

This review summarizes the pathogenic mechanisms involved in renal functional deterioration in patients with atherosclerotic renal artery stenosis.
Abstract
Renal artery stenosis remains an important contributor to renal failure in the elderly population, but uncertainty continues to surround the mechanisms underlying progressive renal dysfunction. Here, we present the current understanding of the pathogenic mechanisms responsible for renal injury in these patients, with emphasis on those involved in disease progression.
Vascular occlusive diseases of the kidney account for an important proportion of secondary hypertension and are increasingly associated with progressive renal dysfunction and cardiovascular complications. Among them, renal artery stenosis (RAS), the narrowing of one or both main renal arteries or their major branches, remains the most frequent primary disease of the renal vasculature (66, 72).
Atherosclerotic renovascular disease is responsible for ∼90% of cases of RAS and affects almost 7% of individuals older than 65 (40). Importantly, these patients tend to develop renovascular hypertension, which accelerates renal injury and predisposes to potentially lethal complications such as myocardial infraction, pulmonary edema, or stroke (18, 56, 58).
Over the last couple of decades, mounting evidence has clarified the pathogenesis of RAS and the molecular mechanisms upregulated beyond the stenotic lesion (1, 46). Activation of the renin-angiotensin-aldosterone system (RAAS), increased generation of reactive oxygen species (ROS), and amplification of profibrotic mechanisms are cardinal deleterious pathways extensively described. Importantly, the interplay of these pathways generates a vicious cycle that potentiates renal damage (FIGURE 1). Nevertheless, since many of these pathways are interdependent and hard to discern, their specific roles in the progression of kidney damage remain to be elucidated.
FIGURE 1.

Schematic of multiple mechanisms responsible for kidney injury in renal artery stenosis
Reduced kidney perfusion activates the renin-angiotensin-aldosterone system, triggering inflammation and oxidative stress. Atherosclerosis also promotes oxidative stress and inflammation, leading to apoptosis, mitochondrial dysfunction, and defective angiogenesis. This in turn promotes microvascular rarefaction, which exacerbates oxidative stress and leads to hypoxia and tubular dysfunction, tubulointerstitial fibrosis, and renal failure.
This review aims to summarize the current understanding of the pathophysiological mechanisms activated in the hypoperfused kidney and the interactions among them. Discussion is largely confined to the contribution of these injurious pathways to progression of kidney disease and its complications.
RAAS Activation
Activation of the RAAS leads to systemic vasoconstriction, salt retention, and stimulation of the sympathetic nervous system, resulting in rapid development of hypertension that aims to restore renal perfusion pressure. However, persistently high levels of angiotensin II activate several mechanisms that promote vascular remodeling and tissue fibrosis within the stenotic kidney (57, 59, 63). For example, studies in cultured mesangial cells have uncovered unique modulatory actions of angiotensin II on extracellular matrix synthesis through upregulation of signal transduction pathways, activation of early growth genes, and generation of cytokines and growth factors (63). Similarly, angiotensin II exerts a strong stimulatory effect on transcription of profibrotic factors like transforming growth-factor (TGF)-β (79) and plasminogen activator inhibitor (PAI)-1, leading to accumulation of extracellular matrix (21). Hence, maladaptive prolonged activation of angiotensin II can directly lead to renal fibrosis.
In addition to its canonical roles, angiotensin II has been implicated in modulation of the immune system through activation of angiotensin receptor (AT)-1 on inflammatory cells. Increased angiotensin II activity upregulates expression of cell adhesion molecules, promoting recruitment and activation of inflammatory cells, including neutrophils, macrophages, and T-lymphocytes (55). Similarly, in renovascular hypertensive mice, angiotensin II promotes evolution of vulnerable plaques via a T-helper switch, independently from its hemodynamic effect (51).
These observations are supported by studies in uninephrectomized, spontaneously hypertensive rats that showed that both angiotensin-converting-enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) decrease renal cell proliferation and suppress infiltration of mononuclear cells, diminishing extracellular matrix deposition and nephrosclerosis (33).
Atherosclerosis
Systemic atheromatous disease is found in many patients with RAS and is commonly associated with risk of progressive extrarenal vascular disease (17). Atheroembolic, atherosclerotic, and hypertensive vascular changes in RAS kidneys correlate with increased prevalence of cardiovascular disease, greater degree of renal dysfunction, and severe dyslipidemia (42). Furthermore, the severity of renovascular disease in the contralateral kidney does not predict the extent of baseline renal dysfunction, underscoring the importance of intrarenal parenchymal damage, rather than the hemodynamic effects of a given stenosis (16).
The impact of atherosclerosis is likely mediated by many mechanisms described in other sections (FIGURE 1) and by its effects on vascular wall and microvascular remodeling. We have previously shown in swine RAS that coexistence of early atherosclerosis and hypoperfusion aggravates renal functional and structural impairment, characterized by pronounced increases in systemic and tissue oxidative stress, inflammation, and fibrosis (10). Importantly, upregulation of these mechanisms in turn potentiates the deleterious effect of hypoperfusion on the kidney, accelerating the progression to chronic renal failure.
Atherosclerosis impacts in the response to revascularization, likely by inducing irreversible vascular and tubulointerstitial remodeling. Although revascularization of a non-atherosclerotic swine RAS kidney improves renal blood flow (RBF) and restores glomerular filtration rate (GFR) (32), both remain impaired when atherosclerosis is superimposed on RAS (31). In addition, tubulointerstitial injury and microvascular rarefaction persist 4 wk after revascularization in the atherosclerotic RAS kidney, implicating these alterations in the blunted improvement in renal function in patients with atherosclerotic renovascular disease (2, 78).
Oxidative Stress
Several lines of evidence highlight the involvement of oxidation-sensitive mechanisms in the pathophysiology of RAS. We have shown that unilateral swine RAS is accompanied by increases in systemic levels of isoprostanes and renal expression of oxidative stress markers like NAD(P)H oxidase and nitrotyrosine (28, 31, 81).
Angiotensin II is a powerful stimulus for ROS generation. Forms of hypertension associated with elevated circulating levels of angiotensin II also show increased superoxide anion radical production (61). These observations are underscored by direct correlations between plasma renin activity and isoprostanes levels in early RAS (45). Being potent vasoconstrictors, isoprostanes sustain renovascular hypertension at the chronic phase, in which PRA may decline. Indeed, Tempol, which promotes ROS metabolism, causes rapid dose-dependent reductions in blood pressure and prevents progression to renal dysfunction in several experimental models of hypertension (53).
Notably, ROS directly impact vascular tone, producing structural and functional consequences in the stenotic kidney. Homologs of the NADPH oxidase subunits are expressed in different regions of the kidney, including the glomeruli, tubules, and vessels, and are upregulated in hypertension (4). ROS may act as direct vasoconstrictors and produce multiple vasoactive and fibrogenic factors. Furthermore, ROS decreases availability of nitric oxide-favoring predominance of vasoconstrictors like angiotensin II and endothelin-1 (81).
However, this deleterious pathway can be modulated by antioxidant intervention. Increased oxidative stress in the stenotic swine kidney can be prevented by antioxidant vitamins, and Tempol improves renal perfusion and oxygenation (54), underscoring involvement of oxidative stress in the pathogenesis of several facets of renal injury in RAS (81).
Microvascular Loss
Remodeling or loss of small vessels in the stenotic kidney accelerate progression of renal injury and are main determinants of the response to revascularization (5, 7). Microvessels (vessels of <500 μm in diameter) are responsible for delivery of blood to the renal parenchyma and possess unique abilities to adapt to local metabolic demands, sustaining renal function in early stages of RAS (47). Therefore, alterations in microvascular structure or function may lead to hypoperfused and hypo-oxygenated regions in the kidney, triggering matrix accumulation, interstitial fibrosis, and renal dysfunction.
Importantly, reduced RBF affects not only the number of microvessels in the stenotic kidney but also their structure and functionality. Indeed, 3D microcomputed tomography images obtained from stenotic-kidney of hypertensive pigs 10 wk after induction of RAS shows significant impairment of the microvascular architecture and spatial density (FIGURE 2). Furthermore, superimposition of atherosclerosis aggravates renal microvascular thickening, paralleling the development of fibrosis in the stenotic kidney (11). These observations were accompanied by declined microvascular quality characterized by augmented vessel tortuosity (27, 28).
FIGURE 2.
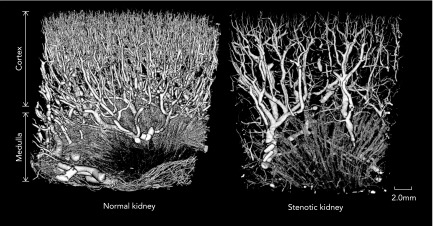
Representative 3D micro-computer tomography images
Representative 3D micro-computer tomography images from a normal (left) and a stenotic (right) swine kidney, showing decreased number of cortical and medullary microvessels in the hypoperfused kidney.
Multiple pathways may contribute to microvascular damage in the stenotic kidney, including oxidative stress, apoptosis, inflammation, and fibrosis. Interestingly, inflammation damages the microvasculature through a mechanism characterized by “defective angiogenesis.” Although inflammatory markers stimulate angiogenesis in early disease (9), neovessels are often tortuous and leaky, allowing inflammatory mediators to extravasate, thereby promoting infiltration of inflammatory cells into the interstitium and aggravating renal parenchymal injury. This stipulation is supported by the coexistence in chronic RAS of increased parenchymal inflammation with reduced microvascular density (80).
Nevertheless, insufficient angiogenesis also contributes to renal microvascular damage. Downregulation of angiogenic factors, possibly due to increased ROS abundance, is associated with decreased spatial density of cortical (31) and medullary (23) microvessels in the stenotic kidney, suggesting that ongoing new vessel formation constitutes a crucial process in preservation of the renal microvasculature. This notion is supported by the observation that intrarenal administration of angiogenic factors like vascular endothelial growth factor (VEGF) can both prevent and reverse stenotic-kidney microvascular damage and is associated with significant improvement in renal function (7, 8).
Renal scarring may also promote microvascular damage and irreversible renal injury. Tubulointerstitial fibrosis is commonly seen in experimental and clinical RAS, associated with hypertensive vascular changes and microvascular rarefaction (42, 81). Presumably, accumulation of extracellular matrix limits the ability of budding microvessels to expand. In contrast, the glomeruli are often well preserved until a late stage of the disease. Importantly, permanent changes in microvascular structure likely affect the response to therapeutic interventions. In swine atherosclerotic RAS, interstitial fibrosis aggravates peritubular capillary rarefaction and reduces oxygen delivery to the microcirculation, compromising renal functional outcomes after revascularization (23). Interestingly, in the cortex, loss of outer cortical microvessels selectively correlates with residual renal function after revascularization (31), implicating this section of the microcirculation in maintenance of GFR. However, given that blood vessels in the kidney cortex often perfuse structures in different levels, the responsible glomeruli may in fact reside in the middle cortex. Further studies are needed to elucidate the role attributed to outer cortical microvessels in regulation of renal function.
Hypoxia and Oxygen Consumption
Few putative mechanisms attributed to “ischemic nephropathy” generate more controversy than the role of hypoxia. For many years, the term “ischemic nephropathy” was used to describe RAS, yet the contribution of hypoxia to progression of the disease has been challenged (71), possibly because global renal oxygen consumption has been assessed mostly from the arterio-venous oxygen gradient and may not adequately reflect regional changes in oxygen availability or metabolism.
To assess intrarenal oxygenation, blood-oxygen-level-dependent magnetic resonance imaging (BOLD-MRI), which evaluates deoxyhemoglobin levels within the kidney, has been applied in experimental and clinical RAS (22, 70, 76). In swine RAS, cortical and medullary hypoxia increased during gradual renal arterial occlusion (41), as well as distal to chronic, significant RAS (FIGURE 3), whereas they are associated with lower cortical and medullary volumes, RBF, and GFR (23).
FIGURE 3.

Blood-oxygen level-dependent magnetic resonance imaging
Blood-oxygen level-dependent magnetic resonance imaging from normal (left) and stenotic (right) swine kidneys. Cortical and medullary hypoxic (red) regions are observed in the poststenotic kidney.
In patients with moderate RAS, despite reduced volume and RBF, cortical and medullary oxygenation is relatively well preserved (35), suggesting that small and gradual reductions in RBF initially trigger compensatory mechanisms that preserve oxygenation by decreasing oxygen consumption. These observations were confirmed in kidneys of rats with mild RAS (62). However, compensation for impaired RBF is limited in patients with severe RAS (34), which results in significant decreases in cortical oxygenation.
In addition to impaired delivery of oxygen, BOLD-MRI offers assessment of functional hypoxia (consequent to changes in renal oxygen consumption) by measuring responses to the loop-diuretic furosemide, which inhibits oxygen-dependent solute transport in the Loop of Henle (76). A blunted increase in stenotic kidney oxygenation after furosemide implies attenuated oxygen consumption related to tubular transport (23). The associated decreased medullary vascular density, impaired angiogenesis, and augmented oxidative stress, inflammation, and tubulointerstitial fibrosis suggest that these pathways might impair the efficiency of tubular reabsorption, limiting oxygen supply and diffusion. The ability of Tempol to improve oxygen utilization efficiency (54) implicates ROS in the pathological mechanism of renal hypoxia. Nevertheless, attenuated tubular oxygen-dependent transport might be initially a defense mechanism that aims to decrease oxygen consumption and prevent hypoxic injury(23, 24, 34), whereas, at advanced disease, failure to respond may also result from severe and potentially irreversible injury to the renal tubules (70).
Apoptosis and Mitochondrial Injury
Angiotensin II induces apoptosis via both AT1 and AT2 receptors through generation of TGF-β, followed by transcription of cell-death genes (3). Additionally, increased numbers of apoptotic cells are associated with an imbalance in the expression of proapoptotic and antiapoptotic mediators in the stenotic kidney (27, 28). Apoptosis, possibly induced by ROS, may also contribute to loss of vascular cells by promoting inflammation and subsequent tissue injury (19). Furthermore, inhibition of apoptosis prevents inflammation and tissue injury in murine renal ischemia-reperfusion injury, implicating apoptosis in inflammation and tissue injury after ischemia (19). Conversely, apoptosis may represent a maladaptive defense mechanism, being less deleterious than necrosis, which may follow severe depletion of ATP and disruption of the cytoskeleton.
Mitochondrial injury often triggers the apoptotic pathway. Opening of the mitochondrial permeability transition pore (mPTP), a channel formed in the inner mitochondrial membrane in response to ischemia, promotes release of cytochrome C into the cytosol, which triggers apoptosis via caspase-3 and -9. Indeed, mPTP inhibitors and mitochondria-targeted peptides attenuate renal damage in experimental ischemia-reperfusion injury (69). In swine atherosclerotic RAS, intravenous infusion of Bendavia at the time of revascularization preserved renal structure and function, emphasizing the role of mPTP dynamics in modulating recovery after revascularization (27). Further studies are needed to explore the ability of such agents to reduce kidney and target organ damage in RAS.
Inflammation
Recent studies have underscored the vital contribution of the innate immune system in the pathogenesis of hypertension. Reports regarding a crucial role of T-cell activation in the pathogenesis of hypertension (38, 50) were followed by demonstrations in angiotensin II-induced hypertensive mice that activated T cells infiltrate the kidney and release inflammatory cytokines (48).
Unilateral clamping of the renal artery in rats is also associated with abundant interstitial chronic inflammatory infiltrates, composed of B-lymphocytes, T-helper lymphocytes, and macrophages (73). Likewise, the stenotic swine kidneys exhibit increased tissue levels of the pro-inflammatory chemokine monocyte chemoattractant protein (MCP-1) and nuclear factor-κB, which were associated with impaired renal function (15, 80). The number of CD45 (B-T cells), CD3 (T cells), and CD163 (macrophages) cells infiltrating the kidney is also increased in atherosclerotic RAS pigs (28). Moreover, we recently demonstrated increased number of inflammatory (M1) and decreased number of reparative (M2) macrophages in the poststenotic swine kidney (29). Importantly, CD68+ macrophages accumulate in kidney obtained from patients with severe RAS, accompanied by robust TGF-β immunoreactivity (36), underscoring the important role of inflammation in the pathogenesis of RAS.
Inflammatory cell infiltration bears several consequences for the kidney and target organs. The poststentotic kidney with reduced RBF and GFR releases multiple inflammatory cytokines that portend renal injury (24, 25). In agreement, systemic C-reactive protein (CRP) levels and peripheral leukocytes are elevated in RAS compared with essential hypertensive patients and healthy controls (64), and are associated with higher prevalence of coronary artery disease and mortality (65). Elevation of systemic neutrophil gelatinase-associated lipocalin levels in RAS patients also implies activation of systemic inflammation (26). Indeed, systemic and kidney-derived cytokines may account for target organ injury in renovascular disease and for some forms of the cardiorenal syndrome (74). It would be important to determine to what extent levels of these cytokines mirror and may serve as indexes of the extent of renal damage.
Fibrogenic Pathways
A great deal of research has been devoted to understanding how renal scarring and tissue remodeling modulate progression to renal dysfunction. Tubulointerstitial fibrosis is the predominant pattern of injury in human RAS and correlates with progression to renal insufficiency (42). Therefore, understanding the profibrotic cascade is critical to prevent disease progression and develop targeted interventions to improve renal function in these patients.
Upregulation of profibrotic factors like TGF-β, PAI-1, or TIMP-1 is commonly observed in experimental RAS (11, 31). TGF-β stimulates expression of MCP-1 in mesangial cells through pathways involving ROS generation, suggesting that this cascade promotes progressive renal disease (14). Indeed, abrogation of TGF-β/Smad3 signaling pathway confers protection against development of fibrosis and atrophy in murine RAS (75). Given that TGF-β expression is elevated in poststenotic kidneys during their remodeling process (although apparently not upon its completion) (36), it might represent a useful therapeutic target.
Expression of TIMP-1 and PAI-1 is significantly upregulated in the RAS kidney, and both localize to the tubular and interstitial compartments (11). Although TIMP-1 inhibits extracellular matrix degradation, leading to accumulation of fibroblasts and collagen deposition (37), increased expression of PAI-1 plays an important role in angiotensin II-mediated hypertensive kidney and heart injury (44, 79). TGF-β released from the stenotic kidney may play a role in cardiac injury in RAS (74). Therefore, treatment approaches oriented to block these pathways might prevent the development of fibrosis and subsequent renal dysfunction.
Novel Treatment Strategies
Recently, there has been growing interest in the promising potential of using cell-based therapies in cardiovascular diseases, and their efficacy, safety, and feasibility in ischemic diseases, including RAS. Intrarenal administration of endothelial progenitor cells (EPC) preserves tissue integrity and improves renal hemodynamic and function in the poststenotic kidney (12, 13). Furthermore, delivery of EPC into the stenotic kidney at the time of revascularization preserves oxygen-dependent tubular function and microvascular architecture, and decreases inflammation and fibrosis, becoming an effective adjunctive therapy to improve renal outcomes in RAS (23).
Mesenchymal stem cells (MSC), mainly isolated from bone marrow or fat tissue, possess unlimited self-renewal capability, unique immunomodulatory properties, and broad differentiation spectrum to produce multiple different cell types (49). Indeed, administration of adipose tissue-derived MSC induced restoration of the renal microcirculation by suppressing inflammatory cytokines and endoplasmic reticulum stress-induced apoptosis (82), normalized RBF and GFR, and decreased stenotic kidney fibrosis. Furthermore, in swine RAS, intrarenal administration of MSC during revascularization attenuated oxidative stress, apoptosis, fibrosis, inflammation, and microvascular remodeling (28). This, in turn, permitted significant improvement in renal function 4 wk after revascularization, underscoring the potential of MSC to restore renal cellular integrity and slow the progression to chronic renal failure.
Endothelin-1 is a potent vasoconstrictor that is activated in renovascular hypertensive rats and acts via its receptor (endothelin A), aggravating tissue injury in the clipped kidney (20). Indeed, endothelin-A blockade preserves microvascular architecture and function, and normalizes RBF and GFR in the poststenotic swine kidney (43). Moreover, endothelin-1 blockers may confer antihypertensive benefits. Further studies are needed to test the efficacy of this tactic in human RAS.
Finally, angiogenic factors like VEGF (7) or hepatocyte growth factor (67) harbor unique vasculo- and renoprotective effects in the stenotic kidney, mainly by preserving the renal microcirculation. Notably, intrarenal delivery of these factors decreased renal scarring independently of changes in blood pressure, becoming attractive candidates for future therapies in patients with RAS.
Lingering Questions
As mentioned before, the role of hypoxia in the pathogenesis of RAS remains controversial. Although BOLD-MRI studies have identified more abundant medullary hypoxic regions in swine RAS compared with normal pigs (41), complex adaptive processes that preserve oxygenation despite substantial reductions in RBF (35) might partly explain the stability of kidney function observed in clinical trials including patients with moderate RAS. Nevertheless, renal adaptation is limited in patients with severe RAS (34), compromising the viability of the stenotic kidney.
One of the most controversial issues in this field is the ability of renal revascularization to recover kidney function in these patients. Although small clinical trials demonstrated lowering blood pressure and improving renal function (60, 77), large randomized clinical trials failed to establish major benefits from revascularizing the poststenotic kidney (2, 78). The recently completed Cardiovascular Outcomes in Renal Atherosclerotic Lesions (CORAL) trial, which evaluates long-term renal and cardiovascular outcomes in RAS patients randomized to optimal medical therapy with or without stenting (52), might provide useful management guidance. Importantly, the mechanisms responsible for the limited renal function response to revascularization remain to be identified, as are indexes to predict its success. We have recently shown in swine RAS that lower basal GFR, decreased tubular oxygen consumption, and preserved endothelial function may predict recovery of GFR after revascularization, whereas elevated release of inflammatory markers is associated with attenuated renal functional recovery, uncovering potentially clinically applicable tools for the identification of patients likely to improve renal function after revascularization (24). Nevertheless, future studies are needed to confirm these results in patients with RAS.
RAS patients frequently have concomitant cardiovascular disease, which contributes to the increased mortality rates (18, 58). However, the mechanism responsible for increased cardiovascular morbidity and mortality in RAS patients remains unclear. In swine RAS, a single intrarenal infusion of EPC improved renal function by reducing renal and systemic oxidative stress and inflammation, preserving remote myocardial microvascular function and architecture, despite sustained hypertension (74). These observations underscore functionally important cross talk between the kidney and heart, possibly mediated by renal injury signals. Therefore, therapeutic strategies aimed to target these pathways may attenuate cardiac remodeling and dysfunction in RAS patients.
Finally, experimental studies have shown the feasibility of several novel treatment strategies to salvage poststenotic kidney function. For example, intrarenal administration of EPC (12, 13, 23, 29) and MSC (28, 82) in swine RAS attenuates microvascular loss and restores stenotic kidney GFR and mitochondrial targeted peptides (27), blockade of vasoconstrictors (43), and angiogenic factors (7, 67), which can ameliorate renal damage. Yet, the best strategy to protect the stenotic kidney in patients with RAS remains to be identified. Further studies are necessary to resolve this question and elucidate the mechanisms by which these novel treatments improve dysfunction in the poststenotic kidney (FIGURE 4).
FIGURE 4.

Proposed mechanisms of action of progenitor/stem cells for repairing the stenotic kidney
Conclusions
RAS remains an important cause of secondary hypertension in the elderly population, but the mechanisms underlying irreversible kidney injury in this disease have not been fully elucidated. Several deleterious pathways have been implicated in the progression of the disease, including oxidative stress, microvascular damage, inflammation, and fibrogenic pathways. Continuous interactions among them amplify renal injury (FIGURE 1), accelerating the progression of kidney damage in RAS. Current clinical management in patients with RAS includes the use of antihypertensive drugs that block the RAAS (ACEIs and ARBs), statins, and antiplatelet therapy (39). However, additional novel treatment approaches that target these deleterious pathways are in the experimental phase. Of these, angiogenic factors (6, 67) and cell-based therapies (12, 23, 28) have demonstrated a potential to address multiple pathways within the kidney to slow progression of renal dysfunction. Development of additional innovative strategies is critically needed, as are new tools to probe kidney injury and indexes to identify patients who are likely to benefit from specific therapeutic interventions.
Footnotes
These studies were supported by grants from the National Institutes of Health (HL-85307, HL-77131, and DK-37608), UL1TR000135, and the American Heart Association.
No conflicts of interest, financial or otherwise, are declared by the author(s).
Author contributions: A.E. and L.O.L. conception and design of research; A.E. performed experiments; A.E. and L.O.L. analyzed data; A.E. and L.O.L. interpreted results of experiments; A.E. prepared figures; A.E. drafted manuscript; A.E. and L.O.L. edited and revised manuscript; A.E. and L.O.L. approved final version of manuscript.
References
- 1. Adamczak M, Wiecek A. Ischemic nephropathy: pathogenesis and treatment. Nefrologia 32: 432–438, 2012 [DOI] [PubMed] [Google Scholar]
- 2. Bax L, Woittiez AJ, Kouwenberg HJ, Mali WP, Buskens E, Beek FJ, Braam B, Huysmans FT, Schultze Kool LJ, Rutten MJ, Doorenbos CJ, Aarts JC, Rabelink TJ, Plouin PF, Raynaud A, van Montfrans GA, Reekers JA, van den Meiracker AH, Pattynama PM, van de Ven PJ, Vroegindeweij D, Kroon AA, de Haan MW, Postma CT, Beutler JJ. Stent placement in patients with atherosclerotic renal artery stenosis and impaired renal function: a randomized trial. Ann Intern Med 150: 840–848, W150–W841, 2009 [DOI] [PubMed] [Google Scholar]
- 3. Bhaskaran M, Reddy K, Radhakrishanan N, Franki N, Ding G, Singhal PC. Angiotensin II induces apoptosis in renal proximal tubular cells. Am J Physiol Renal Physiol 284: F955–F965, 2003 [DOI] [PubMed] [Google Scholar]
- 4. Chabrashvili T, Tojo A, Onozato ML, Kitiyakara C, Quinn MT, Fujita T, Welch WJ, Wilcox CS. Expression and cellular localization of classic NADPH oxidase subunits in the spontaneously hypertensive rat kidney. Hypertension 39: 269–274, 2002 [DOI] [PubMed] [Google Scholar]
- 5. Chade AR. Renovascular disease, microcirculation, and the progression of renal injury: role of angiogenesis. Am J Physiol Regul Integr Comp Physiol 300: R783–R790, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chade AR. VEGF: potential therapy for renal regeneration. F1000 Med Rep 4: 1, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chade AR, Kelsen S. Renal microvascular disease determines the responses to revascularization in experimental renovascular disease. Circ Cardiovasc Interv 3: 376–383, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chade AR, Kelsen S. Reversal of renal dysfunction by targeted administration of VEGF into the stenotic kidney: a novel potential therapeutic approach. Am J Physiol Renal Physiol 302: F1342–F1350, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chade AR, Krier JD, Galili O, Lerman A, Lerman LO. Role of renal cortical neovascularization in experimental hypercholesterolemia. Hypertension 50: 729–736, 2007 [DOI] [PubMed] [Google Scholar]
- 10. Chade AR, Rodriguez-Porcel M, Grande JP, Krier JD, Lerman A, Romero JC, Napoli C, Lerman LO. Distinct renal injury in early atherosclerosis and renovascular disease. Circulation 106: 1165–1171, 2002 [DOI] [PubMed] [Google Scholar]
- 11. Chade AR, Rodriguez-Porcel M, Grande JP, Zhu X, Sica V, Napoli C, Sawamura T, Textor SC, Lerman A, Lerman LO. Mechanisms of renal structural alterations in combined hypercholesterolemia and renal artery stenosis. Arterioscler Thromb Vasc Biol 23: 1295–1301, 2003 [DOI] [PubMed] [Google Scholar]
- 12. Chade AR, Zhu X, Lavi R, Krier JD, Pislaru S, Simari RD, Napoli C, Lerman A, Lerman LO. Endothelial progenitor cells restore renal function in chronic experimental renovascular disease. Circulation 119: 547–557, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chade AR, Zhu XY, Krier JD, Jordan KL, Textor SC, Grande JP, Lerman A, Lerman LO. Endothelial progenitor cells homing and renal repair in experimental renovascular disease. Stem Cells 28: 1039–1047, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheng J, Diaz Encarnacion MM, Warner GM, Gray CE, Nath KA, Grande JP. TGF-beta1 stimulates monocyte chemoattractant protein-1 expression in mesangial cells through a phosphodiesterase isoenzyme 4-dependent process. Am J Physiol Cell Physiol 289: C959–C970, 2005 [DOI] [PubMed] [Google Scholar]
- 15. Cheng J, Zhou W, Warner GM, Knudsen BE, Garovic VD, Gray CE, Lerman LO, Platt JL, Romero JC, Textor SC, Nath KA, Grande JP. Temporal analysis of signaling pathways activated in a murine model of two-kidney, one-clip hypertension. Am J Physiol Renal Physiol 297: F1055–F1068, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cheung CM, Wright JR, Shurrab AE, Mamtora H, Foley RN, O'Donoghue DJ, Waldek S, Kalra PA. Epidemiology of renal dysfunction and patient outcome in atherosclerotic renal artery occlusion. J Am Soc Nephrol 13: 149–157, 2002 [DOI] [PubMed] [Google Scholar]
- 17. Chrysochou C, Kalra PA. Epidemiology and natural history of atherosclerotic renovascular disease. Prog Cardiovasc Dis 52: 184–195, 2009 [DOI] [PubMed] [Google Scholar]
- 18. Conlon PJ, Little MA, Pieper K, Mark DB. Severity of renal vascular disease predicts mortality in patients undergoing coronary angiography. Kidney Int 60: 1490–1497, 2001 [DOI] [PubMed] [Google Scholar]
- 19. Daemen MA, van 't Veer C, Denecker G, Heemskerk VH, Wolfs TG, Clauss M, Vandenabeele P, Buurman WA. Inhibition of apoptosis induced by ischemia-reperfusion prevents inflammation. J Clin Invest 104: 541–549, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Diekmann F, Zart R, Thone-Reineke C, Bauer C, Neumayer HH, Hocher B. Regulation of the renal endothelin system in the two-kidney, one clip renal hypertensive rat. J Cardiovasc Pharmacol 36: S191–S194, 2000 [DOI] [PubMed] [Google Scholar]
- 21. Doller A, Gauer S, Sobkowiak E, Geiger H, Pfeilschifter J, Eberhardt W. Angiotensin II induces renal plasminogen activator inhibitor-1 and cyclooxygenase-2 expression post-transcriptionally via activation of the mRNA-stabilizing factor human-antigen R. Am J Pathol 174: 1252–1263, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ebrahimi B, Gloviczki M, Woollard JR, Crane JA, Textor SC, Lerman LO. Compartmental analysis of renal BOLD MRI data: introduction and validation. Invest Radiol 47: 175–182, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ebrahimi B, Li Z, Eirin A, Zhu XY, Textor SC, Lerman LO. Addition of endothelial progenitor cells to renal revascularization restores medullary tubular oxygen consumption in swine renal artery stenosis. Am J Physiol Renal Physiol 302: F1478–F1485, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eirin A, Ebrahimi B, Zhang X, Zhu XY, Tang H, Crane JA, Lerman A, Textor SC, Lerman LO. Changes in glomerular filtration rate after renal revascularization correlate with microvascular hemodynamics and inflammation in Swine renal artery stenosis. Circ Cardiovasc Interv 5: 720–728, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eirin A, Gloviczki ML, Tang H, Gossl M, Jordan KL, Woollard JR, Lerman A, Grande JP, Textor SC, Lerman LO. Inflammatory and injury signals released from the post-stenotic human kidney. Eur Heart J 34: 540–548, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eirin A, Gloviczki ML, Tang H, Rule AD, Woollard JR, Lerman A, Textor SC, Lerman LO. Chronic renovascular hypertension is associated with elevated levels of neutrophil gelatinase-associated lipocalin. Nephrol Dial Transplant 27: 4153–4161, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eirin A, Li Z, Zhang X, Krier JD, Woollard JR, Zhu XY, Tang H, Herrmann SM, Lerman A, Textor SC, Lerman LO. A mitochondrial permeability transition pore inhibitor improves renal outcomes after revascularization in experimental atherosclerotic renal artery stenosis. Hypertension 60: 1242–1249, 2012 [DOI] [PubMed] [Google Scholar]
- 28. Eirin A, Zhu XY, Krier JD, Tang H, Jordan KL, Grande JP, Lerman A, Textor SC, Lerman LO. Adipose tissue-derived mesenchymal stem cells improve revascularization outcomes to restore renal function in swine atherosclerotic renal artery stenosis. Stem Cells 30: 1030–1041, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eirin A, Zhu XY, Li Z, Ebrahimi B, Zhang X, Korsmo MJ, Chade AR, Grande JP, Ward CJ, Simari RD, Lerman A, Textor SC, Lerman LO. Endothelial outgrowth cells shift macrophage phenotype and improve kidney viability in swine renal artery stenosis. Arterioscler Thromb Vasc Biol 33: 1006–1013, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Eirin A, Zhu XY, Urbieta-Caceres VH, Grande JP, Lerman A, Textor SC, Lerman LO. Persistent kidney dysfunction in swine renal artery stenosis correlates with outer cortical microvascular remodeling. Am J Physiol Renal Physiol 300: F1394–F1401, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Favreau F, Zhu XY, Krier JD, Lin J, Warner L, Textor SC, Lerman LO. Revascularization of swine renal artery stenosis improves renal function but not the changes in vascular structure. Kidney Int 78: 1110–1118, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Geiger H, Fierlbeck W, Mai M, Ruchti H, Schonfeld V, Dammrich J, Hugo C, Neumayer HH. Effects of early and late antihypertensive treatment on extracellular matrix proteins and mononuclear cells in uninephrectomized SHR. Kidney Int 51: 750–761, 1997 [DOI] [PubMed] [Google Scholar]
- 34. Gloviczki ML, Glockner JF, Crane JA, McKusick MA, Misra S, Grande JP, Lerman LO, Textor SC. Blood oxygen level-dependent magnetic resonance imaging identifies cortical hypoxia in severe renovascular disease. Hypertension 58: 1066–1072, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gloviczki ML, Glockner JF, Lerman LO, McKusick MA, Misra S, Grande JP, Textor SC. Preserved oxygenation despite reduced blood flow in poststenotic kidneys in human atherosclerotic renal artery stenosis. Hypertension 55: 961–966, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gloviczki ML, Keddis MT, Garovic VD, Friedman H, Herrmann S, McKusick MA, Misra S, Grande JP, Lerman LO, Textor SC. TGF expression and macrophage accumulation in atherosclerotic renal artery stenosis. Clin J Am Soc Nephrol 8: 546–553, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gomez DE, Alonso DF, Yoshiji H, Thorgeirsson UP. Tissue inhibitors of metalloproteinases: structure, regulation and biological functions. Eur J Cell Biol 74: 111–122, 1997 [PubMed] [Google Scholar]
- 38. Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hackam DG, Spence JD, Garg AX, Textor SC. Role of renin-angiotensin system blockade in atherosclerotic renal artery stenosis and renovascular hypertension. Hypertension 50: 998–1003, 2007 [DOI] [PubMed] [Google Scholar]
- 40. Hansen KJ, Edwards MS, Craven TE, Cherr GS, Jackson SA, Appel RG, Burke GL, Dean RH. Prevalence of renovascular disease in the elderly: a population-based study. J Vasc Surg 36: 443–451, 2002 [DOI] [PubMed] [Google Scholar]
- 41. Juillard L, Lerman LO, Kruger DG, Haas JA, Rucker BC, Polzin JA, Riederer SJ, Romero JC. Blood oxygen level-dependent measurement of acute intra-renal ischemia. Kidney Int 65: 944–950, 2004 [DOI] [PubMed] [Google Scholar]
- 42. Keddis MT, Garovic VD, Bailey KR, Wood CM, Raissian Y, Grande JP. Ischaemic nephropathy secondary to atherosclerotic renal artery stenosis: clinical and histopathological correlates. Nephrol Dial Transplant 25: 3615–3622, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kelsen S, Hall JE, Chade AR. Endothelin-A receptor blockade slows the progression of renal injury in experimental renovascular disease. Am J Physiol Renal Physiol 301: F218–F225, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Knier B, Cordasic N, Klanke B, Heusinger-Ribeiro J, Daniel C, Veelken R, Hartner A, Hilgers KF. Effect of the plasminogen-plasmin system on hypertensive renal and cardiac damage. J Hypertens 29: 1602–1612, 2011 [DOI] [PubMed] [Google Scholar]
- 45. Lerman LO, Nath KA, Rodriguez-Porcel M, Krier JD, Schwartz RS, Napoli C, Romero JC. Increased oxidative stress in experimental renovascular hypertension. Hypertension 37: 541–546, 2001 [DOI] [PubMed] [Google Scholar]
- 46. Lerman LO, Textor SC, Grande JP. Mechanisms of tissue injury in renal artery stenosis: ischemia and beyond. Prog Cardiovasc Dis 52: 196–203, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Levy BI. Microvascular plasticity and experimental heart failure. Hypertension 47: 827–829, 2006 [DOI] [PubMed] [Google Scholar]
- 48. Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 55: 500–507, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marigo I, Dazzi F. The immunomodulatory properties of mesenchymal stem cells. Semin Immunopathol 33: 593–602, 2011 [DOI] [PubMed] [Google Scholar]
- 50. Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, Gordon FJ, Harrison DG. Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension. Circ Res 107: 263–270, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mazzolai L, Duchosal MA, Korber M, Bouzourene K, Aubert JF, Hao H, Vallet V, Brunner HR, Nussberger J, Gabbiani G, Hayoz D. Endogenous angiotensin II induces atherosclerotic plaque vulnerability and elicits a Th1 response in ApoE−/− mice. Hypertension 44: 277–282, 2004 [DOI] [PubMed] [Google Scholar]
- 52. Murphy TP, Cooper CJ, Dworkin LD, Henrich WL, Rundback JH, Matsumoto AH, Jamerson KA, D'Agostino RB. The Cardiovascular Outcomes with Renal Atherosclerotic Lesions (CORAL) study: rationale and methods. J Vasc Interv Radiol 16: 1295–1300, 2005 [DOI] [PubMed] [Google Scholar]
- 53. Onuma S, Nakanishi K. Superoxide dismustase mimetic tempol decreases blood pressure by increasing renal medullary blood flow in hyperinsulinemic-hypertensive rats. Metabolism 53: 1305–1308, 2004 [DOI] [PubMed] [Google Scholar]
- 54. Palm F, Onozato M, Welch WJ, Wilcox CS. Blood pressure, blood flow, and oxygenation in the clipped kidney of chronic 2-kidney, 1-clip rats: effects of tempol and Angiotensin blockade. Hypertension 55: 298–304, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pastore L, Tessitore A, Martinotti S, Toniato E, Alesse E, Bravi MC, Ferri C, Desideri G, Gulino A, Santucci A. Angiotensin II stimulates intercellular adhesion molecule-1 (ICAM-1) expression by human vascular endothelial cells and increases soluble ICAM-1 release in vivo. Circulation 100: 1646–1652, 1999 [DOI] [PubMed] [Google Scholar]
- 56. Pelta A, Andersen UB, Just S, Baekgaard N. Flash pulmonary edema in patients with renal artery stenosis: the Pickering Syndrome. Blood Press 20: 15–19, 2011 [DOI] [PubMed] [Google Scholar]
- 57. Peters H, Noble NA. Angiotensin II and L-arginine in tissue fibrosis: more than blood pressure. Kidney Int 51: 1481–1486, 1997 [DOI] [PubMed] [Google Scholar]
- 58. Pillay WR, Kan YM, Crinnion JN, Wolfe JH. Prospective multicentre study of the natural history of atherosclerotic renal artery stenosis in patients with peripheral vascular disease. Br J Surg 89: 737–740, 2002 [DOI] [PubMed] [Google Scholar]
- 59. Pimentel JL, Jr, Sundell CL, Wang S, Kopp JB, Montero A, Martinez-Maldonado M. Role of angiotensin II in the expression and regulation of transforming growth factor-beta in obstructive nephropathy. Kidney Int 48: 1233–1246, 1995 [DOI] [PubMed] [Google Scholar]
- 60. Plouin PF, Chatellier G, Darne B, Raynaud A. Blood pressure outcome of angioplasty in atherosclerotic renal artery stenosis: a randomized trial. Essai Multicentrique Medicaments vs Angioplastie (EMMA) Study Group. Hypertension 31: 823–829, 1998 [DOI] [PubMed] [Google Scholar]
- 61. Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest 97: 1916–1923, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rognant N, Guebre-Egziabher F, Bacchetta J, Janier M, Hiba B, Langlois JB, Gadet R, Laville M, Juillard L. Evolution of renal oxygen content measured by BOLD MRI downstream a chronic renal artery stenosis. Nephrol Dial Transplant 26: 1205–1210, 2011 [DOI] [PubMed] [Google Scholar]
- 63. Ruiz-Ortega M, Gomez-Garre D, Alcazar R, Palacios I, Bustos C, Gonzalez S, Plaza JJ, Gonzalez E, Egido J. Involvement of angiotensin II and endothelin in matrix protein production and renal sclerosis. J Hypertens Suppl 12: S51–S58, 1994 [PubMed] [Google Scholar]
- 64. Saeed A, Herlitz H, Nowakowska-Fortuna E, Nilsson U, Alhadad A, Jensen G, Mattiasson I, Lindblad B, Gottsater A, Guron G. Oxidative stress and endothelin-1 in atherosclerotic renal artery stenosis and effects of renal angioplasty. Kidney Blood Press Res 34: 396–403, 2011 [DOI] [PubMed] [Google Scholar]
- 65. Safak E, Wilke C, Derer W, Busjahn A, Gross M, Moeckel M, Mueller DN, Luft FC, Dechend R. Long-term follow-up of patients with atherosclerotic renal artery disease. J Am Soc Hypertens 7: 24–31, 2013 [DOI] [PubMed] [Google Scholar]
- 66. Safian RD, Textor SC. Renal-artery stenosis. N Engl J Med 344: 431–442, 2001 [DOI] [PubMed] [Google Scholar]
- 67. Stewart N, Chade AR. Renoprotective effects of hepatocyte growth factor in the stenotic kidney. Am J Physiol Renal Physiol 304: F625–F633, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng FY, Zhao Z, Ganger M, Tow CY, Seshan SV. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol 22: 1041–1052, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Textor SC, Glockner JF, Lerman LO, Misra S, McKusick MA, Riederer SJ, Grande JP, Gomez SI, Romero JC. The use of magnetic resonance to evaluate tissue oxygenation in renal artery stenosis. J Am Soc Nephrol 19: 780–788, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Textor SC, Wilcox CS. Ischemic nephropathy/azotemic renovascular disease. Semin Nephrol 20: 489–502, 2000 [PubMed] [Google Scholar]
- 72. Textor SC, Wilcox CS. Renal artery stenosis: a common, treatable cause of renal failure? Annu Rev Med 52: 421–442, 2001 [DOI] [PubMed] [Google Scholar]
- 73. Truong LD, Farhood A, Tasby J, Gillum D. Experimental chronic renal ischemia: morphologic and immunologic studies. Kidney Int 41: 1676–1689, 1992 [DOI] [PubMed] [Google Scholar]
- 74. Urbieta-Caceres VH, Zhu XY, Jordan KL, Tang H, Textor K, Lerman A, Lerman LO. Selective improvement in renal function preserved remote myocardial microvascular integrity and architecture in experimental renovascular disease. Atherosclerosis 221: 350–358, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Warner GM, Cheng J, Knudsen BE, Gray CE, Deibel A, Juskewitch JE, Lerman LO, Textor SC, Nath KA, Grande JP. Genetic deficiency of Smad3 protects the kidneys from atrophy and interstitial fibrosis in 2K1C hypertension. Am J Physiol Renal Physiol 302: F1455–F1464, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Warner L, Glockner JF, Woollard J, Textor SC, Romero JC, Lerman LO. Determinations of renal cortical and medullary oxygenation using blood oxygen level-dependent magnetic resonance imaging and selective diuretics. Invest Radiol 46: 41–47, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Webster J, Marshall F, Abdalla M, Dominiczak A, Edwards R, Isles CG, Loose H, Main J, Padfield P, Russell IT, Walker B, Watson M, Wilkinson R. Randomised comparison of percutaneous angioplasty vs continued medical therapy for hypertensive patients with atheromatous renal artery stenosis. Scottish and Newcastle Renal Artery Stenosis Collaborative Group. J Hum Hypertens 12: 329–335, 1998 [DOI] [PubMed] [Google Scholar]
- 78. Wheatley K, Ives N, Gray R, Kalra PA, Moss JG, Baigent C, Carr S, Chalmers N, Eadington D, Hamilton G, Lipkin G, Nicholson A, Scoble J. Revascularization versus medical therapy for renal-artery stenosis. N Engl J Med 361: 1953–1962, 2009 [DOI] [PubMed] [Google Scholar]
- 79. Wolf G, Mueller E, Stahl RA, Ziyadeh FN. Angiotensin II-induced hypertrophy of cultured murine proximal tubular cells is mediated by endogenous transforming growth factor-beta. J Clin Invest 92: 1366–1372, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhu XY, Chade AR, Krier JD, Daghini E, Lavi R, Guglielmotti A, Lerman A, Lerman LO. The chemokine monocyte chemoattractant protein-1 contributes to renal dysfunction in swine renovascular hypertension. J Hypertens 27: 2063–2073, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhu XY, Chade AR, Rodriguez-Porcel M, Bentley MD, Ritman EL, Lerman A, Lerman LO. Cortical microvascular remodeling in the stenotic kidney: role of increased oxidative stress. Arterioscler Thromb Vasc Biol 24: 1854–1859, 2004 [DOI] [PubMed] [Google Scholar]
- 82. Zhu XY, Urbieta-Caceres V, Krier JD, Textor SC, Lerman A, Lerman LO. Mesenchymal stem cells and endothelial progenitor cells decrease renal injury in experimental swine renal artery stenosis through different mechanisms. Stem Cells 31: 117–125, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]