

Abstract
A detailed understanding of the molecular and cellular mechanisms that underlie epitope preferences in T cell priming is important for vaccines designed to elicit a broad T cell response. Protein vaccinations generally elicit CD4 T cell responses that are skewed toward a small fraction of epitopes, a phenomenon known as immunodominance. This characteristic of T cell responses, that limits the diversity of CD4 T cell recognition, is generally attributed to intracellular antigen processing. However, we recently discovered that immunodominance hierarchies persist even after vaccination with synthetic peptides. In this study, we probed the regulatory mechanisms that cause diminished CD4 T cell responses to subdominant peptides after such multi-peptide immunization in mice. We have found that the delivery of subdominant and dominant epitopes on separate dendritic cells rescues expansion of less favored CD4 T cells. Furthermore, through the use of genetic models and inhibitors, we have found that selective losses in CD4 T cell responses are mediated by an IFN-γ-induced pathway, involving indoleamine 2,3-dioxygenase (IDO), and that regulatory T cell (Treg) activities may also regulate preferences in CD4 T cell specificity. We propose that after multi-peptide immunization, the expansion and differentiation of dominant T cells initiate complex regulatory events that determine the final peptide specificity of the elicited CD4 T cell response.
INTRODUCTION
Recently, there has been tremendous progress in both epitope discovery and the development of predictive algorithms to identify antigenic peptides that can participate in protective T cell responses toward both pathogenic organisms and neoplastic tissue (reviewed in (1–6)). With the steadily increasing number of known pathogen and cancer-derived epitopes comes great potential for the use of synthetic peptides for vaccination and immunotherapy. Peptide-based immunotherapy has been most commonly explored in the treatment of various forms of cancer (7–10), since the early identification of the MAGE peptide by Boon and colleagues (11). Synthetic peptides have significant advantages over other vaccine modalities, including minimal toxicity, chemical stability, indefinite storage and easy characterization for purity and composition using well-established technology (reviewed in (12)). Also, peptides are free of risk from bacterial or viral contamination and concerns regarding genetic integration. Synthetic peptides can be used in dendritic cell-based immunization strategies or in conjunction with adjuvants to elicit more vigorous T cell priming. Although peptide-based vaccines for cancer immunotherapy have focused on stimulating CD8 T cells, more recent efforts have advocated inclusion of CD4 T cell epitopes that promote more robust priming and long-term protective CD8 T cell responses (reviewed in (13)).
In addition to safety and convenience, a theoretical advantage of peptide-based vaccines is that they avoid the complications of antigen processing and the barrier of immunodominance. Protein vaccines and viral vectors require intracellular proteolysis of antigen prior to association of the derived peptides with MHC molecules, unlike synthetic peptides that can bind directly to MHC proteins at the cell surface. The responses to complex antigens are characterized by dramatic asymmetries in the specificity of the elicited T cell repertoire, where only a few dominant specificities are detected (14–19). Typically, cryptic and subdominant peptide epitopes only elicit robust T cell responses when administered as single peptides. Accordingly, T cell hierarchies have been thought to be primarily a consequence of intracellular and presentation events within antigen presenting cells (APC). Earlier studies by our laboratory identified the parameters that determine immunodominance in CD4 T cell responses to protein antigens and found that the immunogenicity of a peptide can be both predicted and regulated by the kinetic stability of the peptide:MHC class II (pMHC) complex (18, 20). Biochemical studies revealed that dominant peptides persist on class II molecules with a t1/2 of 100–200 hours, while cryptic peptides typically display off-rates of only 2–10 hours. Our subsequent studies revealed that DM editing favors export and cell surface presentation of high stability pMHC complexes on APC, allowing recruitment of a disproportionate fraction of the immune response (21). These and other studies (22–27) support the view that intracellular DM editing selects the peptides that can be presented by class II molecules and that can thus elicit CD4 T cells.
The preceding model predicts that antigen in the form of peptides should avoid issues of immunodominance. However, the most recent studies by our group revealed that the preference of CD4 T cell responses to high stability pMHC complexes persists, even when the antigen is introduced in the form of peptides, which do not require antigen processing or DM editing for presentation (28). After multi-peptide immunization, responses to low stability pMHC complexes were found to initially prime CD4 T cells, but the T cells failed to expand further at later time points. The study reported here was initiated to identify the mechanisms that favor such CD4 T cell responses to dominant peptides, at the expense of simultaneous responses to subdominant peptides. We have identified a network of suppression that involves IFN-γ, IDO, and regulatory T cells (Tregs) that together can reduce CD4 T cell responses to subdominant peptides when immune responses to dominant peptides are occurring simultaneously. The mechanisms revealed here are relevant for both clinical applications of peptide-based vaccines intended to elicit a broad CD4 T cell response and for our understanding of the normal homeostatic mechanisms that regulate CD4 T cell priming in secondary lymphoid tissue.
MATERIALS AND METHODS
Mice
BALB/c mice (6–8 weeks) were purchased from National Cancer Institute (NCI). C57BL/6J, Foxp3GFP, IFN-γKO, and IFN-γRKO mice were purchased from The Jackson Laboratories and maintained in our facility. NOS2KO mice were purchased from Taconic Farms. Foxp3DTR breeders were obtained with a material transfer agreement (MTA) from Dr. Alexander Rudensky (MSKCC, New York, NY). Thy1.1 mice were generous gifts from Dr. Deborah J. Fowell (University of Rochester, Rochester, NY). Animal handling was performed in accordance with University Committee on Animal Care at the University of Rochester (UCAR).
IFA immunization
For peptide/IFA immunizations, mice were immunized in both hind footpads with 50 μl of emulsion containing 12.5 nmoles of subdominant peptides and 2.5 nmoles of dominant peptides with 20 ng LPS (Sigma-Aldrich) per footpad.
Flt3L-secreting cells
Lung epithelial Line 1 carcinoma cells (29) were transfected with a pHβ Apr-1-neo vector expressing the human Flt3L under the β-actin promoter (data not shown). The subclone with the best Flt3L secretors was maintained with G418 selection until ready for use.
Dendritic cell immunization
To expand splenic dendritic cells (DC) in vivo, 5×105 Flt3L-secreting cells were injected intradermally into BALB/c mice on the flank and the tumor was allowed to grow for 10–14 days before mice were euthanized. Purification of DC was conducted using serum-free DMEM containing 1% penicillin/streptomycin. Intact spleens were injected with 0.3 mg/ml Liberase DL (Roche) in PBS containing Ca2+ and Mg2+ and were incubated at 37°C for 20 min. before making single cell suspensions. The spleens were treated with ACK lysis buffer (0.15 M NH4Cl/1 mM KHCO3/0.1 mM Na2-EDTA, pH 7.2) prior to purification of CD11c+ cells by using a positive selection kit (Miltenyi Biotech). DC were pulsed with 100 μM subdominant peptides and 10 μM dominant peptides with 0.5 μg/ml LPS at 37°C for 1 hour, washed with PBS and injected into both hind footpads at 1×106 cells in 50 μl PBS per footpad. For CFSE labeling, DC were resuspended at 3×106/ml in PBS +1.5 μM CFSE (Molecular Probes) and incubated at room temperature for 8 min. before washing with serum-free media.
Cell isolation
Unless otherwise indicated, draining popliteal or inguinal lymph nodes (dLNs) were treated with 1 mg/ml Collagenase D in PBS containing Ca2+ and Mg2+ for 20 min. at 37°C prior to making single cell suspensions.
Elispot assay
CD4 T cells were purified by a negative selection kit (Miltenyi Biotec) and were plated at several dilutions with 5×105 syngeneic splenic APC and 10 μM peptides overnight at 37°C. IL-2 and IFN-γ spots were quantified with an Immunospot Reader. The total number of T cell responders per LN was calculated from spots obtained per CD4 T cells plated and by back calculating to the starting LN cell count and percentage CD4 in the LN.
Diphtheria toxin treatment
At the indicated time points, Foxp3DTR mice were administered with 50 μg diphtheria toxin (DT) (Sigma-Aldrich) per kg body weight in 100 μl PBS intraperitoneally (30). DT was reconstituted according to manufacturer’s protocol.
IDO inhibition
At the time of immunization and for up to 10 days thereafter, mice were either given pH balanced, sterile animal-grade water containing 2 mg/ml 1-methyl-D-tryptophan (1-MT) (Sigma-Aldrich) with aspartame or aspartame only control water in light-sensitive water bottles (31). The solution was changed every 7 days.
Treg suppression assay
Foxp3GFP+ cells were sorted by flow cytometry from dLNs of immunized Foxp3GFP mice based on expression of GFP. Sorted Tregs were plated at 1×105 and serial 2-fold dilutions in 96-well U-bottom plates with 1×105 CD4+CD62L+CD25− target T cells, and 1×105 T cell-depleted splenic APC from Thy1.1 mice. Target T cells were labeled with 5 μM CFSE (Molecular Probes) at 1×107 cells/ml PBS +5% FBS for 5 min. at room temperature. Cells were resuspended in RPMI-1640 containing glutamine, 1% penicillin/streptomycin, 10% heat-inactivated FBS, and 2-ME. Cocultures were stimulated with 1 μg/ml anti-CD3 mAb (2C11) for 72 hours at 37°C.
Reagents
Purified rat anti-mouse IL-2 (JES6-1A12), biotinylated rat anti-mouse IL-2 (JES6-5H4), purified rat anti-mouse IFN-γ (AN-18), biotinylated rat anti-mouse IFN-γ (XMG1.2), purified rat anti-mouse IL-4 (11B11), biotinylated rat anti-mouse IL-4 (BVD6-24G2) were purchased from BD Biosciences. Peptides were purchased from BioPeptides. The hybridoma producing monoclonal antibody J1j.10 was acquired from American Type Culture Collection was used to complement-deplete T cells from spleens.
RESULTS
CD4 T cell responses to subdominant peptides are suppressed when there are simultaneous responses to dominant peptides
We have previously established that after multi-peptide immunization, when there are simultaneous CD4 T cell responses to dominant peptides, CD4 T cells specific for subdominant peptides initiate proliferation, but abort expansion midway through the response, typically at day 5 or 6 (28). Figure 1A shows the kinetics of CD4 T cell responses to a representative subdominant CD4 T cell epitope OVA327-339A>S when it is administered as a single peptide or co-administered with dominant CD4 T cell epitopes (MalE102-115 and Myo107-118). Peptides were emulsified in IFA with added LPS and administered subcutaneously in BALB/c mice and expansion of antigen-specific T cells was enumerated by quantifying the number of CD4 T cells that secrete IL-2 or IFN-γ within draining lymph nodes (dLNs) over time. Expansion of CD4 T cells specific for subdominant peptides was robust when introduced as a single peptide, but when two dominant peptides were co-introduced, CD4 T cells specific for the subdominant peptide halted expansion early (day 6 post immunization) (Fig. 1A). In contrast, CD4 T cells specific for the two dominant peptides continued to expand to until day 8 and then declined (Fig. 1B and 1C). In the kinetic studies shown in Figure 1, we noted that expansion of CD4 T cells specific for dominant peptides is detectable at day 4 and peaks with 4–6 times more antigen-specific T cells at day 6 as compared to the CD4 T cells specific for subdominant epitopes. Data that quantify the number of peptide-reactive cells at day 6 producing IFN-γ or IL-2 under conditions of multi-peptide immunization are shown in Figure 1D. Thus, early in the response, dominant CD4 T cells are more abundant than CD4 T cells specific for subdominant responses. Earlier experiments by our laboratory showed that delivering subdominant peptides in a separate emulsion from dominant peptides bypassed the suppression (28), suggesting that the induced loss in responses required that CD4 T cells of different specificities be in close proximity. We therefore considered that there could be competition between different CD4 T cell specificities for APC interactions (32–35) and that over time, adhesion, costimulatory molecules, or APC themselves could become increasingly limiting as dominant CD4 T cells expand.
Figure 1. Expansion of CD4 T cells specific for cryptic peptides fails to progress when there are ongoing CD4 T cell responses to dominant peptides.

BALB/c mice were immunized with peptide/IFA emulsions containing OVA peptide alone (filled) or in combination with dominant MalE and Myo peptides (open). CD4 T cells were purified from pooled popliteal LNs of 2 mice/group and were restimulated with OVA peptide (A), MalE peptide (B), or Myo peptide (C) in IFN-γ Elispot assays, with the peptide used for restimulation indicated above each panel. (D) CD4 T cells were restimulated with the indicated peptides at day 6 post immunization and total number of cytokine-producing cells for the indicated peptide are shown for IFN-γ (left) or IL-2 (right). Data are represented as mean and s.d. between 4 independent experiments; p-values were calculated using 1-way ANOVA.
To rigorously evaluate if suppression required simultaneous presentation of the different epitopes on the same APC, we developed a dendritic cell (DC) priming regimen, which allowed manipulation of APC that will present peptides to CD4 T cells. A serum-free method was used to expand and purify DC from donors to avoid loading DC with foreign antigens present in FBS. This regimen involved the expansion of splenic DC in vivo through intradermal injection of Flt3L-secreting carcinoma cells in donor mice. CD11c+ DC were purified from the spleens of Flt3L-treated mice, pulsed with peptides, and then used to prime naive syngeneic mice. Expansion of CD4 T cell responses was assessed at several time points after immunization (Fig. 2). These experiments confirmed that DC-based priming elicited a similar pattern of response as did emulsion-based immunization: Mice immunized with DC pulsed with subdominant peptide alone elicited a readily detectable response. However, when dominant peptides were co-introduced on the same DC, although the CD4 T cells specific for the subdominant peptides rapidly initiated a response, expansion failed to progress past day 5 (Fig. 2A). Therefore, under competitive conditions, CD4 T cells specific for the subdominant peptide accumulated to levels only one third of that achieved when the peptide was introduced alone (Fig. 2D). As before, CD4 T cell responses to dominant peptides attained higher T cell numbers early in the immune response, by day 5 (Fig. 2B and 2C). This pattern of loss in responses due to competition was seen at various doses of peptide-pulsed DC (5×105–2×106/footpad, data not shown). We next tested whether the suppression required the display of the two types of peptides on the same APC, by pulsing subdominant and dominant peptides on separate populations of DC that were then co-introduced subcutaneously at the same tissue site. These studies, shown in Figure 2D, revealed that expansion of CD4 T cells specific for subdominant peptides is rescued when subdominant peptides are presented by different DC (filled bar vs. hatched bar). There was no significant difference in the response to the subdominant peptide introduced alone compared to the response elicited when there were bystander responses initiated to dominant peptides on separate cohorts of APC (Figure 2D; unfilled bar vs. hatched bar). Thus, loss of CD4 T cell responses requires simultaneous engagement of CD4 T cells specific for dominant and subdominant peptides on the same DC, supporting the hypothesis that suppression is due to highly localized events.
Figure 2. Use of DC-based priming regimen to evaluate the requirement for simultaneous presentation of subdominant and dominant peptides by the same DC.

(A–C) BALB/c mice were immunized with DC loaded with OVA peptide alone (filled) or in combination with dominant MalE and Myo peptides (open). CD4 T cells were purified from pooled popliteal LNs of 3–5 mice/group and were restimulated with OVA peptide (A), MalE peptide (B), or Myo peptide (C) in IL-2 Elispot assays at the indicated time points post immunization, with the peptide used for restimulation indicated above each panel. (D) Mice were immunized with DC loaded with either OVA peptide alone (unfilled bar), OVA in combination with dominant peptides (filled bar), or OVA on separate DC (hatched bar). CD4 T cells were purified from pooled popliteal LNs of 3–5 mice/group at day 9 and restimulated with OVA peptide in IL-2 Elispot assays. Data is represented as the mean and s.e.m. between 4–5 independent experiments; statistical analysis was conducted using 1-way ANOVA.
There are at least two non-mutually exclusive possibilities to explain the preceding result. First, it is known that membrane fragments containing surface-bound molecules such as pMHC and costimulatory molecules can be transferred or snatched from the APC onto T cells after immunological synapse formation and upon cellular detachment, a process known as trogocytosis (reviewed in (36–38)). We speculated that as the dominant CD4 T cells expand over time, APC may lose stimulatory properties through loss of pMHC ligands and costimulatory molecules via this process of membrane transfer. This “membrane snatching” by T cells would render the peptide-bearing APC less able to sustain continued engagement of CD4 T cells specific for the subdominant peptide, halting their progression program. The second possibility is that APC contact with dominant CD4 T cells induces local suppressive mechanisms that act toward CD4 T cells specific for the subdominant peptides. We have evaluated both of these possibilities.
We first explored whether trogocytosis of molecules on the peptide-bearing APC contributed to the loss in continued expansion of CD4 T cells specific for subdominant peptides. Although trogocytosis is antigen-specific and TCRs have to initially engage cognate pMHC ligands, T cells may also snatch non-cognate pMHC ligands if these molecules are present within the transferred membrane fragment. This process would be compounded as dominant daughter CD4 T cells expand and interact with APC. Under these conditions, requisite cell surface molecules on APC that are required for continued expansion of CD4 T cells specific for subdominant peptides may become limiting. This idea is supported by published data that suggest that non-cognate and endogenous pMHC molecules accumulate within the immunological synapse (39). To evaluate trogocytosis, the level of several cell surface molecules on peptide-loaded DC was quantified over time within dLNs over the course of the immune response. The expression of MHC class II I-Ad and ICAM-1 molecules on the cell surface of DC was monitored ex vivo and we compared three separate immunization conditions: 1) DC with no peptide 2) DC pulsed with subdominant peptide 3) DC pulsed with both subdominant and dominant peptides on the same DC. Peptide-pulsed DC were labeled with CFSE prior to immunization to facilitate their identification. Draining LNs were isolated from recipient mice at several time points after immunization and analyzed by flow cytometry at various time points (Fig. 3). The expression level of MHC class II and ICAM molecules was indistinguishable over time among the three experimental groups, indicating that the loss of molecules from the cell surface over time occurs independently of antigen-specific CD4 T cell interaction. The same was observed for CD80 and CD86 (data not shown). These results suggest that trogocytosis does not account for the selective loss in responses to less-favored peptides during competitive T cell responses. We also assessed whether the absolute numbers of peptide-bearing DC suffered greater losses when there are ongoing responses to dominant peptides. The frequency and absolute number of CFSE-labeled DC were compared between peptide-pulse conditions in the dLN over time. These studies revealed that the number of DC accumulating and retained in the LN was independent of peptide, as were the rates of loss of DC in LNs (Fig. 3B). We have also analyzed trogocytosis in APC that have migrated from the tissue site after IFA-based immunizations by labeling the emulsion with CFSE, which labeled DC that emigrate from the site of immunization to the dLN and found the same results (data not shown). Altogether, increased loss of DC or key cell surface molecules on DC does not detectably contribute to diminished CD4 T cell responses to subdominant peptides when responses to dominant peptides are ongoing.
Figure 3. Cell surface expression of MHCII and ICAM-1 on DC is not further reduced after multiple peptide immunization.
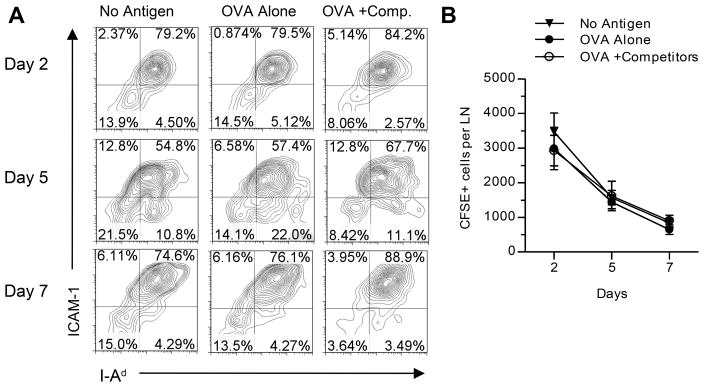
BALB/c mice (4–5 mice/group) were immunized with CFSE-labeled DC loaded with no antigen, OVA peptide alone, or OVA peptide in combination with dominant MalE and Myo peptides. (A) Expression of ICAM-1 and I-Ad on gated CFSE+ cells in pooled dLNs. (B) Absolute number of CFSE bright cells per LN represented as the mean and s.e.m. between four independent experiments.
Treg actvity is increased under competitive conditions
One alternate explanation for the loss in CD4 T cell responses to subdominant peptides is that regulatory CD4 T cells (Tregs) are more suppressive when simultaneous responses to dominant peptides are ongoing. Various mechanisms of Treg suppression, either mediated by cell contact-dependent or soluble mechanisms, have been proposed and the specific mechanism in vivo may depend on the organ and inflammatory context (reviewed in (40–43, 44). Tregs can suppress target CD4 T cells by inhibiting T cell proliferation, T effector cytokine production, inducing T cell cytolysis, physically blocking DC, and downregulating costimulatory molecules on DC (41). We therefore assessed whether Treg activity is increased numerically or functionally after multi-peptide immunization compared to single peptide immunization. Foxp3GFP reporter mice were utilized to quantify Tregs after peptide immunization and to easily sort them for functional assays. By tracking the expansion of Tregs based on expression of Foxp3 within the CD4 T cell compartment, we found that both the frequency (Fig. 4A) and absolute number of Tregs (Fig. 4B) were similar between mice immunized with either subdominant peptide alone or co-administered with dominant peptides at all time points assessed. Because there was no significant increase in Treg numbers in dLNs, we examined whether the function of Tregs changes when multiple peptide epitopes are introduced. An in vitro suppression assay was used to assess Treg suppressive activity (45, 46). Foxp3GFP mice were immunized with subdominant peptides alone or with the addition of dominant peptides and at day 6 post immunization, flow cytometry was used to isolate Tregs from dLNs. Serial dilutions of Tregs were cocultured with CFSE-labeled polyclonal naive target T cells and irradiated splenic APC. After stimulation of cocultures with anti-CD3 for 72 hours, proliferation of CFSE-labeled target CD4 T cells was assessed. Figure 5A shows that, although modest and variable from experiment to experiment, the suppressive activity of Tregs was enhanced by the presence of dominant peptides in the immunogen. Therefore, Tregs appear to have somewhat more functional activity on per cell basis in LNs where immune responses to multiple epitopes take place. Based on this result, we asked whether suppression of T cell responses to subdominant peptides would be alleviated if Tregs were depleted. Mice expressing the human diphtheria toxin (DT) receptor under the control of the Foxp3 promoter (Foxp3DTR) were immunized, and DT was administered at days 3 and 4 post immunization. DT administration at days 3 and 4 depleted Tregs for at least 6 days, as indicated by the lack of Foxp3DTR-GFP expression within the CD4 T cell compartment at day 10 (Supplemental Fig. 1). We compared T cell responses to subdominant peptides at the peak of the immune response. The experiments displayed in Figure 5B show that at day 10, CD4 T cell responses to subdominant peptides were partially rescued when immunized Foxp3DTR mice were depleted of Tregs by treatment with DT. Collectively, these data suggest that when the immune system is confronted with multiple epitopes, more suppressive Tregs are elicited and these act to selectively diminish expansion of some peptide specificities.
Figure 4. Multiple peptide immunization does not selectively increase Treg expansion.
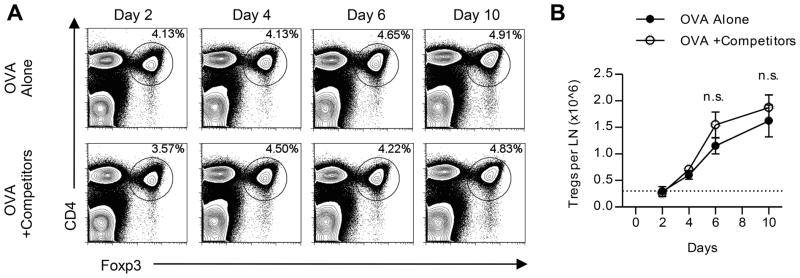
Foxp3GFP mice were immunized with peptide/IFA emulsions containing OVA peptide alone or in combination with dominant MalE and Myo peptides (+Competitors). (A) Frequency of Foxp3+ CD4 T cells in unfractionated popliteal LNs from a representative mouse of two is shown for each group over time. (B) Absolute number of Tregs per popliteal LN was quantified from the experiment in A. Data are representative of three independent experiments. Error bars represent the mean and s.d. between two individual mice per group; statistical analysis was conducted using 2-way ANOVA.
Figure 5. Treg activity is enhanced by the presence of dominant peptides.
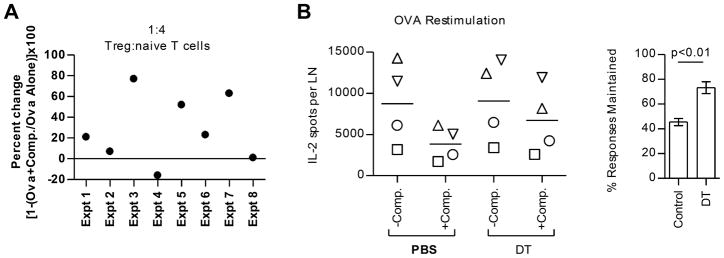
(A) Tregs were flow-sorted from pooled popliteal LNs of Foxp3GFP mice (2 mice/group) that were immunized 6 days prior. Tregs were co-cultured with irradiated APCs and CFSE-labeled target T cells (Thy1.1) in the presence of 1μg/ml anti-CD3 for 72 hrs. Shown is the change in the frequency of Thy1.1+ responder CD4 T cells that have proliferated in cocultures with Tregs sorted from the OVA +Competitors group compared to cocultures with Tregs sorted from the OVA alone group. Data from eight experiments are shown. (B) Foxp3DTR mice (2–3 mice/group) were immunized with peptide/IFA emulsions containing OVA peptide competitor peptides. Mice alone or OVA peptide in combination with MalE, NP261-274, and NP310-325 were administered DT or PBS control at days 3 and 4 post immunization. At day 10, CD4 T cells were purified from pooled inguinal and popliteal LNs and restimulated with OVA peptide. Data from four independent experiments are shown, with each experiment represented by a different symbol (B; left panel). The % of the OVA-specific response maintained in the presence of the competitor peptides relative to no competition with and without DT treatment is displayed in the bar graph (B; right panel).
IFN-γ mediates suppression of CD4 T cell responses to subdominant peptides
Because depletion of Tregs did not lead to complete rescue of responses to subdominant peptides, we explored other potential mechanisms responsible for the loss of responses. We considered the possibility that as CD4 T cells specific for dominant peptides expand and differentiate, they produce cytokines that downregulate the ongoing response in the same dLN. The main cytokines detected under the conditions of priming used here are IL-2 and IFN-γ ((28) and this study). Although IFN-γ is historically considered a pro-inflammatory cytokine, there is also evidence that it can have suppressive effects (47–50). To test the potential involvement of IFN-γ in the loss of responses to subdominant peptides, a genetic approach was adopted. CD4 T cell responses were compared in wild-type (WT) mice and mice deficient in IFN-γ (Fig. 6). These studies revealed that the magnitude of the CD4 T cell responses specific for subdominant peptides in competitive conditions are partially recovered in mice lacking IFN-γ (Fig. 6A). Responses to dominant peptides were not detectably altered by the presence of IFN-γ in the host (Fig. 6B and 6C).
Figure 6. IFN-γ mediates the loss of cryptic CD4 T cell responses after multiple peptide immunization.

WT BALB/c and IFN-γKO mice (3–5 mice/group) were immunized with peptide/IFA emulsions containing OVA peptide alone (−Comp.) or OVA peptide in combination with dominant MalE and Myo peptides (+Comp.). At day 10 post immunization, CD4 T cells were purified from popliteal LNs of individual mice and were restimulated with OVA peptide (A), MalE peptide (B), or Myo peptide (C) in IL-2 Elispot assays. The right panel of (A) shows the % of the OVA peptide-specific response maintained when in competition among the four experiments in WT or IFN-γKO mice. The right panels of (B) and (C) show the relative response to dominant peptides of IFN-γKO compared to WT mice. Data from 4 independent experiments were overlaid (each represented by a different color). p-values in the left panel of (A) were calculated using 1-way ANOVA followed by Bonferroni post test. p-values in the right panel of (A) and the left panel of (B) and (C) were calculated from Student’s t test.
IFN-γ might have negative effects on either APC function or neighboring CD4 T cells. Therefore, we explored the cell type responsive to IFN-γ. To determine if expression of IFN-γ receptors on DC could account for the suppression, we used the DC-based priming strategy. WT and IFN-γ receptor-deficient (IFNγ-RKO) DC were expanded in vivo by using Flt3L as before and isolated DC were pulsed with peptide and used to prime WT mice (Fig. 7). Strikingly, CD4 T cell responses initiated by IFN-γRKO DC were no longer affected by bystander responses to the dominant peptides. When IFN-γRKO DC were presenting the antigenic peptides, subdominant peptides were as capable of recruiting and expanding CD4 T cells in the presence of dominant CD4 T cells as in their absence. These experiments suggest that suppression of CD4 T cell responses to subdominant peptides by ongoing CD4 T cell responses can be accounted for by the action of IFN-γ on antigen-bearing DC.
Figure 7. IFN-γ responsiveness in DC is necessary for the suppression of subdominant CD4 T cell responses.

In four independent experiments (represented by different symbols), WT BALB/c mice (3–5 pooled mice/group) were immunized with WT or IFN-γRKO DC loaded with OVA peptide alone (−Comp.) or OVA peptide in combination with dominant MalE and Myo peptides (+Comp.). CD4 T cells were purified from pooled popliteal LNs at day 9 and tested for reactivity to OVA peptide (A), MalE peptide (B), or Myo peptide (C). The left panels show the number of peptide-reactive cells per LN. In (A), the right panel shows the % of the OVA response maintained when in competition. The right panels of (B) and (C) show percent change in T cell responses to the dominant peptides in mice immunized with IFN-γRKO DCs compared to mice immunized with WT DCs. p-values were calculated using 1-way ANOVA in the left panel of (A) and Student’s t test in the right panel of (A) and for (B) and (C).
We next sought to identify potentially suppressive molecules that may be induced in DC, in response to IFN-γ signaling. It is known that IFN-γ can suppress CD4 T cell responses by activating a tryptophan-catabolizing enzyme, IDO (51–56). IDO catalyzes the rate-limiting step in degradation of the essential amino acid tryptophan. The depletion of tryptophan and accumulation of tryptophan metabolites can promote tolerogenic DC function, proliferative arrest in T cells, and induction of Tregs (56–59). This pathway of regulation is thought to ordinarily function as part of a negative feedback loop that regulates uncontrolled activation of the adaptive immune response (60, 61). To explore the participation of this pathway of regulation, IDO enzymatic activity was blocked throughout the immune response by administering 1-MT in the drinking water (60). As before, at day 10, dLNs were isolated and peptide-specific cells were quantified by cytokine Elispot assays. These studies, shown in Figure 8, revealed that CD4 T cell responses to subdominant peptides, when in the presence of bystander responses, partially recovered when an IDO inhibitor was present. These results suggested that CD4 T cell responses to subdominant peptides are suppressed by an IDO-dependent mechanism. We have also combined treatments of Treg depletion and 1-MT treatment (Supplemental Fig. 2), as well as IFN-γKO and 1-MT treatment (Supplemental Fig. 3) and did not see additive effects, suggesting that these mediators are part of the same pathway of regulation.
Figure 8. Involvement of the IDO pathway in regulating the selectivity of CD4 T cell responses.

In three independent experiments (each represented by a different color), BALB/c mice (5 mice/group) were immunized with peptide/IFA emulsions containing OVA peptide alone (−Comp.) or OVA peptide in combination with dominant MalE and Myo peptides (+Comp.) and were orally administered 1-MT or control. At day 10 post immunization, CD4 T cells were purified from popliteal LNs of individual mice and were restimulated with OVA peptide (A), MalE peptide (B), or Myo peptide (C). The left panels show the total number of peptide-specific T cells. The right panels show the % of the OVA-specific response maintained when in competition (A) or the percent change in T cell responses in mice treated with 1-MT compared to control (B) and (C); p-values shown were calculated from 1-way ANOVA followed by Bonferroni post test in the left panel of (A) or Student’s t test in the right panel of (A) and for (B) and (C).
DISCUSSION
There is a great deal of interest in identifying the epitopes that CD4 T cells focus on during immune responses and the forces that shape preferences in T cell priming in response to pathogen infection and vaccination. Progress in our understanding of this issue would allow the use of these epitopes in vaccines to elicit broadly reactive CD4 T cells. The current study addressed the mechanisms that underlie peptide epitope preferences of CD4 T cell responses to multi-peptide vaccination. Our studies revealed that CD4 T cell responses to less favored pMHC complexes can fully expand if subdominant and dominant peptides are loaded on separate DC. We considered the possibility that expansion of dominant CD4 T cells would diminish the potency of APC through trogocytosis, thus limiting the amount of molecules available for priming subdominant CD4 T cells. However, we found that the expression level of key costimulatory molecules was not affected by the number of epitopes contained in the immunogen. This raised the possibility that dominant T cell responses actively suppress cryptic or subdominant CD4 T cell responses after multi-peptide immunization, an issue not experimentally addressed in the field.
We then evaluated the possibility that suppressive mechanisms might be induced by robust responses to dominant peptides and found strong evidence for a network of regulation that involves IFN-γ, IDO, and Tregs. Our studies revealed that expression of IFN-γ by the host is necessary for restricting the CD4 T cell responses to only dominant peptides. This possibility is further supported by our results that when peptide-pulsed APC are deficient in the receptor for IFN-γ, CD4 T cell responses to subdominant peptides were restored. Although Treg numbers were similar, their suppressive activity was slightly increased when dominant peptides were co-administered. Furthermore, the depletion of Tregs enhanced responses to subdominant peptides. These results collectively suggest that continued expansion of CD4 T cell responses to cryptic or subdominant peptides is inhibited by suppressive cells and molecules that arise when there are stronger responses to co-introduced dominant peptides.
The key findings made here suggest a complex pathway of immunoregulation (illustrated in Fig. 9). CD4 T cells reactive to dominant peptides that bind with high stability to the MHC class II molecule expand rapidly, differentiate and produce IFN-γ. Under the conditions of priming used here, typical of pathogens that ligate TLR4, expansion of CD4 T cells is associated with differentiation into IFN-γ-producing cells. During the same time frame, expansion of CD4 T cells for subdominant peptides is initiated, but lags behind the dominant T cells, possibly due to lower TcR signaling by unstable pMHC ligands. By day 4–6 after immunization, dominant CD4 T cells have completed their expansion and secrete IFN-γ in the local environment. IFN-γ binds to IFN-γ receptors on peptide-bearing DC leading to induction of IDO and kynurenines, activation of Tregs, and attenuation of DC stimulatory capacity. Under these conditions, expansion of CD4 T cells specific for subdominant and cryptic peptides discontinues. Inflammation in the dLN may induce IFN-γ production by NK cells or CD8 T cells. However, we did not observe a difference in T cell responses when NK cell depletion experiments were conducted and we have determined that our antigenic peptides do not initiate antigen-specific CD8 T cell responses (data not shown).
Figure 9. Ongoing CD4 T cell responses to dominant peptides induce suppressive activities in the local microenvironment that reduce the abundance of CD4 T cells specific for subdominant peptides.

Peptides introduced either subcutaneously or on autologous DC initially activate all peptide-specific CD4 T cells equally whether in the presence or absence of other antigenic peptides. Within the first few days of DC arrival, CD4 T cells specific for dominant peptides expand more quickly than CD4 T cells specific for subdominant peptides. As dominant CD4 T cells expand, they differentiate into effector cells and produce IFN-γ. The presence of IFN-γ in the local environment alters the function of DCs that are co-engaging CD4 T cells specific for subdominant peptides. In response to IFN-γ, DC rapidly activate IDO, which in turn increases tryptophan catabolism and production of kynurenines that can arrest cell division of subdominant CD4 T cells and induce the generation of antigen-specific Tregs.
There is likely to be at least one more, yet unidentified participating cell or molecule that is induced by IFN-γ in DC because we were not able to completely replicate the rescue of subdominant responses in experiments in which IDO inhibitors were used or when Tregs were depleted, as seen with immunization with IFN-γR deficient DC. One candidate molecule is iNOS. IFN-γ has also been shown to induce iNOS expression in DCs and nonhematopoietic stromal cells of lymphoid organs that can negatively regulate T cell survival and proliferation (62–64). To test the possible involvement of induced iNOS activity in the suppression of subdominant T cell responses after peptide immunization, we compared competitive T cell responses in NOS2KO and WT mice (data not shown). These experiments revealed that deficiency in iNOS had no effect on T cell responses, suggesting that iNOS either may not be optimally expressed in cells during our experimental priming conditions or that its activity was not sufficient to suppress T cell expansion.
A critical issue to understand is why CD4 T cell responses to subdominant peptides are selectively sensitive to suppression by this immunoregulatory network. There are several interesting possibilities. First, in vivo, there is likely to be a finite proliferative program for most CD4 T cells. In our experimental system, the expansion and differentiation of CD4 T cell responses to dominant peptides is completed prior to the induction of suppressive pathways. Sensitivity to the suppression may thus be determined by generation time. If subdominant CD4 T cells are relatively slower to expand, they would immediately be susceptible to inhibitory pathways initiated prematurely by ongoing dominant responses. An alternative possibility is that protection from suppression is dependent on productive TCR signaling, and diminishing epitope density of cryptic and subdominant peptides over time leaves T cells vulnerable to this suppression. In this way, halted expansion of T cells to subdominant peptides may be a consequence of their inefficient signaling. This would potentially be a mechanism to “weed out” less useful T cells, those that are specific for rapidly decaying pMHC complexes.
The current study has clinical implications for DC-based cancer immunotherapy approaches. DC-based approaches are currently used clinically to induce T cell responses to tumor-associated antigens (reviewed in (65)). Our findings suggest that DC immunization strategies can be successful if subdominant peptides are loaded on separate DC from dominant peptides, which allows the unperturbed expansion of subdominant CD4 T cells and sequesters subdominant CD4 T cells from the effects of ongoing dominant CD4 T cell responses.
Supplementary Material
Acknowledgments
This work was supported by grants HHSN27220201200005C, HHSN266200700008C and R01AI51542, and 5T32AI007285 from the National Institutes of Health.
We thank Dr. Jason M. Weaver for his efforts in generating the Flt3L-secreting cell line. We thank Dr. Deborah J. Fowell and Scott Leddon for helpful discussions and for critically reading this manuscript.
References
- 1.Scharnagl NC, Klade CS. Experimental discovery of T-cell epitopes: combining the best of classical and contemporary approaches. Expert Rev Vaccines. 2007;6:605–615. doi: 10.1586/14760584.6.4.605. [DOI] [PubMed] [Google Scholar]
- 2.Nielsen M, Lund O, Buus S, Lundegaard C. MHC class II epitope predictive algorithms. Immunology. 2010;130:319–328. doi: 10.1111/j.1365-2567.2010.03268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang P, Sidney J, Dow C, Mothe B, Sette A, Peters B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol. 2008;4:e1000048. doi: 10.1371/journal.pcbi.1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dudek NL, Perlmutter P, Aguilar MI, Croft NP, Purcell AW. Epitope discovery and their use in peptide based vaccines. Curr Pharm Des. 2010;16:3149–3157. doi: 10.2174/138161210793292447. [DOI] [PubMed] [Google Scholar]
- 5.Sette A, Fikes J. Epitope-based vaccines: an update on epitope identification, vaccine design and delivery. Current Opinion in Immunology. 2003;15:461–470. doi: 10.1016/s0952-7915(03)00083-9. [DOI] [PubMed] [Google Scholar]
- 6.Deluca DS, Blasczyk R. The immunoinformatics of cancer immunotherapy. Tissue Antigens. 2007;70:265–271. doi: 10.1111/j.1399-0039.2007.00914.x. [DOI] [PubMed] [Google Scholar]
- 7.Slingluff CL., Jr The present and future of peptide vaccines for cancer: single or multiple, long or short, alone or in combination? Cancer J. 2011;17:343–350. doi: 10.1097/PPO.0b013e318233e5b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mocellin S, Pilati P, Nitti D. Peptide-based anticancer vaccines: recent advances and future perspectives. Curr Med Chem. 2009;16:4779–4796. doi: 10.2174/092986709789909648. [DOI] [PubMed] [Google Scholar]
- 9.Kanodia S, Kast WM. Peptide-based vaccines for cancer: realizing their potential. Expert Rev Vaccines. 2008;7:1533–1545. doi: 10.1586/14760584.7.10.1533. [DOI] [PubMed] [Google Scholar]
- 10.Mocellin S. Peptides in melanoma therapy. Curr Pharm Des. 2012;18:820–831. doi: 10.2174/138161212799277734. [DOI] [PubMed] [Google Scholar]
- 11.Traversari C, van der Bruggen P, Luescher IF, Lurquin C, Chomez P, Van Pel A, De Plaen E, Amar-Costesec A, Boon T. A nonapeptide encoded by human gene MAGE-1 is recognized on HLA-A1 by cytolytic T lymphocytes directed against tumor antigen MZ2-E. J Exp Med. 1992;176:1453–1457. doi: 10.1084/jem.176.5.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Purcell AW, McCluskey J, Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nat Rev Drug Discov. 2007;6:404–414. doi: 10.1038/nrd2224. [DOI] [PubMed] [Google Scholar]
- 13.Wiesel M, Oxenius A. From crucial to negligible: functional CD8(+) T-cell responses and their dependence on CD4(+) T-cell help. Eur J Immunol. 2012;42:1080–1088. doi: 10.1002/eji.201142205. [DOI] [PubMed] [Google Scholar]
- 14.Deng H, Fosdick L, Sercarz E. The involvement of antigen processing in determinant selection by class II MHC and its relationship to immunodominance. APMIS. 1993;101:655–662. doi: 10.1111/j.1699-0463.1993.tb00161.x. [DOI] [PubMed] [Google Scholar]
- 15.Chen W, McCluskey J. Immunodominance and immunodomination: critical factors in developing effective CD8+ T-cell-based cancer vaccines. Adv Cancer Res. 2006;95:203–247. doi: 10.1016/S0065-230X(06)95006-4. [DOI] [PubMed] [Google Scholar]
- 16.Yewdell JW. Confronting complexity: real-world immunodominance in antiviral CD8+ T cell responses. Immunity. 2006;25:533–543. doi: 10.1016/j.immuni.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 17.Sant AJ, Chaves FA, Krafcik FR, Lazarski CA, Menges P, Richards K, Weaver JM. Immunodominance in CD4 T-cell responses: implications for immune responses to influenza virus and for vaccine design. Expert Rev Vaccines. 2007;6:357–368. doi: 10.1586/14760584.6.3.357. [DOI] [PubMed] [Google Scholar]
- 18.Sant AJ, Chaves FA, Jenks SA, Richards KA, Menges P, Weaver JM, Lazarski CA. The relationship between immunodominance, DM editing, and the kinetic stability of MHC class II:peptide complexes. Immunol Rev. 2005;207:261–278. doi: 10.1111/j.0105-2896.2005.00307.x. [DOI] [PubMed] [Google Scholar]
- 19.Ma C, Whiteley PE, Cameron PM, Freed DC, Pressey A, Chen SL, Garni-Wagner B, Fang C, Zaller DM, Wicker LS, Blum JS. Role of APC in the selection of immunodominant T cell epitopes. J Immunol. 1999;163:6413–6423. [PubMed] [Google Scholar]
- 20.Lazarski CA, Chaves FA, Jenks SA, Wu S, Richards KA, Weaver JM, Sant AJ. The kinetic stability of MHC class II:peptide complexes is a key parameter that dictates immunodominance. Immunity. 2005;23:29–40. doi: 10.1016/j.immuni.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 21.Lazarski CA, Chaves FA, Sant AJ. The impact of DM on MHC class II-restricted antigen presentation can be altered by manipulation of MHC-peptide kinetic stability. J Exp Med. 2006;203:1319–1328. doi: 10.1084/jem.20060058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Ham SM, Gruneberg U, Malcherek G, Broker I, Melms A, Trowsdale J. Human histocompatibility leukocyte antigen (HLA)-DM edits peptides presented by HLA-DR according to their ligand binding motifs. J Exp Med. 1996;184:2019–2024. doi: 10.1084/jem.184.5.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schulze MS, Wucherpfennig KW. The mechanism of HLA-DM induced peptide exchange in the MHC class II antigen presentation pathway. Curr Opin Immunol. 2012;24:105–111. doi: 10.1016/j.coi.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blum JS, Ma C, Kovats S. Antigen-presenting cells and the selection of immunodominant epitopes. Crit Rev Immunol. 1997;17:411–417. [PubMed] [Google Scholar]
- 25.Nanda NK, Sant AJ. DM determines the cryptic and immunodominant fate of T cell epitopes. J Exp Med. 2000;192:781–788. doi: 10.1084/jem.192.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karlsson L. DM and DO shape the repertoire of peptide-MHC-class-II complexes. Curr Opin Immunol. 2005;17:65–70. doi: 10.1016/j.coi.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 27.Nanda NK, Bikoff EK. DM peptide-editing function leads to immunodominance in CD4 T cell responses in vivo. J Immunol. 2005;175:6473–6480. doi: 10.4049/jimmunol.175.10.6473. [DOI] [PubMed] [Google Scholar]
- 28.Weaver JM, Chaves FA, Sant AJ. Abortive activation of CD4 T cell responses during competitive priming in vivo. Proc Natl Acad Sci U S A. 2009;106:8647–8652. doi: 10.1073/pnas.0811584106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McAdam AJ, Pulaski BA, Storozynsky E, Yeh KY, Sickel JZ, Frelinger JG, Lord EM. Analysis of the effect of cytokines (interleukins 2, 3, 4, and 6, granulocyte-monocyte colony-stimulating factor, and interferon-gamma) on generation of primary cytotoxic T lymphocytes against a weakly immunogenic tumor. Cell Immunol. 1995;165:183–192. doi: 10.1006/cimm.1995.1204. [DOI] [PubMed] [Google Scholar]
- 30.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 31.Hou DY, Muller AJ, Sharma MD, DuHadaway J, Banerjee T, Johnson M, Mellor AL, Prendergast GC, Munn DH. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 2007;67:792–801. doi: 10.1158/0008-5472.CAN-06-2925. [DOI] [PubMed] [Google Scholar]
- 32.Kedl RM, Kappler JW, Marrack P. Epitope dominance, competition and T cell affinity maturation. Current Opinion in Immunology. 2003;15:120–127. doi: 10.1016/s0952-7915(02)00009-2. [DOI] [PubMed] [Google Scholar]
- 33.Ge Q, Bai A, Jones B, Eisen HN, Chen J. Competition for self-peptide-MHC complexes and cytokines between naive and memory CD8+ T cells expressing the same or different T cell receptors. Proc Natl Acad Sci U S A. 2004;101:3041–3046. doi: 10.1073/pnas.0307339101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blair DA, Lefrancois L. Increased competition for antigen during priming negatively impacts the generation of memory CD4 T cells. Proc Natl Acad Sci U S A. 2007;104:15045–15050. doi: 10.1073/pnas.0703767104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Willis RA, Kappler JW, Marrack PC. CD8 T cell competition for dendritic cells in vivo is an early event in activation. Proc Natl Acad Sci U S A. 2006;103:12063–12068. doi: 10.1073/pnas.0605130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahmed KA, Xiang J. Mechanisms of cellular communication through intercellular protein transfer. J Cell Mol Med. 2011;15:1458–1473. doi: 10.1111/j.1582-4934.2010.01008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caumartin J, Lemaoult J, Carosella ED. Intercellular exchanges of membrane patches (trogocytosis) highlight the next level of immune plasticity. Transpl Immunol. 2006;17:20–22. doi: 10.1016/j.trim.2006.09.032. [DOI] [PubMed] [Google Scholar]
- 38.Joly E, Hudrisier D. What is trogocytosis and what is its purpose? Nat Immunol. 2003;4:815. doi: 10.1038/ni0903-815. [DOI] [PubMed] [Google Scholar]
- 39.Gascoigne NR, Zal T, Yachi PP, Hoerter JA. Co-receptors and recognition of self at the immunological synapse. Curr Top Microbiol Immunol. 2010;340:171–189. doi: 10.1007/978-3-642-03858-7_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fehervari Z, Sakaguchi S. Development and function of CD25+CD4+ regulatory T cells. Curr Opin Immunol. 2004;16:203–208. doi: 10.1016/j.coi.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 41.Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol. 2010;11:7–13. doi: 10.1038/ni.1818. [DOI] [PubMed] [Google Scholar]
- 42.Yamaguchi T, Wing JB, Sakaguchi S. Two modes of immune suppression by Foxp3(+) regulatory T cells under inflammatory or non-inflammatory conditions. Semin Immunol. 2011;23:424–430. doi: 10.1016/j.smim.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 43.Shevach EM. Biological functions of regulatory T cells. Adv Immunol. 2011;112:137–176. doi: 10.1016/B978-0-12-387827-4.00004-8. [DOI] [PubMed] [Google Scholar]
- 44.Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol. 2008;9:239–244. doi: 10.1038/ni1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sojka DK, Fowell DJ. Regulatory T cells inhibit acute IFN-gamma synthesis without blocking T-helper cell type 1 (Th1) differentiation via a compartmentalized requirement for IL-10. Proc Natl Acad Sci U S A. 2011;108:18336–18341. doi: 10.1073/pnas.1110566108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sojka DK, Hughson A, Sukiennicki TL, Fowell DJ. Early kinetic window of target T cell susceptibility to CD25+ regulatory T cell activity. J Immunol. 2005;175:7274–7280. doi: 10.4049/jimmunol.175.11.7274. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Y, Apilado R, Coleman J, Ben-Sasson S, Tsang S, Hu-Li J, Paul WE, Huang H. Interferon gamma stabilizes the T helper cell type 1 phenotype. J Exp Med. 2001;194:165–172. doi: 10.1084/jem.194.2.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li X, McKinstry KK, Swain SL, Dalton DK. IFN-gamma acts directly on activated CD4+ T cells during mycobacterial infection to promote apoptosis by inducing components of the intracellular apoptosis machinery and by inducing extracellular proapoptotic signals. J Immunol. 2007;179:939–949. doi: 10.4049/jimmunol.179.2.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Y, Janeway CA., Jr Interferon gamma plays a critical role in induced cell death of effector T cell: a possible third mechanism of self-tolerance. J Exp Med. 1990;172:1735–1739. doi: 10.1084/jem.172.6.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Refaeli Y, Van Parijs L, Alexander SI, Abbas AK. Interferon gamma is required for activation-induced death of T lymphocytes. J Exp Med. 2002;196:999–1005. doi: 10.1084/jem.20020666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Munn DH. Indoleamine 2,3-dioxygenase, tumor-induced tolerance and counter-regulation. Curr Opin Immunol. 2006;18:220–225. doi: 10.1016/j.coi.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 52.Terness P, Bauer TM, Rose L, Dufter C, Watzlik A, Simon H, Opelz G. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med. 2002;196:447–457. doi: 10.1084/jem.20020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. 2002;196:459–468. doi: 10.1084/jem.20020121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Munn DH, Sharma MD, Lee JR, Jhaver KG, Johnson TS, Keskin DB, Marshall B, Chandler P, Antonia SJ, Burgess R, Slingluff CL, Jr, Mellor AL. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science. 2002;297:1867–1870. doi: 10.1126/science.1073514. [DOI] [PubMed] [Google Scholar]
- 55.Boasso A, Herbeuval JP, Hardy AW, Anderson SA, Dolan MJ, Fuchs D, Shearer GM. HIV inhibits CD4+ T-cell proliferation by inducing indoleamine 2,3-dioxygenase in plasmacytoid dendritic cells. Blood. 2007;109:3351–3359. doi: 10.1182/blood-2006-07-034785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, Servillo G, Brunacci C, Calvitti M, Bicciato S, Mazza EM, Boon L, Grassi F, Fioretti MC, Fallarino F, Puccetti P, Grohmann U. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol. 2011;12:870–878. doi: 10.1038/ni.2077. [DOI] [PubMed] [Google Scholar]
- 57.Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, Mellor AL. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol. 2009;183:2475–2483. doi: 10.4049/jimmunol.0900986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mellor AL, Baban B, Chandler P, Marshall B, Jhaver K, Hansen A, Koni PA, Iwashima M, Munn DH. Cutting edge: induced indoleamine 2,3 dioxygenase expression in dendritic cell subsets suppresses T cell clonal expansion. J Immunol. 2003;171:1652–1655. doi: 10.4049/jimmunol.171.4.1652. [DOI] [PubMed] [Google Scholar]
- 59.Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, Mellor AL. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22:633–642. doi: 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 60.Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4:762–774. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 61.Johnson BA, 3rd, Baban B, Mellor AL. Targeting the immunoregulatory indoleamine 2,3 dioxygenase pathway in immunotherapy. Immunotherapy. 2009;1:645–661. doi: 10.2217/IMT.09.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lukacs-Kornek V, Malhotra D, Fletcher AL, Acton SE, Elpek KG, Tayalia P, Collier AR, Turley SJ. Regulated release of nitric oxide by nonhematopoietic stroma controls expansion of the activated T cell pool in lymph nodes. Nat Immunol. 2011;12:1096–1104. doi: 10.1038/ni.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lu L, Bonham CA, Chambers FG, Watkins SC, Hoffman RA, Simmons RL, Thomson AW. Induction of nitric oxide synthase in mouse dendritic cells by IFN-gamma, endotoxin, and interaction with allogeneic T cells: nitric oxide production is associated with dendritic cell apoptosis. J Immunol. 1996;157:3577–3586. [PubMed] [Google Scholar]
- 64.Bogdan C. Nitric oxide and the immune response. Nat Immunol. 2001;2:907–916. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 65.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12:265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.