

Abstract
Genomic, proteomic, and other omic-based approaches are now broadly used in biomedical research to facilitate the understanding of disease mechanisms and identification of molecular targets and biomarkers for therapeutic and diagnostic development. While the Omics technologies and bioinformatics tools for analyzing Omics data are rapidly advancing, the functional analysis and interpretation of the data remain challenging due to the inherent nature of the generally long workflows of Omics experiments. We adopt a strategy that emphasizes the use of curated knowledge resources coupled with expert-guided examination and interpretation of Omics data for the selection of potential molecular targets. We describe a downstream workflow and procedures for functional analysis that focus on biological pathways, from which molecular targets can be derived and proposed for experimental validation.
Keywords: Proteomics, Genomics, Bioinformatics, Biological pathways, Cell signaling, Databases, Molecular targets, Biomarkers
1. Introduction
Biomarkers are referred to as biological entities or characteristics that can be used to indicate the states of healthy or diseased cells, tissues, or individuals. Nowadays, biomarkers are mostly molecular makers, such as genes, proteins, metabolites, glycans, and other molecules, that can be used for disease diagnosis, prognosis, prediction of therapeutic responses, as well as therapeutic development (1–3). Over the past decade, high-throughput technologies, such as genomic microarrays, proteomic and metabolomic mass spectrometry, have been used to generate large amount of data from single experiments that allow for global comparison of changes in molecular profiles that underlie particular cellular phenotypes. As a result, the omics-based approaches, coupled with computational and bioinformatics methods, provide unprecedented opportunities to speed up the biomarker discovery and now are widely used to facilitate diagnostic and therapeutic developments for many diseases and particularly in cancers (4–10). Potential biomarkers have been identified at various molecular levels, including genetic, mRNA, protein/peptide, as well as epigenetic (11), miRNA (12), glycans (13), and metabolites (4). For example, using DIGE-based proteomics potential biomarkers (e.g., PPA2 and Ezrin) were identified to be useful for the diagnosis of metastatic prostate cancer (14), and a proteolytic fragment of alpha1-antitrypsin (BF5) was identified as a potential diagnostic and prognostic marker for inflammatory breast cancer as well as a target for potential therapeutic intervention (15, 16). Epigenetic marker, such as PITX2 DNA methylation, is reported as a robust assay for paraffin-embedded tissue for outcome prediction in early breast cancer patients treated by adjuvant tamoxifen therapy (11). In addition, microRNAs, such as miR-500, were identified as a potential diagnostic marker for hepatic cell carcinoma (17).
Increasingly, pathway and network-based analyses are applied to Omics data to gain more insight into the underlying biological function and processes, such as cell signaling and metabolic pathways and gene regulatory networks (18, 19). For example, 12 core signaling pathways were shown to be altered in human pancreatic cancers through genomic analyses (18). Network modeling linked breast cancer susceptibility to the centrosome dysfunction (20), and led to the identification of a proliferation/differentiation switch in cellular networks of multicellular organisms (21). These approaches have led to a new trend in identifying biomarkers in recent years, namely, pathway and network-based biomarker discovery, which identify panels of, instead of single, biomarkers for practical use in diagnostic and therapeutic developments (22–24). Protein networks have been shown to provide a powerful source of information for disease classification and to help in predicting disease causing genes (25, 26). Network approaches have also been used for improving the prediction of cancer outcome (27, 28), providing novel hypotheses for pathways involved in tumor progression (28), and exploring cancer-associated genes (29).
In this chapter, we focus on the methodology for the identification of molecular targets through functional Omics data analysis particularly of biological pathways, which can provide more mechanistic insights into the underlying phenotypes and may facilitate therapeutics development. We adopt a strategy that emphasizes the use of curated knowledge resources, and describe a workflow and procedures, coupled with expert-guided analysis and interpretation, for the selection of potential molecular targets.
2. Materials
Despite the rapid advancement of the high-throughput technologies and the bioinformatics tools, the functional analysis and interpretation of Omics data remain challenging due to high variation, low reproducibility, and noise of the data. Although many algorithms and tools have been developed to address these challenges, much is inherent to the long workflows of the Omics experiments, e.g., from sample preparation and raw data acquisition, to data processing and analysis. Many statistical and machine learning methods have been developed for better partitioning or clustering of genes (30–33), however, understanding of the biological meaning and functional interpretation of the group of genes/proteins are critical downstream steps in the Omics workflow, and are necessary for the design of therapeutic strategies. This downstream functional analysis relies heavily on existing knowledge annotated for genes or proteins and frequently requires expert-guided analysis for appropriate interpretation.
2.1. Bioinformatics Databases
Annotations of genes and proteins integrated from multiple bioinformatics databases are the basis for functional analysis and interpretation of Omics data (34). Numerous gene and protein databases, varying in size and scope, have been developed to provide functional annotations for genes and gene products, as archived for the past decade, e.g., in the “Molecular Biology Database Collection” in the journal Nucleic Acids Research (35). The number of databases and database entries is rapidly growing, e.g., in 2009 the journal archived a total of 1,170 databases, nearly 100 more than in 2008. These databases are divided into 14 general categories, including databases of DNA, RNA and protein sequences, structure, genomics, proteomics, metabolic, and signaling pathways. Databases most relevant to Omics data analyses include: (1) Gene and protein databases, such as UniProt (36) for protein-based annotations, Entrez Gene (37) and model organism databases (e.g., Mouse Genome Database) (38) for gene-based annotations; (2) GO annotations, such as GOA for annotation of gene products with Gene Ontology (GO) terms (39); (3) Biological pathways, such as KEGG (40) and Pathway Interaction Database (PID) (41) for annotations of proteins involved in metabolic and signaling pathways; Pathway Commons has been developed as a single point of access for diverse pathway databases; and (4) Protein–protein interaction (PPI) databases, such as IntAct (42) and MINT (43) for annotations of proteins involved in physical interaction of proteins.
2.2. Data Mapping and Integration Tools
Mapping different Omics data types (e.g., gene, mRNA, peptide/protein, metabolite) to the common biological entities (e.g., proteins) is an essential step for deriving comprehensive annotations for functional Omics data analysis (34). Omics data mapping is accomplished most commonly by ID (database entry identifier) mapping that allows different but related biological entities to be mapped to the IDs of common entities (e.g., proteins). One of the most common issues in protein mapping is that the relation between different types of biological entity could be one-to-one (e.g., one gene ID to one protein ID) or one-to-many (e.g., one gene ID to two or more protein IDs), and this is not only caused by the difference between genes and proteins (e.g., one gene encodes several protein isoforms), but can also result from database redundancy (see Note 1). UniProt Knowledgebase (UniProtKB) is the main section of UniProt with comprehensive and high-quality protein sequence annotations (44), and iProClass is an integrated database for all UniProt protein sequences with value-added annotations integrated from over 100 other databases (45). The UniProt and iProClass databases thus serve as the underlying infrastructure for protein ID mapping (different IDs mapped to UniProtKB protein IDs) and data integration for experimental Omics data. ID mapping based on the two databases allows ~32 commonly used, heterogeneous IDs to be converted from each other and the ID mapping services are available online both at the Protein Information Resource (PIR) (http://pir.georgetown.edu) and UniProt (http://www.uniprot.org). ID mapping data files are also available at PIR for download to perform data mapping offline. Other ID mapping tools include DAVID gene ID conversion tool (http://david.abcc.ncifcrf.gov/conversion.jsp) (46) and Protein Identifier Cross-Reference Service (PICR, http://www.ebi.ac.uk/Tools/picr) (47).
2.3. Functional Profiling and Pathway Analysis Tools
Various bioinformatics tools are available for functional profiling of Omics data based on annotations of genes and proteins, such as PIR batch retrieval and functional categorization tool (http://pir.georgetown.edu/pirwww/search/batch.shtml), iProXpress (http://pir.georgetown.edu/iproxpress) (34), DAVID (48), and BABELOMICS (http://babelomics.bioinfo.cipf.es) (49). Annotations used for profiling by these tools include GO terms, pathways, keywords, sequence features, and families, among which GO terms and pathways are the most commonly used: GO has become a common annotation standard, and pathways provide more insightful biological meaning for the data. Moreover, many concepts in other annotations, such as keywords, have been covered by GO terms. While most of these tools allow profiling of single gene/protein list or two for comparison, iProXpress provides comparative profiling of multiple data sets (or data groups) for cross-data sets comparison, a very useful feature that accommodates many real-world data analysis issues.
For pathway analysis, mapping experimental data to metabolic and signaling pathways is a key for functional interpretation of the Omics data. Curated canonical pathway maps are available in many pathway databases, however, few public Omics analysis tools integrate the maps into their systems to allow experimental data superimposed onto the pathway maps. Several commercial pathway analysis systems are available, such as Ingenuity IPA (http://www.ingenuity.com) and GeneGO MetaCore (http://www.genego.com). Although these tools differ in features, such as visualization of canonical pathways and presentation of experimental data mapped onto the pathways, they all have one feature in common, i.e., integration of additional pathway and functional association data manually curated from literature into the systems in addition to the publicly available data in pathway databases, such as KEGG and PID.
2.4. Literature Text Mining Tools
Despite the extensive use of annotations from current knowledgebase for functional analysis of Omics data, annotations of genes and proteins lag far behind the rapid growth of literature due to the ever-expanding sequence data and the laborious nature of manual curation. In nearly all Omics experiments, varying numbers of identified genes or proteins lack sufficient annotations in databases to be functionally analyzed, and in such cases literature becomes the critical source for deriving functional information. Although literature data have been used solely or combined with other Omics data to generate gene/protein association networks (50–52), currently no literature mining tools have been integrated into any pipelined Omics system in a fashion that computationally extracted data are directly used as annotations for functional data analysis. Nonetheless, literature text mining is an important component of the data analysis workflow, and has been used to assist pathway analysis, such as ResNet of Pathway Studio (53) (http://www.ariadnegenomics.com/products/databases/ariadne-resnet). A variety of text mining tools are available to assist in mining relevant gene or protein data from literature, and this coupled with manual search of PubMed are often necessary for functional Omics data analyses (see Note 2).
3. Methods
The pathway and network-based Omics data analysis approach aims to delineate molecular maps that underlie the changes in biological samples under investigation, and to aid in discovery of molecular targets and biomarkers for diagnostic and therapeutic developments. Below we describe practical procedures applied to analyses of Omics data related to cell signaling and metabolic pathways, as well as organelle biogenesis.
3.1. Omics Data Analysis Workflow
We focus on downstream analytical steps of the Omics workflow leading to functional interpretation of Omics data. The workflow begins with a list of gene or protein identifiers or peptide sequences as results from upstream data processing and analysis, e.g., gene clusters or differentially expressed genes or proteins and follows steps 1–6 depicted in Fig. 1: The genes or proteins in the list are then mapped to UniProtKB protein identifiers (step 1). Next, functional annotations are derived for the list of genes or proteins (step 2) based on integrated data from multiple bioinformatics databases (step 4), including text mining of literature for information that has not yet been annotated in databases (step 5). Steps 4 and 5 make maximal use of public knowledge resources. Functional analyses are often conducted using several approaches (step 3) based on different types of knowledge annotated in bioinformatics databases, i.e., GO profiling, molecular networks, and biological pathways. Among them, GO profiling, while revealing limited biological insights into Omics data, usually covers most of the genes/proteins under analysis (see Note 3). By contrast, while giving more biological insights, pathway analysis is limited by low coverage of proteins annotated in known canonical pathways (see Note 4). In between the GO profiling and pathway mapping is molecular network analysis of interactions or functional associations between genes or proteins. Finally, molecular targets are inferred from the functional analysis (step 6).
Fig. 1.

A downstream functional analysis workflow for molecular target and biomarker discovery from Omics data.
3.2. Omics Data Grouping
Omics experiments are often carried out under various experimental conditions, from which differential patterns of gene or protein expressions are to be analyzed and potential molecular targets are sought. To assist the subsequent bioinformatics analysis, genes or proteins associated with different experimental conditions are divided into appropriate data groups and assigned with proper notations (Table 1). Although there is no fixed scheme for assignment, the notations usually clearly distinguish the key conditions under which each experiment is carried out and/or data are collected for given studies. There are additional considerations in Omics data grouping in the case of proteomic data (see Note 5).
Table 1.
Proteomics data grouping based on experimental design and methods
| Common types | Examples | |
|---|---|---|
| Experimental group | Treatment | −/+ Radiation; −/+ Estrogen (E2) |
| Time course | One time point or multiple (30 m, 1 h, 3 h, 9 h…) | |
| Cell types | ATCL8 and AT5BIVA (Ref. 54); MCF-7 and MCF-7:5C (Ref. 59) | |
| Immunoprecipitation (IP) | Phosphotyrosine (pY) IP; AIB1 IP | |
| Sample separation | 1D or 2D gel electrophoresis | |
| Mass spectrometry (MS) | Single MS; tandem MS (MS/MS) | |
| Data type | Proteomics; mRNA expression microarray | |
| Changes | Increased or decreased | |
| Notations for groups | A_8_3h_increase – Increased on 2D-gel at 3h postradiation in ATCL8 cells | |
| MS2AIB1_A – Identified in lane A using anti-AIB1 IP and MS/MS (MS2) in MCF-7 cells (–) E2 | ||
| Experimental notes | “ATCL8 6.413, 8-pep, 3 h” – Increased by 6.413-fold on 2D-gel and identified with 8 peptides, at 3 h in ATCL8 cells | |
| “B11 30K 24K 100 CI90” – Identified in lane B of 1D-gel, band 11 of MW 30 kDa, calculated MW 24 kDa, a score 100 and CI > 90% |
3.3. Omics Data Mapping and Integration
Since the UniProt and iProClass databases are the data warehouse of the iProXpress system and serving as the underlying infrastructure for Omics data mapping and integration, the list of genes or proteins from Omics data are mapped to UniProtKB protein entries, referred to as protein mapping, to obtain functional annotations. Protein mapping is primarily based on gene/protein identifiers. For gene expression microarray data, commonly used gene identifiers include Entrez Gene ID, NCBI gi number, and Refseq ID. For mass spectrometry (MS) proteomic data, depending on the database selected for protein identification by the search engine (e.g., MASCOT), the commonly used identifiers include UniProtKB, IPI, NCBI nr, Refseq, etc. Gene and protein IDs are mapped to UniProtKB entries based on comprehensive ID mapping tools available at PIR or UniProt, which converts commonly used gene and protein IDs (such as NCBI's gi number and Entrez Gene ID) to UniProtKB IDs and vice versa. After protein mapping, all gene or protein IDs from one or more data sets or experimental groups are integrated into a master list of UniProtKB identifiers (ACs or IDs), each associated with corresponding experimental groups and notes (Table 1). This master list of proteins is the basis for the subsequent functional annotation and analysis using the iProXpress system.
Frequently, UniProtKB entry matches are not found for a fraction of input gene or protein identifiers, resulting from updates of database identifiers or deletion of entries occurring to most databases, especially when analyzing legacy data in which mixed database identifiers are often used. In such cases, the mapping can be based on sequence comparison or name mapping if the sequence is not available. For genes, the sequence identity and taxonomy information may be used to map the gi numbers to UniProtKB IDs in addition to the mapping bridged by EMBL/GenBank protein accessions (34). For MS proteomic data, peptide sequences are matched against all sequences in UniProtKB (see Note 6).
When gene microarray and MS proteomic experiments are conducted on the same biological samples under identical or similar conditions, the two Omics data sets are compared after data being merged through protein mapping. Direct comparison of expression at both mRNA and protein levels can provide stronger evidences for the underlying changes. For example, the 2D-gel/MS proteomics study identified 412 and 771 proteins that potentially changed in response to radiation treatment in ATM (Ataxia Telangiectasia Mutated) mutated (ATM−) and wild type (ATM+) cells, respectively, while the gene microarray study identified 103 and 131 significantly changed genes in the two cells, respectively (54). Among those genes/proteins, only 13 were commonly identified, including RRM2, the catalytic subunit of ribonucleoside-diphosphate reductase (RR), a rate-limiting enzyme required for synthesis of dNDP and thus of DNA synthesis in human (55). However, care should be taken in mapping data from genes to proteins due to one-to-many relations and redundancy existing in the UniProt database (see Note 1).
3.4. The Omics Data Annotation and Functional Profiling
3.4.1. Metadata Annotation
As discussed above, the experimental groups in which the genes or proteins are identified, as well as additional experimental information are annotated for all proteins with proper notations. The annotated data groups are used for direct comparative analysis between selected groups of interest, such as cell types, treatment types and time course, as well as Omics data types. The metadata annotation can also be used for limiting functional profiling to proteins in selected groups using the iProXpress interface (see below).
3.4.2. Functional Annotation
After protein mapping, rich annotations are described for given Omics data sets in the so-called protein information matrix (Table 2) that captures salient features of proteins, such as functions, pathways, and protein–protein interactions, derived from comprehensive protein annotations integrated into the UniProt and iProClass databases. The matrix allows for browsing and search of rich protein information through the iProXpress Web interface, which facilitates detailed examination of the Omics data (Fig. 2). Among protein annotations, GO terms, including molecular function, biological process and cellular component, and pathways, such as KEGG, are most commonly used for functional profiling.
Table 2.
Major categories of a protein information matrix
| Major category | Example data sourcesa |
|---|---|
| General information | |
| Protein name | UniProtKB, RefSeq |
| Taxonomy | NCBI Taxon |
| Gene name | UniProtKB |
| Keywords | UniProtKB |
| Function | UniProtKB |
| Subunit | UniProtKB |
| Tissue specificity | UniProtKB |
| Bibliography | UniProtKB, SGD, GeneRIF |
| Gene-related information | |
| Genome/gene | GenBank, Entrez Gene, MGI |
| Gene expression | GEO, CleanEx |
| Genetic variation/disease | HapMap, OMIM |
| Gene regulation | ISG |
| Protein function-related information | |
| Ontology | GOA |
| Enzyme/function | KEGG, BRENDA, MetaCyc |
| Pathway | KEGG, EcoCyc, PID, Reactome |
| Complex/interaction | IntAct, DIP |
| Protein expression | Swiss-2DPAGE, PMG |
| Structure | PDB, SCOP, CATH |
| Feature and posttranslational modifications | UniProtKB, RESID, PhosphoSite |
| Protein family | PIRSF, Pfam, COG, InterPro |
Detailed data sources are available at http://pir.georgetown.edu/cgi-bin/iproclass_stat
Fig. 2.
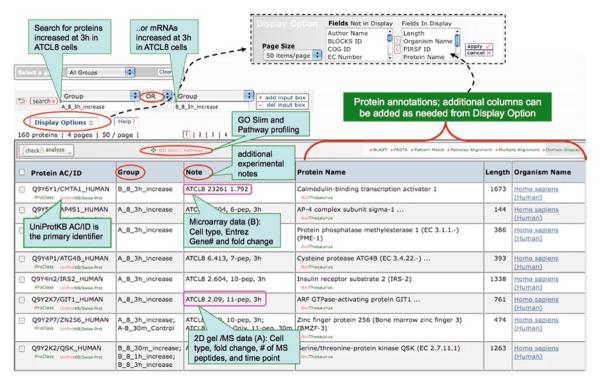
iProXpress interface for browsing, searching, and functional profiling of Omics data. As an example, the interface displays the proteomic data sets derived from 2D gel and mass spectrometry as well as the gene expression microarray data sets from ATM− (AT5BIVA) and ATM+ (ATCL8) human fibroblast cells (54).
3.4.3. GO Profiling
Gene Ontology profiling is primarily based on GO slim, a cut-down version of GO terms at high levels of GO hierarchy (http://www.geneontology.org/GO.slims). GO slims are usually derived from terms at second and third levels of the GO hierarchy, though varying from sources to sources in the selection of additional terms from deeper levels. GO profiling provides a general view of biology underlying the Omics data and can suggest significant functional categories of genes or proteins that can be further investigated. For example, 26 genes are found upregulated in ionizing radiation treated ATM+ cells, which were identified from gene expression microarray data and were profiled using GO biological process (Fig. 3). The profile shows high representation of proteins in GO categories, such as “cell communication,” “response to stimulus,” and “cell proliferation,” in which several proteins are known to be involved in radiation-induced responses, e.g., BRCA1, p53, HDAC1, and STAT3. Because GO slims are terms of high level, genes/proteins profiled under given GO categories often overlap to varying degrees, e.g., the above mentioned proteins are common in three or more of the top five GO categories (Fig. 3). However, some terms are too broad, such as “regulation of biological process” or “biological regulation” to reveal meaningful biological information (see Note 7).
Fig. 3.

GO biological process profiling of upregulated genes in ATCL8 cells (ATM+) at 30 min postirradiation. A total of 26 differentially expressed genes are profiled and the GO categories are ranked based on the number of proteins annotated with the corresponding GO terms (frequency); categories with only one protein are partially displayed at the bottom. Encircled in dashed line are top six categories of GO terms that cover 77% of the proteins (20/26), e.g., five genes appearing in three to five GO categories (in the box).
3.4.4. Pathway Profiling
Due to the overall low coverage of pathway annotations for a given organism, relatively large numbers of proteins are usually missed in pathway profiling for any Omics data set. Nonetheless, it could provide significant insights into the underlying biology, particularly when used for cross-data set comparative profiling. For example, in our previous study, comparison of nine organelle proteomes, including mitochondria, endoplasmic reticulum (ER), and seven other lysosome-related organelles (56), the pathway profiles based on KEGG pathways show that “oxidative phosphorylation pathway” is prevalent in mitochondria while “N-glycan biosynthesis pathway” is in the ER (Fig. 4), which are consistent with the well-established functions of the two organelles. Pathway profiling also led to the identification of “purine metabolism pathway” that showed notable differences between radiation-treated vs. untreated ATM− and ATM+ cells (Fig. 5a) (54).
Fig. 4.

Comparative profiling of organellar proteomes using KEGG pathways. Proteomes of nine organelles (56) are profiled using KEGG pathways. Although only a small portion of the proteome is covered by the KEGG pathways, the profiles show striking contrast between organelles, e.g., endoplasmic reticulum (ER) and mitochondria (Mit) enriched for “oxidative phosphorylation” and “N-Glycan biosynthesis” pathways (encircled on the left), respectively.
Fig. 5.

(a) KEGG pathway profiling of radiation-induced protein expression changes in ATM mutated (ATM−) and ATM wild type (ATM+) cells at 3 h postirradiation. The “purine metabolism” pathway is encircled and it shows that the most differentially changed proteins (up- or downregulated in response to radiation in the two cells) is found in this pathway. This profile is a partial display, with the rest having small number of proteins and no striking differences between groups. The figure is adapted from Hu et al. (54). (b) Mapping of radiation-induced protein changes onto the purine metabolic pathway. Enzymes in the KEGG reference map are represented using Enzyme Commission numbers (EC#, e.g., 1.17.4.1). Enzymes labeled with a diamond shape are those identified in human, and all others without such a label are those known to be absent in human. Enzymes with up-tilted arrows, upregulated in ATM+ cells; those with down-tilted arrows, downregulated in ATM− cells; the enzyme with double down-tilted arrows are downregulated in both cells. Upper left, biochemical steps surrounding dADP/dATP; upper right, biochemical steps surrounding dGDP/dGTP; bottom, illustration of the rate-limiting step in dATP or dGTP synthesis from the reduction of ADP or GDP, respectively, catalyzed by RRM2 in human.
3.5. Pathway Mapping and Visualization
One key step in functional Omics data analysis is pathway mapping, a process that maps genes/proteins detected by Omics experiments to corresponding proteins annotated in canonical pathways. Various software tools are available for pathway mapping, including iProXpress, DAVID, and commercial tools, such as IPA (http://www.ingenuity.com) and MetaCore (http://www.genego.com). Visualization of the mapped pathways greatly facilitates the comparative analysis and understanding of the underlying differences across experimental groups, thus being critical for identifying potential molecular targets. Visualization of mapped pathways is provided as part of several software systems, e.g., mapped proteins in canonical pathways are highlighted by a distinct color (for one experimental condition as in IPA) or labeled with experimental conditions under which they were detected (as in MetaCore). Recently, KEGG developed a standalone tool, KegArray, for mapping gene expression profiles to pathways and genomes (57).
Different pathway tools should be used to maximize the identification of potential pathway-based targets because pathways annotated in different databases vary in their contents and boundaries (see Note 8). We used iProXpress, KEGG, IPA, and MetaCore pathway tools for mapping and/or visualization of metabolic and signaling pathways in several proteomic and functional genomic studies, including those on organelle biogenesis (58), radiation-induced DNA damage repair (54), and estrogen-induced apoptosis in breast cancer cells (59). Pathway mapping could lead to the identification of specific steps in which the proteins participate and the roles they may play.
3.6. Literature Mining
For genes or proteins of interest that were derived from the Omics data based on differential expression and/or functional profiling, but do not have annotated pathway information, literature mining is used to uncover their potential associations with or pathways for the underlying phenotypes. Various text mining tools are available to assist literature mining (see Note 2).
3.7. Practical Applications
3.7.1. Examples
We use the functional analysis of Omics data generated from radiation-treated ATM− and ATM+ cells (54) as an example to illustrate the Omics workflow described above. ATM, a serine–threonine protein kinase, plays critical roles in stress-induced responses, such as DNA damage repair and cell cycle regulation. Using human fibroblast cell lines expressing mutated ATM gene (AT5BIVA cell, ATM−) or wild type ATM (ATCL8 cell, ATM+), the study aims to better understand ATM-mediated pathways in response to ionizing radiation, which could facilitate identification of molecular targets for therapeutic interventions, such as increasing radiation or drug sensitivities of cancers. The two cell lines are subjected to global expression profiling using gene microarray and 2D-gel and MS proteomics. Below are the steps used for the analysis.
Proteins identified from the MASCOT search engine (http://www.matrixscience.com) output files are compiled into one protein list and annotated with corresponding experimental groups (e.g., cells and time points). The database searched by MASCOT is SwissProt (a manually annotated portion of the UniProtKB). The differentially changed genes (up- or downregulated genes from the microarray) are mapped to UniProtKB accessions (ACs) from Entrez Gene IDs.
Some UniProtKB ACs in the protein list from the MASCOT output might need replacement by new ACs (but usually still identify the same protein sequence) if the bioinformatics analysis is conducted at a time later than MS when UniProtKB has newer releases, in which protein sequences may be updated/corrected or redundant sequences be merged. Updated ID mapping files can be downloaded at ftp://ftp.pir.georgetown.edu/databases/idmapping/idmapping.tb.gz and used to obtain an updated experimental protein list. Alternatively, online ID mapping is available at PIR (http://pir.georgetown.edu) or UniProt (http://www.uniprot.org).
Functional annotations of the protein list are derived from the iProClass database containing comprehensive annotations, which is available for download at ftp://ftp.pir.georgetown.edu/databases/iproclass/iproclass.xml.gz. An output data file that contains all identified proteins, corresponding groups and experimental notes, as well as functional annotations is generated.
The data file is browsed, searched, and profiled using the iProXpress interface: http://pir.georgetown.edu/iproxpress (data set: http://pir.georgetown.edu/cgi-bin/textsearch_iprox.pl?data=gu1). Boolean searches (AND, OR, NOT) can be used to display specific experimental groups or proteins pertinent to certain annotations, e.g., using “A_8_3h_increase” OR “B_8_3h_increase” as “group” query displays proteins that are increased at protein (2D-gel/MS) or mRNA (microarray) level 3 h after radiation in ATCL8 cells (ATM+), resulting in 160 proteins (Fig. 2). While providing many analytic functions, the interface mainly provides functionalities for profiling the list of proteins using GO Slims and KEGG pathways.
The GO or pathway profiles are examined and compared across experimental groups for the entire or selected proteins from the list, and most differential GO categories or pathways are examined. Comparison could also be made on merged or de-merged groups using the interface, e.g., experimental repeats could be merged as a single group based on experimental conditions. GO and pathway profiles can also be generated for single list of proteins/genes using PIR batch retrieval at http://pir.georgetown.edu/pirwww/search/batch.shtml, but without metadata annotations (Fig. 3).
The iProXpress interface is used for pathway profiling and shows that the purine metabolism pathway is significantly and differentially represented in radiation treated or untreated ATM−/ATM+ cells (Fig. 5a). Pathway mapping using KEGG is conducted at http://www.genome.jp/kegg/tool/color_ pathway.html, which allows to input enzymes of interests (using EC numbers, e.g., 1.17.4.1) and to generate pathway maps with input enzymes highlighted in colors corresponding to different experimental groups (Fig. 5b).
For pathway analysis using Ingenuity IPA, the entire protein list from this study is loaded and “my list” of genes/proteins is created for specific experimental groups. Pathway profiles are examined, and pathway maps are analyzed regarding the positions and relations of specific genes/proteins of interest (e.g., certain experimental groups) in the pathway, e.g., p53, BRCA1, and Chk1, increased in ATM+ cell after irradiation, are mapped to the G2/M DNA damage check point regulation pathway (Fig. 6a). Since one protein could appear in multiple canonical pathways, the pathway maps should be examined carefully with expert guidance. In addition to canonical pathways, gene/protein networks could be generated based on functional associations annotated in the knowledgebase of Ingenuity IPA (Fig. 6b), which provides further evidence for the ATM-mediated radiation response pathways that involve p53, BRCA1, HDAC1, and RRM2.
Fig. 6.


(a) Ingenuity pathway profiling and mapping of genes/proteins from ATM−/ATM+ cells with or without ionizing radiation treatment. The analysis is performed using Ingenuity IPA. Top, top-ranked pathway profiles (well above the threshold p-value), in which the ratio of genes/proteins detected in the experiment over the total number of proteins annotated in the pathway, is given as gray squares. Purine metabolism (encircled on the left) is shown as the third top pathway in the study. Bottom, pathway map of cell cycle G2/M DNA damage check point regulation. BRCA1 and p53 are upregulated at mRNA level 30 min after irradiation in ATCL8 cells (labeled with a dark triangle shape). Chk1, identified from 2D gel/MS, was increased at 3 h after irradiation in ATCL8 cells (encircled with a dashed line). (b) Gene networks linking RRM2 with DNA damage repair pathway proteins. Functional networks showing RRM2 connected to other major DNA repair and cell cycle proteins, such as p53, BRCA1, and HDAC1. Networks are generated using the Ingenuity IPA tool, and are merged from three subnetworks, one containing RRM2 and HDAC1, one with p53, and the third with BRCA1. The protein or gene nodes labeled with a dark triangle shape are those differentially expressed in the study. The lines (edges) connecting nodes indicate associations between proteins or genes, which encompass interaction, binding, activation, inhibition, etc. Solid lines (edges) are for direct and dashed ones for indirect associations. The figure is adapted from Hu et al. (54).
In summary, through functional profiling and pathway mapping, this example shows that purine metabolism is significantly represented and differentially changed in the ATM− and ATM+ cells in response to radiation. The increased expression of RRM2 at both mRNA and protein levels, and of p53, BRCA1, HDAC1, and Chk1 at the mRNA level in ATM+ but not in ATM− cells, strongly suggest that RRM2 is a downstream target of the ATM-mediated radiation response pathways and is required for radiation-induced DNA repair. This is supported by a recent report that upregulation of RRM2 transcription in response to DNA damage in human involves ATR/ATM-Chk1-E2F1 pathway (60). RRM2 is also known to play roles in cell proliferation, tumorigenicity, metastasis, and drug resistance (61). Increased expression of RRM2 has been linked to increased drug resistance, and its decrease in expression is linked to the reversal of drug-resistance in cancer cells (61, 62). RRM2 is a potential therapeutic target for cancers, e.g., targeting RRM2 for sensitizing cancer cells to drug effects through enhancing camptothecin (CPT)-induced DNA damage in breast cancer cells (60).
3.7.2. Pitfalls
Omics-based molecular target and biomarker identifications remain challenging, and many limitations exist, e.g., see review in ref. 63.
Proteomics data coverage bias. Missing (or false negative) identifications are common to mass spectrometry-based proteomics, thus experimental repeats including those at the level of sample preparation often improve the protein identification rate. The coverage bias also partially accounts for the relatively small percentage of overlaps between proteomics and gene expression microarray data from identical biological samples (54, 64).
Limitations of knowledgebases. Although our approach heavily relies on the annotations in knowledgebases, these curated databases have several limitations. Common shortcomings that might affect the analysis include database entry redundancy, insufficient annotations, and high proportion of electronically derived annotations. For example, database entry redundancy can cause ambiguous ID mapping (see Note 1), and insufficient annotations can limit the power of functional interpretation of Omics data. In the case of GO annotation, the vast majority of GO terms (~90%) annotated for gene products are inferred from electronic annotation (IEA) (see http://www.geneontology.org/GO.current.annotations.shtml), thus cautions should be exercised when using GO slim profiling.
Lack of tissue and/or isoform specificity in pathway annotations. A potential bias in interpretation of pathway mapping results could come from the fact that pathway annotations currently take little consideration of tissue specificities of genes or proteins in the pathway. Thus, specific steps of a pathway may not be actually active in given tissues/cells from which the Omics data may be generated. In some cases, this may occur because protein isoforms or splice variants have been annotated as a protein class or a canonical protein sequence, respectively, in the pathway while they may be expressed differentially in different tissues/cells.
Variations in pathway annotations. Because biological pathways are inherently complex and dynamic, pathway annotations in different pathway databases vary significantly in pathway models and in a number of other aspects, e.g., specific protein forms, dynamic complex formation, subcellular locations, and pathway cross talks (pathway boundaries, also see Note 8). Pathway Commons is an effort to provide a link between the disparate pathway databases.
4. Notes
Gene IDs such as Entrez Gene numbers are often mapped to multiple UniProt protein entries, some of which result from protein isoforms that need to be merged under the entry of the same protein precursor, but most result from sequence redundancy in the database. For example, UniProtKB has two sections, UniProtKB/Swiss-Prot and UniProtKB/TrEMBL; the former is manually annotated with minimal redundancy and the latter is computationally annotated with more redundancy, including fragments of the same gene products. If the complete proteome annotation is available for an organism (e.g., human), in most cases one can limit the ID mapping to UniProtKB/Swiss-Prot and check any remaining unmapped IDs. Redundant sequence entries can be resolved using UniRef100 and/or UniRef90, which cluster sequences of 100 or 90% identity into one group for the selection of the appropriate entries (http://www.uniprot.org/help/uniref).
Although PubMed is the primary tool to access literature citation, some literature mining tools are available to help mining relevant protein data, such as PPI (e.g., MetaServer, http://bcms.bioinfo.cnio.es) and protein phosphorylation (e.g., RLIMS-P, http://pir.georgetown.edu/pirwww/iprolink/rlimsp.shtml). In addition, gene or protein synonyms can be identified using BioThesaurus (http://pir.georgetown.edu/iprolink/biothesaurus), which help identify more relevant literature from PubMed for a given gene/protein.
GO annotations have high coverage for a given genome, e.g., currently >88% of human proteins in UniProtKB/Swiss-Prot are annotated with GO terms (Table 3). Overall, the vast majority of GO terms (~90%) are annotated based on computational inference (evidence code IEA, Inferred from Electronic Annotation: http://www.geneontology.org/GO.evidence.shtml). Manual annotation of GO remains laborious.
In general, only a small percentage of a proteome has been annotated with pathways, thus depending on the data sets being analyzed, the coverage of pathways for the given Omics data vary. For human, currently only about one quarter of proteins are covered by pathway databases, including KEGG, PID, and Reactome (Table 3). Although integrated into Pathway Commons (http://www.pathwaycommons.org), PPI data are not part of annotated pathways, but can be used to generate protein interaction networks.
Another aspect of dividing experimental data relates to dividing proteins identified from mass spectrometry, such as MALDITOF into groups of proteins identified with high (>90%) or low (<90%) confidence intervals (CI) assigned from statistical processing of MASCOT search results by software, e.g., GPS Explorer™, to increase the probability of true target identifications. The low CI values could result from factors, such as the size of database for the search engine, protein abundance, and the type of mass spectrometry instruments. Furthermore, MS proteomic data often require additional filtering for appropriate analysis. For example, a number of proteins that are deemed to be nonspecific (e.g., keratins) are frequently detected for the underlying experiment, which could be caused by factors such as sample contamination and/or detection bias toward high abundant proteins, thus often are removed from analysis. For proteins identified from 1D gel that migrate at apparent molecular weight (MW) highly deviating from the calculated MW could be removed, albeit with caveats that protein degradation or aggregation may have occurred at or before gel electrophoresis. These practices are currently applied to an ongoing study on investigating E2-induced cell apoptosis pathways in breast cancer cells (59).
A two-step procedure is generally used for the peptide mapping: direct sequence mapping and reducing redundancy using UniRef90 clusters (http://www.uniprot.org/help/uniref) (65). Sequences in UniProtKB with 90% or more sequence identity are grouped in a UniRef90 cluster. Proteins within a UniRef90 cluster are more likely to have the same function. For the peptide matched to more than one UniProtKB sequences, if the matching sequences are in the same UniRef90 cluster, then the peptide is mapped to the representative sequence of the cluster.
Some GO terms nearly always appear in high frequencies for any given list of proteins, such as “GO:0065007: biological regulation,” thus reveal little specific functions for the list of proteins being profiled. Statistical testing is provided in such cases to obtain functional enrichment of GO terms by tools, such as DAVID (http://david.abcc.ncifcrf.gov/summary.jsp). In some cases, a pie chart using GO terms is used to depict the functional categories of the list of proteins. This should be interpreted with caution because the GO categories are not mutually exclusive, especially with regard to the molecular functions and biological processes. A list of proteins can be categorized also based on keywords, functions, and other information from literature, as well as guided by experts.
Biological pathways are inherently complex and cross talk between pathways is frequent. Pathways are often annotated using different models in different pathway databases. Among the differences, the pathway boundary for the same core pathway differs most notably in different databases, depending on what additional proteins to be included that are known to interact with the core pathway. For example, 62 proteins are included in TGF beta signaling in PID database (http://pid.nci.nih.gov), while 40 are found in Reactome (http://reactome.org). Thus, the combined pathway data from different databases have better coverage on proteins to be analyzed even when they share the same core pathways.
Table 3.
Numbers of UniProtKB/Swiss-Prot entries with functional annotations
| Organism (Taxon ID) | # Total entry | Ontology |
Pathway |
PPI |
||
|---|---|---|---|---|---|---|
| Gob | KEGG | PID | Reactome | IntAct | ||
| Mammal (40674) | 64,813 | 59,865 | 14,289 | 1,652 | 3,834 | 8,281 |
| Humana (9606) | 20,328 | 18,049 (88.8)c | 4,925 (24.2) | 1,649 (8.1) | 3,790 (18.6) | 6,423 (31.6) |
| Mousea (10090) | 16,204 | 14,955 (92.3) | 3,685 (22.7) | N/A | N/A | 1,467 (9.1) |
| Rat (10116) | 7,449 | 7,060 | 2,415 | N/A | N/A | 304 |
All numbers are derived from iProClass database as of November 24, 2009
Complete human proteome has been annotated in UniProtKB/Swiss-Prot (Human Proteome Initiative project), and the mouse proteome also has high coverage when compared to rat and other mammals. N/A not applicable because only human proteins and pathways are annotated in PID and the Reactome pathway databases
GO annotations are with all evidence codes, including IEA
Numbers in parenthesis are the percentage of proteins annotated in the categories over the total number of entries for the corresponding species. GO gene ontology, PPI protein–protein interaction
Acknowledgments
The work has been supported in part by Federal funds from the National Cancer Institute (NCI), National Institutes of Health (NIH), under Contract No. HHSN261200800001E (Z.Z.H.), by NCI grant P01CA074175 (A.D.), by NIH grant U01-HG02712 (C.W.), and by the Department of Defense Breast Cancer Research Program W81XWH-06-10590 Center of Excellence Grant (A.W., A.T.R.). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
References
- 1.Ransohoff DF. Cancer. Developing molecular biomarkers for cancer. Science. 2003;299:1679–80. doi: 10.1126/science.1083158. [DOI] [PubMed] [Google Scholar]
- 2.Riesterer O, Milas L, Ang KK. Use of molecular biomarkers for predicting the response to radiotherapy with or without chemotherapy. J Clin Oncol. 2007;25:4075–83. doi: 10.1200/JCO.2007.11.8497. [DOI] [PubMed] [Google Scholar]
- 3.Kim YS, Maruvada P, Milner JA. Metabolomics in biomarker discovery: future uses for cancer prevention. Future Oncol. 2008;4:93–102. doi: 10.2217/14796694.4.1.93. [DOI] [PubMed] [Google Scholar]
- 4.Tainsky MA. Genomic and proteomic biomarkers for cancer: a multitude of opportunities. Biochim Biophys Acta. 2009;1796:176–93. doi: 10.1016/j.bbcan.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanash S. Integrated global profiling of cancer. Nat Rev Cancer. 2004;4:638–44. doi: 10.1038/nrc1414. [DOI] [PubMed] [Google Scholar]
- 6.Souchelnytskyi S. Proteomics of TGF-beta signaling and its impact on breast cancer. Expert Rev Proteomics. 2005;2:925–35. doi: 10.1586/14789450.2.6.925. [DOI] [PubMed] [Google Scholar]
- 7.Walgren JL, Thompson DC. Application of proteomic technologies in the drug development process. Toxicol Lett. 2004;149:377–85. doi: 10.1016/j.toxlet.2003.12.047. [DOI] [PubMed] [Google Scholar]
- 8.Tugwood JD, Hollins LE, Cockerill MJ. Genomics and the search for novel biomarkers in toxicology. Biomarkers. 2003;8:79–92. doi: 10.1080/1354750031000070103. [DOI] [PubMed] [Google Scholar]
- 9.Merrick BA, Bruno ME. Genomic and proteomic profiling for biomarkers and signature profiles of toxicity. Curr Opin Mol Ther. 2004;6:600–7. [PubMed] [Google Scholar]
- 10.Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, Yu J, Laxman B, Mehra R, Lonigro RJ, Li Y, Nyati MK, Ahsan A, Kalyana-Sundaram S, Han B, Cao X, Byun J, Omenn GS, Ghosh D, Pennathur S, Alexander DC, Berger A, Shuster JR, Wei JT, Varambally S, Beecher C, Chinnaiyan AM. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature. 2009;457:910–4. doi: 10.1038/nature07762. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 11.Martens JW, Margossian AL, Schmitt M, Foekens J, Harbeck N. DNA methylation as a biomarker in breast cancer. Future Oncol. 2009;5:1245–56. doi: 10.2217/fon.09.89. [DOI] [PubMed] [Google Scholar]
- 12.Ruan K, Fang X, Ouyang G. MicroRNAs: novel regulators in the hallmarks of human cancer. Cancer Lett. 2009;285:116–26. doi: 10.1016/j.canlet.2009.04.031. [DOI] [PubMed] [Google Scholar]
- 13.Brooks SA. Strategies for analysis of the glycosylation of proteins: current status and future perspectives. Mol Biotechnol. 2009;43:76–88. doi: 10.1007/s12033-009-9184-6. [DOI] [PubMed] [Google Scholar]
- 14.Pang J, Liu WP, Liu XP, Li LY, Fang YQ, Sun QP, Liu SJ, Li MT, Su ZL, Gao X. Profiling protein markers associated with lymph node metastasis in prostate cancer by DIGE-based proteomics analysis. J Proteome Res. 2010;9(1):216–26. doi: 10.1021/pr900953s. [DOI] [PubMed] [Google Scholar]
- 15.Li J, Zhao J, Yu X, Lange J, Kuerer H, Krishnamurthy S, Schilling E, Khan SA, Sukumar S, Chan DW. Identification of biomarkers for breast cancer in nipple aspiration and ductal lavage fluid. Clin Cancer Res. 2005;11:8312–20. doi: 10.1158/1078-0432.CCR-05-1538. [DOI] [PubMed] [Google Scholar]
- 16.Zhou J, Trock B, Tsangaris TN, Friedman NB, Shapiro D, Brotzman M, Chan-Li Y, Chan DW, Li J. A unique proteolytic fragment of alpha1-antitrypsin is elevated in ductal fluid of breast cancer patient. Breast Cancer Res Treat. 2010;123(1):73–86. doi: 10.1007/s10549-009-0625-5. [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto Y, Kosaka N, Tanaka M, Koizumi F, Kanai Y, Mizutani T, Murakami Y, Kuroda M, Miyajima A, Kato T, Ochiya T. MicroRNA-500 as a potential diagnostic marker for hepatocellular carcinoma. Biomarkers. 2009;14:529–38. doi: 10.3109/13547500903150771. [DOI] [PubMed] [Google Scholar]
- 18.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu X, Gerstein M, Snyder M. Getting connected: analysis and principles of biological networks. Genes Dev. 2007;21:1010–24. doi: 10.1101/gad.1528707. [DOI] [PubMed] [Google Scholar]
- 20.Pujana MA, Han JD, Starita LM, Stevens KN, Tewari M, Ahn JS, Rennert G, Moreno V, Kirchhoff T, Gold B, Assmann V, Elshamy WM, Rual JF, Levine D, Rozek LS, Gelman RS, Gunsalus KC, Greenberg RA, Sobhian B, Bertin N, Venkatesan K, Ayivi-Guedehoussou N, Solé X, Hernández P, Lázaro C, Nathanson KL, Weber BL, Cusick ME, Hill DE, Offit K, Livingston DM, Gruber SB, Parvin JD, Vidal M. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat Genet. 2007;39:1338–49. doi: 10.1038/ng.2007.2. [DOI] [PubMed] [Google Scholar]
- 21.Xia K, Xue H, Dong D, Zhu S, Wang J, Zhang Q, Hou L, Chen H, Tao R, Huang Z, Fu Z, Chen YG, Han JD. Identification of the proliferation/differentiation switch in the cellular network of multicellular organisms. PLoS Comput Biol. 2006;2:e145. doi: 10.1371/journal.pcbi.0020145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bertagnolli MM. The forest and the trees: pathways and proteins as colorectal cancer biomarkers. J Clin Oncol. 2009;27(35):5866–7. doi: 10.1200/JCO.2009.24.8013. [DOI] [PubMed] [Google Scholar]
- 23.Zhang DY, Ye F, Gao L, Liu X, Zhao X, Che Y, Wang H, Wang L, Wu J, Song D, Liu W, Xu H, Jiang B, Zhang W, Wang J, Lee P. Proteomics, pathway array and signaling network-based medicine in cancer. Cell Div. 2009;4:20. doi: 10.1186/1747-1028-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ptitsyn AA, Weil MM, Thamm DH. Systems biology approach to identification of biomarkers for metastatic progression in cancer. BMC Bioinformatics. 2008;9(Suppl 9):S8. doi: 10.1186/1471-2105-9-S9-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ideker T, Sharan R. Protein networks in disease. Genome Res. 2008;18:644–52. doi: 10.1101/gr.071852.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loscalzo J, Kohane I, Barabasi AL. Human disease classification in the postgenomic era: a complex systems approach to human pathobiology. Mol Syst Biol. 2007;3:124. doi: 10.1038/msb4100163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Auffray C. Protein subnetwork markers improve prediction of cancer outcome. Mol Syst Biol. 2007;3:141. doi: 10.1038/msb4100183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chuang HY, Lee E, Liu YT, Lee D, Ideker T. Network-based classification of breast cancer metastasis. Mol Syst Biol. 2007;3:140. doi: 10.1038/msb4100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang E, Lenferink A, O'Connor-McCourt M. Cancer systems biology: exploring cancer-associated genes on cellular networks. Cell Mol Life Sci. 2007;64:1752–62. doi: 10.1007/s00018-007-7054-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Do JH, Choi DK. Clustering approaches to identifying gene expression patterns from DNA microarray data. Mol Cells. 2008;25:279–88. [PubMed] [Google Scholar]
- 31.Kerr G, Ruskin HJ, Crane M, Doolan P. Techniques for clustering gene expression data. Comput Biol Med. 2008;38:283–93. doi: 10.1016/j.compbiomed.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 32.Weeraratna AT, Taub DD. Microarray data analysis: an overview of design, methodology, and analysis. Methods Mol Biol. 2007;377:1–16. doi: 10.1007/978-1-59745-390-5_1. [DOI] [PubMed] [Google Scholar]
- 33.Handl J, Knowles J, Kell DB. Computational cluster validation in postgenomic data analysis. Bioinformatics. 2005;21:3201–12. doi: 10.1093/bioinformatics/bti517. [DOI] [PubMed] [Google Scholar]
- 34.Huang H, Hu ZZ, Arighi CN, Wu CH. Integration of bioinformatics resources for functional analysis of gene expression and proteomic data. Front Biosci. 2007;12:5071–88. doi: 10.2741/2449. [DOI] [PubMed] [Google Scholar]
- 35.Galperin MY, Cochrane GR. Nucleic Acids Research annual Database Issue and the NAR online Molecular Biology Database Collection in 2009. Nucleic Acids Res. 2009;37(Database issue):D1–4. doi: 10.1093/nar/gkn942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.UniProt Consortium The Universal Protein Resource (UniProt) 2009. Nucleic Acids Res. 2009;37(Database issue):D169–74. doi: 10.1093/nar/gkn664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2005;33(Database issue):D54–8. doi: 10.1093/nar/gki031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bult CJ, Kadin JA, Richardson JE, Blake JA, Eppig JT, The Mouse Genome Database Group The Mouse Genome Database: enhancements and updates. Nucleic Acids Res. 2010;38(Database issue):D586–92. doi: 10.1093/nar/gkp880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barrell D, Dimmer E, Huntley RP, Binns D, O'Donovan C, Apweiler R. The GOA database in 2009 – an integrated Gene Ontology Annotation resource. Nucleic Acids Res. 2009;37(Database issue):D396–403. doi: 10.1093/nar/gkn803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36(Database issue):D480–4. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schaefer CF, Anthony K, Krupa S, Buchoff J, Day M, Hannay T, Buetow KH. PID: the Pathway Interaction Database. Nucleic Acids Res. 2009;37(Database issue):D674–9. doi: 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aranda B, Achuthan P, Alam-Faruque Y, Armean I, Bridge A, Derow C, Feuermann M, Ghanbarian AT, Kerrien S, Khadake J, Kerssemakers J, Leroy C, Menden M, Michaut M, Montecchi-Palazzi L, Neuhauser SN, Orchard S, Perreau V, Roechert B, van Eijk K, Hermjakob H. The IntAct molecular interaction database in 2010. Nucleic Acids Res. 2010;38(Database issue):D525–31. doi: 10.1093/nar/gkp878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ceol A, Chatr Aryamontri A, Licata L, Peluso D, Briganti L, Perfetto L, Castagnoli L, Cesareni G. MINT, the molecular interaction database: 2009 update. Nucleic Acids Res. 2010;38(Database issue):D532–9. doi: 10.1093/nar/gkp983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O'Donovan C, Redaschi N, Yeh LS. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 2004;32:D115–9. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu CH, Huang H, Nikolskaya A, Hu Z, Barker WC. The iProClass integrated database for protein functional analysis. Comput Biol Chem. 2004;28:87–96. doi: 10.1016/j.compbiolchem.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 46.Huang da W., Sherman BT, Stephens R, Baseler MW, Lane HC, Lempicki RA. DAVID gene ID conversion tool. Bioinformation. 2008;2:428–30. doi: 10.6026/97320630002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Côté RG, Jones P, Martens L, Kerrien S, Reisinger F, Lin Q, Leinonen R, Apweiler R, Hermjakob H. The Protein Identifier Cross-Referencing (PICR) service: reconciling protein identifiers across multiple source databases. BMC Bioinformatics. 2007;8:401. doi: 10.1186/1471-2105-8-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sherman BT, Huang da W., Tan Q, Guo Y, Bour S, Liu D, Stephens R, Baseler MW, Lane HC, Lempicki RA. DAVID Knowledgebase: a gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis. BMC Bioinformatics. 2007;8:426. doi: 10.1186/1471-2105-8-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Al-Shahrour F, Carbonell J, Minguez P, Goetz S, Conesa A, Tárraga J, Medina I, Alloza E, Montaner D, Dopazo J. Babelomics: advanced functional profiling of transcriptomics, proteomics and genomics experiments. Nucleic Acids Res. 2008;36(Web Server issue):W341–6. doi: 10.1093/nar/gkn318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y, Agarwal P. A pathway-based view of human diseases and disease relationships. PLoS One. 2009;4:e4346. doi: 10.1371/journal.pone.0004346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ozgür A, Vu T, Erkan G, Radev DR. Identifying gene-disease associations using centrality on a literature mined gene-interaction network. Bioinformatics. 2008;24:i277–85. doi: 10.1093/bioinformatics/btn182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li S, Wu L, Zhang Z. Constructing biological networks through combined literature mining and microarray analysis: a LMMA approach. Bioinformatics. 2006;22:2143–50. doi: 10.1093/bioinformatics/btl363. [DOI] [PubMed] [Google Scholar]
- 53.Nikitin A, Egorov S, Daraselia N, Mazo I. Pathway studio – the analysis and navigation of molecular networks. Bioinformatics. 2003;19:2155–7. doi: 10.1093/bioinformatics/btg290. [DOI] [PubMed] [Google Scholar]
- 54.Hu ZZ, Huang H, Cheema A, Jung M, Dritschilo A, Wu CH. Integrated bioinformatics for radiation-induced pathway analysis from proteomics and microarray data. J Proteomics Bioinform. 2008;1:47–60. doi: 10.4172/jpb.1000009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nordlund P, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 56.Hu ZZ, Valencia JC, Huang H, Chi A, Shabanowitz J, Hearing VJ, Appella E, Wu CH. Comparative bioinformatics analyses and profiling of lysosome-related organelle proteomes. Int J Mass Spectrom. 2007;259:147–60. doi: 10.1016/j.ijms.2006.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wheelock CE, Wheelock AM, Kawashima S, Diez D, Kanehisa M, van Erk M, Kleemann R, Haeggström JZ, Goto S. Systems biology approaches and pathway tools for investigating cardiovascular disease. Mol Biosyst. 2009;5:588–602. doi: 10.1039/b902356a. [DOI] [PubMed] [Google Scholar]
- 58.Chi A, Valencia JC, Hu ZZ, Watabe H, Yamaguchi H, Mangini NJ, Huang H, Canfield VA, Cheng KC, Yang F, Abe R, Yamagishi S, Shabanowitz J, Hearing VJ, Wu C, Appella E, Hunt DF. Proteomic and bioinformatic characterization of the biogenesis and function of melanosomes. J Proteome Res. 2006;5:3135–44. doi: 10.1021/pr060363j. [DOI] [PubMed] [Google Scholar]
- 59.Hu ZZ, Kagan B, Huang H, Liu H, Jordan VC, Riegel A, Wellstein A, Wu C. Pathway and Network Analysis of E2-Induced Apoptosis in Breast Cancer Cells. 100th AACR Conference; Denver, CO. Apr, 2009. pp. 18–22. Abstract #3285. [Google Scholar]
- 60.Zhang YW, Jones TL, Martin SE, Caplen NJ, Pommier Y. Implication of checkpoint kinase-dependent up-regulation of ribonucleotide reductase R2 in DNA damage response. J Biol Chem. 2009;284:18085–95. doi: 10.1074/jbc.M109.003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou B, Yen Y. Characterization of the human ribonucleotide reductase M2 subunit gene; genomic structure and promoter analyses. Cytogenet Cell Genet. 2001;95:52–59. doi: 10.1159/000057017. [DOI] [PubMed] [Google Scholar]
- 62.Zhou B, Tsai P, Ker R, Tsai J, Ho R, Yu J, Shih J, Yen Y. Overexpression of transfected human ribonucleotide reductase M2 subunit in human cancer cells enhances their invasive potential. Clin Exp Metastasis. 1998;16:43–9. doi: 10.1023/a:1006559901771. [DOI] [PubMed] [Google Scholar]
- 63.Ransohoff DF. Promises and limitations of biomarkers. Recent Results Cancer Res. 2009;181:55–9. doi: 10.1007/978-3-540-69297-3_6. [DOI] [PubMed] [Google Scholar]
- 64.Waters KM, Pounds JG, Thrall BD. Data merging for integrated microarray and proteomic analysis. Brief Funct Genomic Proteomic. 2006;5:261–72. doi: 10.1093/bfgp/ell019. [DOI] [PubMed] [Google Scholar]
- 65.Wu CH, Apweiler R, Bairoch A, Natale DA, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Mazumder R, O'Donovan C, Redaschi N, Suzek B. The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic Acids Res. 2006;34(Database issue):D187–91. doi: 10.1093/nar/gkj161. [DOI] [PMC free article] [PubMed] [Google Scholar]