, Pegoraro V, Ronchetti D, Mosca L, Deliliers GL, Motta V, Bertazzi PA, Baccarelli A, Neri A. Differential repetitive DNA methylation in multiple myeloma molecular subgroups. Carcinogenesis. 2009a;30:1330–1335. doi: 10.1093/carcin/bgp149. [DOI] [PubMed] [Google Scholar]
- Bollati V, Schwartz J, Wright R, Litonjua A, Tarantini L, Suh H, Sparrow D, Vokonas P, Baccarelli A. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev. 2009b;130:234–239. doi: 10.1016/j.mad.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burczynski ME, Dorner AJ. Transcriptional profiling of peripheral blood cells in clinical pharmacogenomic studies. Pharmacogenomics. 2006;7:187–202. doi: 10.2217/14622416.7.2.187. [DOI] [PubMed] [Google Scholar]
- Chouliaras L, Rutten BP, Kenis G, Peerbooms O, Visser PJ, Verhey F, van Os J, Steinbusch HW, van den Hove DL. Epigenetic regulation in the pathophysiology of Alzheimer's disease. Prog Neurobiol. 2010;90:498–510. doi: 10.1016/j.pneurobio.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002;21:5400–5413. doi: 10.1038/sj.onc.1205651. [DOI] [PubMed] [Google Scholar]
- Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y, Jorm A, Mathers C, Menezes PR, Rimmer E, Scazufca M. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366:2112–2117. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuso A, Nicolia V, Pasqualato A, Fiorenza MT, Cavallaro RA, Scarpa S. Changes in Presenilin 1 gene methylation pattern in diet-induced B vitamin deficiency. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.02.013. [DOI] [PubMed] [Google Scholar]
- Gladkevich A, Kauffman HF, Korf J. Lymphocytes as a neural probe: potential for studying psychiatric disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:559–576. doi: 10.1016/j.pnpbp.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res. 1990;31:545–548. [PubMed] [Google Scholar]
- James SJ, Pogribny IP, Pogribna M, Miller BJ, Jernigan S, Melnyk S. Mechanisms of DNA damage, DNA hypomethylation, and tumor progression in the folate/methyl-deficient rat model of hepatocarcinogenesis. J Nutr. 2003;133:3740S–3747S. doi: 10.1093/jn/133.11.3740S. [DOI] [PubMed] [Google Scholar]
- Jiang YH, Bressler J, Beaudet AL. Epigenetics and human disease. Annu Rev Genomics Hum Genet. 2004;5:479–510. doi: 10.1146/annurev.genom.5.061903.180014. [DOI] [PubMed] [Google Scholar]
- Jones PA. DNA methylation and cancer. Cancer Res. 1986;46:461–466. [PubMed] [Google Scholar]
- Lee C, Wevrick R, Fisher RB, Ferguson-Smith MA, Lin CC. Human centromeric DNAs. Hum Genet. 1997;100:291–304. doi: 10.1007/s004390050508. [DOI] [PubMed] [Google Scholar]
- Lonneborg A. Biomarkers for Alzheimer disease in cerebrospinal fluid, urine, and blood. Mol Diagn Ther. 2008;12:307–320. doi: 10.1007/BF03256296. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Petronis A. Human morbid genetics revisited: relevance of epigenetics. Trends Genet. 2001;17:142–146. doi: 10.1016/s0168-9525(00)02213-7. [DOI] [PubMed] [Google Scholar]
- Pogribny IP, Beland FA. DNA hypomethylation in the origin and pathogenesis of human diseases. Cell Mol Life Sci. 2009;66:2249–2261. doi: 10.1007/s00018-009-0015-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpa S, Cavallaro RA, D'Anselmi F, Fuso A. Gene silencing through methylation: an epigenetic intervention on Alzheimer disease. J Alzheimers Dis. 2006;9:407–414. doi: 10.3233/jad-2006-9406. [DOI] [PubMed] [Google Scholar]
- Sellebjerg F, Jensen CV, Christiansen M. Intrathecal IgG synthesis and autoantibody-secreting cells in multiple sclerosis. J Neuroimmunol. 2000;108:207–215. doi: 10.1016/s0165-5728(00)00292-7. [DOI] [PubMed] [Google Scholar]
- Sharp FR, Xu H, Lit L, Walker W, Apperson M, Gilbert DL, Glauser TA, Wong B, Hershey A, Liu DZ, Pinter J, Zhan X, Liu X, Ran R. The future of genomic profiling of neurological diseases using blood. Arch Neurol. 2006;63:1529–1536. doi: 10.1001/archneur.63.11.1529. [DOI] [PubMed] [Google Scholar]
- Wang SC, Oelze B, Schumacher A. Age-specific epigenetic drift in late-onset Alzheimer's disease. PLoS One. 2008;3:e2698. doi: 10.1371/journal.pone.0002698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32:e38. doi: 10.1093/nar/gnh032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawia NH, Lahiri DK, Cardozo-Pelaez F. Epigenetics, oxidative stress, and Alzheimer disease. Free Radic Biol Med. 2009;46:1241–1249. doi: 10.1016/j.freeradbiomed.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.