

Abstract
Physical tethering of membrane proteins to the cortical actin cytoskeleton provides functional organization to the plasma membrane and contributes to diverse cellular processes including cell signaling, vesicular trafficking, endocytosis, and migration. For these processes to occur, membrane protein tethering must be dynamically regulated in response to environmental cues. In this study, we describe a novel biochemical scheme for isolating the complement of plasma membrane proteins that are physically tethered to the actin cytoskeleton. We utilized this method in combination with tandem liquid chromatography/mass spectrometry (LC–MS/MS) to demonstrate that cytoskeletal tethering of membrane proteins is acutely regulated by epidermal growth factor (EGF) in normal human kidney (HK2) cells. Our results indicate that several proteins known to be involved in EGF signaling, as well as other proteins not traditionally associated with this pathway, are tethered to the cytoskeleton in dynamic fashion. Further analysis of one hit from our proteomic survey, the receptor phosphotyrosine phosphatase PTPRS, revealed a correlation between cytoskeletal tethering and endosomal trafficking in response to EGF. This finding parallels previous indications that PTPRS is involved in the desensitization of EGFR and provides a potential mechanism to coordinate localization of these two membrane proteins in the same compartment upon EGFR activation.
Keywords: tethering, actin cytoskeleton, EGFR, proteomics, mass spectrometry, PTPRS
INTRODUCTION
The transmembrane proteome comprises a diverse collection of proteins involved in myriad cellular functions including nutrient transport, adhesion, cellular motility, metabolism, and cell signaling. These functions are regulated by an equally diverse array of mechanisms that include post-translational modification, endocytosis, and oligomerization/aggregation within the membrane. All of these regulatory mechanisms involve interactions of transmembrane proteins with the underlying cortical actin cytoskeleton, which both organizes and stabilizes the membrane proteome into distinct functional domains.1
Interactions between transmembrane proteins and the actin cytoskeleton can be classified as passive or active.2 Passive interactions involve cytoskeleton-mediated organization of the membrane into distinct “corrals”, which primarily serve to limit lateral diffusion of proteins within the lipid bilayer.3 Active interactions, on the other hand, involve physical tethering of transmembrane proteins to cortical actin filaments (F-actin).4 While active tethering similarly limits the lateral mobility of transmembrane proteins, it is also critical for the maintenance of cell–cell contacts and membrane microdomains, such as lipid rafts and focal adhesions, and for vesicular trafficking at the cell surface.5
Active tethering of transmembrane proteins is facilitated either by direct interactions of proteins with F-actin or, more commonly, by adaptor proteins that link the cytoplasmic domains of transmembrane proteins to F-actin. Adaptor proteins involved in cytoskeletal tethering interact with distinct sets of substrates within the membrane proteome, allowing for coordinated tethering of multiple transmembrane proteins in response to signals that govern adaptor protein abundance or activity.6 Temporal and spatial coordination of transmembrane protein tethering by adaptor proteins clearly plays an important role in complex cellular processes such as signal transduction, though the mechanisms by which the relatively limited set of adaptor proteins, which include proteins of the ezrin–radixin–moesin (ERM) family, are directed to specific membrane complexes is incompletely understood.6a
Cytoskeletal tethering plays in important role in both the positive and negative regulation of epidermal growth factor receptor (EGFR), depending on which adaptor protein is involved in the process.6c Tethering by ERM proteins mediates efficient internalization and endocytic sorting of activated EGFR, which is required for endosomal signaling to various EGFR effectors and also mediates receptor downregulation.7 Conversely, interaction of EGFR with merlin, a closely related FERM family protein that cannot interact with F-actin, sequesters EGFR into an insoluble membrane compartment from which it cannot signal or become internalized.8 Loss of EP50, another protein involved in tethering complexes, disturbs both of these processes, leading to accumulation of activated EGFR on the cell surface due to defects in receptor-mediated endocytosis.7b Together these studies point out both the complexity and importance of cytoskeletal tethering to growth factor signaling from the cell surface.
While it is clear that ligand-mediated activation of EGFR promotes its tethering to the cytoskeleton, it is less obvious whether signaling through this receptor also promotes the tethering of other proteins associated with endocytosis and signal transduction to downstream effectors. To answer this question, we devised an unbiased proteomic strategy to globally evaluate composition of the tethered membrane proteome before and after stimulation of cells with EGF. In this approach, adherent cells are first surface biotinylated using an amine-reactive cross-linking reagent and then extracted with detergents to remove soluble cytoplasmic and membrane proteins. The remaining insoluble/cytoskeletal fraction is then homogenized, affinity purified with streptavidin beads, and analyzed by mass spectrometry. Though aspects of this approach have been previously utilized to isolate specific protein pools of membrane proteins for proteomic analysis, the combination of membrane biotinylation and cytoskeletal extraction we describe has not been previously applied to differential protein tethering to the cytoskeleton.9
Quantitative comparison of the “membrane tetherome” before and after EGF stimulation suggests that EGFR activation leads to the association of several different proteins with the cytoskeleton, including those involved in signal transduction and vesicular trafficking. At the same time, EGFR signaling also promotes the dissociation of another subset of proteins from the cytoskeleton, including those that mediate cytoskeletal dynamics and RNA processing. This experimental approach provides a relatively simple means to identify and quantify components of the tethered membrane proteome and will allow us to decipher how specific plasma membrane complexes are organized by the underlying cortical cytoskeleton in response to different environmental or signaling conditions.
EXPERIMENTAL SECTION
Cell Culture
HK2 normal human renal epithelial cells were obtained from ATCC. Cells were grown in normal tissue culture-treated dishes in antibiotic-free RPMI-1640 media (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (FBS) under standard growth conditions of 37 °C and 5% CO2. Cells were passaged at 85–90% confluency every 3–4 days to maintain continuous logarithmic growth. Treatments with recombinant EGF were performed in basal RPMI-1640 media after 16–20 h of serum starvation. Treatment with methyl-beta-cyclodextrin (MβCD) was performed at 5 mM concentration for 30 min to disrupt lipid raft microdomains in the plasma membrane.
Adenoviral Expression of PTPRS
Recombinant adenoviral vectors containing PTPRS were generated by recombining the full-length open reading frame of PTPRS into pAd/CMV/V5-DEST using the GATEWAY cloning system (Invitrogen). Adenoviral particles were produced by transfecting this vector into the 293A packaging line, harvesting a crude viral lysate and amplifying the stock by one additional round of infection in 293A cells. The resulting viral supernatant was titered and frozen prior to further use. HK2 cells plated to 6 cm plates and glass coverslips were infected at a multiplicity of infection (MOI) of 5 for 24 h prior to media change. At 32–48 h after infection, the cells were stimulated with EGF and analyzed by immunoblot or immunofluorescent staining.
Antibodies
Antibodies for EGFR, ERM, CD44, Grb2, clathrin, pEGFRY1068, pERMT567, and actin were obtained from Cell Signaling Technologies (Danvers, MA). The mouse monoclonal antibody to tubulin was obtained from Sigma-Aldrich (St. Louis, MO), and the rabbit polyclonal antibody to 14-3-3 epsilon was obtained from Epitomics (Burlingame, CA). The mouse monoclonal antibody to EEA1 was purchased from BD Biosciences (San Diego, CA). The mouse monoclonal antibody to PTPRS was kindly provided by Dr. Michel Tremblay (McGill University, Montreal, Quebec, Canada).
Immunofluorescent Staining and Microscopy
HK2 cells were seeded to coverslips and allowed to adhere for 24 h. Cells were then starved overnight in serum-free RPMI-1640 media prior to treatment with recombinant human EGF (100 ng/mL) for the indicated times. For imaging of total protein content, cells were fixed with 3.7% formaldehyde in phosphate-buffered saline (PBS) and permeabilized with 0.2% TritonX-100 in PBS. Imaging of the cytoskeletal fraction only was performed by treating cells with solublization buffer (10 mM PIPES, 50 mM KCl, 20 mM EGTA, 3 mM MgCl2, 2 M glycerol, 2 mM NaF, 2 mM Na3(VO)4, 1% TritonX-100) supplemented with 1× protease inhibitor cocktail (Sigma) on ice for 5 min, followed by fixation with formaldehyde as above. After blocking with 5% normal goat serum and 3% bovine serum albumin in PBS, the coverslips were incubated at 4 °C overnight with anti-EGFR (1:100), anti-EEA1 (1:200), or anti-PTPRS (1:150) diluted in block buffer. After washing in PBS/0.02% TritonX-100, coverslips were incubated for one hour with AlexaFluor-488 or AlexaFluor-546 coupled secondary antibodies (Invitrogen). After a final round of washing, cells were costained with DAPI to detect nuclei, and coverslips were mounted on glass slides with antifade gel mounting medium. Images were obtained using a Nikon Ti-E inverted fluorescence microscope equipped with DAPI, FITC, and Texas Red filter sets and processed using the NIS Elements software package (Nikon Instruments, Melville, NY).
Cytoskeletal Fractionation
HK2 cells were plated to 6-well dishes (35 mm diameter) at a density of 1.5 × 105 cells per well and allowed to adhere overnight in complete media. Cells were then starved overnight in serum-free RPMI-1640 media prior to treatment with recombinant human EGF (100 ng/mL) for the indicated times. Cell lysates were fractionated according to previously described methods.9c Briefly, cells were first washed on ice and then incubated with ice-cold solublization buffer (10 mM PIPES, 50 mM KCl, 20 mM EGTA, 3 mM MgCl2, 2 M glycerol, 2 mM NaF, 2 mM Na3(VO)4, 1% TritonX-100) supplemented with 1× protease inhibitor cocktail (Sigma) on ice for 5 min with gentle rocking. The soluble fraction was removed to a fresh tube on ice, and the remaining insoluble material was washed twice with cold detergent-free solublization buffer to remove any remaining soluble protein. The cytoskeletal fraction was then scraped into extraction buffer (25 mM Tris-HCl, 300 mM NaCl, 30 mM MgCl2, 2 mM NaF, 2 mM Na3(VO)4, 1% NP-40, 0.2% Brij35, 0.2% sodium deoxycholate, 2 mM dithiothreitol) supplemented with 1× protease inhibitor cocktail (Sigma) and sheared ten times through a 27 gauge needle to homogenize the cytoskeletal proteins. Equal volumes of protein lysate were diluted with Laemmli buffer and denatured by boiling for immunoblot analysis.
Immunoblotting
Protein samples were separated on Tris-glycine polyacrylamide gels and transferred overnight to nitrocellulose membranes in a wet transfer apparatus (Hoefer, Holliston, MA). Membranes were blocked in 3% nonfat dry milk in Tris-buffered saline/ 0.1% Tween (TBS-T) and probed with primary antibodies overnight at 4 °C. After washing in TBS-T buffer and incubation with a horseradish peroxidase-coupled secondary antibody, membranes were incubated in enhanced chemiluminescent reagent, exposed to film, and developed for signal using a X-omat film processor (Kodak, Rochester, NY).
Surface Protein Purification
HK2 cells were plated to eight 15 cm dishes at a density of 3.0 × 106 cells per plate and allowed to adhere overnight in complete media. Cells were then starved overnight in serum-free RPMI-1640 media prior to biotinylation on ice for 30 min with 0.25 mg/mL (0.41 mM) sulfo-NHS-SS-biotin (Thermo/Pierce, Rockford, IL). After the surface biotinylation reaction was quenched with 50 mM glycine, cells were washed twice with cold RPMI-1640 media and treated with or without recombinant human EGF (100 ng/mL) in prewarmed RPMI-1640 media for 5 min. Four dishes of cells were used for each condition (± EGF). Fractionation of the cells was performed as above, except that the Extraction Buffer did not contain DTT in order to maintain the biotin cross-links. After removal of insoluble debris by centrifugation for 10 min at 10000g, cytoskeletal fraction lysates were quantified by Bradford assay, and equal amounts of protein (4 mg) were incubated with pre-equilibrated neutravidin beads (Thermo/Pierce) overnight at 4 °C with constant rotation. Beads were pelleted at 200g for 1 min and washed twice with DTT-free extraction buffer and twice with Tris-buffered saline (TBS) containing 5% glycerol. Bound proteins were eluted from the beads by incubating with TBS/5% glycerol buffer containing 50 mM DTT for 2 h at room temperature with constant agitation. The final eluate was stored at −80 °C until analysis by immunoblot and LC–MS/MS.
Mass Spectrometry and Protein Identification
Tether enriched samples were quantified using Qubit fluorometry (Invitrogen). For each sample (purified fractions ± EGF), 20 μg of purified eluate was separated on a 4–12% Bis Tris NuPage gel (Invitrogen) in the MOPS buffer system. The 20 μg gel lane was excised into 20 equally sized segments, and gel pieces were processed using a ProGest robot (DigiLab, Holliston, MA). The trypsin digestion protocol involved washing the gel pieces with 25 mM ammonium bicarbonate followed by acetonitrile, reduction with 10 mM DTT at 60 °C followed by alkylation with 50 mM iodoacetamide (IA) at RT. Reduced and alkylated samples were digested with sequencing grade trypsin (Promega, Madison, WI) at 37 °C for 4 h. The digestion was quenched with formic acid, and the supernatant was analyzed directly by LC–MS/MS without further processing.
LC–MS/MS analysis was performed with a NanoAcquity HPLC system (Waters, Milford, MA) interfaced to a LTQ Orbitrap Velos mass spectrometer (ThermoFisher, Waltham, MA). Peptides were loaded on a trapping column and eluted over a 75 μm analytical column at 350 nL/min; both columns were packed with Jupiter Proteo resin (Phenomenex, Torrance, CA). The mobile phases consisted of HPLC grade H2O (A) and HPLC grade acetonitrile (B), both containing 0.1% (v/v) formic acid. The gradient started at 2% B, reached 50% B in 18 min, 80% B in the next 0.5 min, and 98% A in the final 1 min (see Supporting Information Table S3 for HPLC gradient details). The mass spectrometer was operated in data-dependent mode, with MS performed in the Orbitrap at 60 000 fwhm resolution and MS/MS performed in the LTQ. The 15 most abundant ions were selected for MS/MS. Charge state deconvolution and deisotoping were not performed.
Peptide fragmentation data were searched using a local copy of Mascot (Matrix, Boston, MA). Mascot was configured to search the SwissProt database (human, 41016 entries) assuming the digestion enzyme trypsin was used. Searches were performed with a fragment ion mass tolerance of 0.80 Da and a parent ion tolerance of 10.0 PPM. Carbamidomethylation of cysteine was specified in Mascot as a fixed modification. Gln → pyro-Glu of the n-terminus, deamidated of asparagine and glutamine, oxidation of methionine, acetyl of the n-terminus, and CAMthiopropanoyl of lysine and the n-terminus were specified in Mascot as variable modifications. CAMthiopropanoyl modifications were included to account for changes to peptide mass that result from biotinylation with NHS-SS-biotin.10 Data were searched with a maxiumum of 2 missed cleavage events allowed.
The resulting Mascot DAT files were parsed into the Scaffold v3.2 software package (Proteome Software Inc., Portland, OR) to validate MS/MS-based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 95.0% probability as specified by the Peptide Prophet algorithm.11 Protein probabilities were assigned by the Protein Prophet algorithm and were accepted if they could be established at greater than 99.0% probability and contained at least 4 identified peptides.12 Proteins that contained similar peptides and could not be differentiated on the basis of MS/MS analysis alone were grouped to satisfy the principles of parsimony. Quantification of protein abundance was performed using the label-free spectral counting method as previously described.13 The quantitative value assigned by Scaffold represents a normalized spectral count. Normalization was performed by calculating the average number of spectral counts for all samples and then multiplying the spectral counts in each individual sample by the average divided by the individual sample's sum.
RESULTS
EGF Stimulation Causes Redistribution of EGFR to the Cytoskeleton
To demonstrate that EGF promotes the tethering of its receptor to the cytoskeleton, we treated immortalized human renal epithelial cells (HK2) with recombinant EGF (100 ng/mL) for different times and then stained formaldehyde-fixed cells for EGFR before and after extraction of the membrane and cytosol. In the absence of EGF stimulation, cells fixed with formaldehyde display diffuse EGFR staining across the entire cell surface with a mild concentration in the perinuclear region (Figure 1A). This entire population of receptors is completely removed by detergent extraction prior to fixation, indicating that EGFR is not tethered to the cytoskeleton in the inactive state (Figure 1A).4a Treatment of HK2 cells with EGF leads to a rapid (0–5 min) redistribution of EGFR into dense aggregates that progressively concentrate in the perinuclear region, consistent with receptor internalization and trafficking to the endosomal compartment (Figure 1B).14 At the same time, receptors progressively associate with the cytoskeletal fraction and become visible by immunostaining in detergent-extracted cells (Figure 1B). Costaining of HK2 cells with antibodies for EGFR and the early endosomal marker EEA1 clearly demonstrates that EGFR localizes to endosomes upon activation by EGF, though tethering to the cytoskeleton is not a general property of all endosomal proteins, as EEA1 is not retained in the cytoskeletal fraction after detergent extraction (Supporting Information Figure S1).
Figure 1.

The epidermal growth factor receptor associates with the detergent-insoluble cytoskeletal fraction upon activation by its ligand. (A) HK2 cells were plated to glass coverslips and serum-starved for 24 h. They were then either fixed directly or extracted with buffer containing 1% TritonX-100 prior to fixation. Cells were labeled with antibodies to EGFR and detected with AlexaFluor-488 coupled secondary antibody (green) and also costained with Hoescht stain (blue) to detect nuclei. Images were captured using an epifluorescent inverted microscope at 60× magnification. Scale bars indicate 20 μm. (B) HK2 cells were cultured and serum-starved as before, then treated for the indicated time with 100 ng/mL of recombinant human EGF prior to detergent extraction and fixation. The remaining cytoskeletal fraction was stained for EGFR as before to demonstrate the progressive tethering of activated receptor to the cytoskeleton. Images are shown at 60× magnification with inset bars indicating 20 μm. (C) HK2 cells were plated at fixed density, serum-starved, and then stimulated for the indicated time with 100 ng/mL of recombinant human EGF. Cells were then biochemically fractionated, and lysates were analyzed by immunoblot for the indicated total or phosphorylated proteins. (D) HK2 cells were plated at fixed density, serum-starved, and pretreated with 5 mM MβCD prior to stimulation for 5 min ±100 ng/mL of recombinant human EGF. Cells were fractionated and analyzed by immunoblot as in C.
Redistribution of activated EGFR to the cytoskeletal fraction can also be demonstrated by biochemical separation of HK2 cells into detergent-soluble and -insoluble fractions.9c Prior to EGF stimulation, EGFR is primarily detected in the detergent-soluble fraction of HK2 cells. EGF stimulation causes a rapid (0–5 min) increase in EGFR concentration in the insoluble cytoskeletal fraction, which is sustained with receptor activation (Figure 1C). Concentrations of EGFR in both soluble and cytoskeletal fractions decrease in parallel by 60 min poststimulation because of receptor degradation. The identities of the detergent-soluble and insoluble fractions are indicated by probing HK2 protein lysates for activated ezrin–radixin–moesin (ERM) family proteins, which are directly tethered to the actin cytoskeleton when phosphorylated at the C-terminus (pERMT567). While ERM proteins can be detected in both soluble and insoluble fractions, the activated/phosphorylated form is highly enriched in the insoluble fraction (Figure 1C). Similarly, actin itself is also enriched in the cytoskeletal fraction.
The detergent extraction method we used to fractionate HK2 cells is similar, albeit not identical, to procedures used in lipid raft microdomain isolation.9a,15 To demonstrate that redistribution of EGFR to a detergent-insoluble fraction after binding to EGF is a result of cytoskeletal tethering and not lipid raft aggregation, we pretreated HK2 cells for 30 min with 5 μM methyl-beta-cyclodextrin (MβCD) prior to stimulation with EGF. Treatment of cells with this reagent depletes cholesterol from the plasma membrane and thereby disrupts lipid raft microdomains.16 As expected, MβCD had little effect on the activation and redistribution of EGFR to the detergent-insoluble fraction upon EGF stimulation (Figure 1D). Conversely, MβCD treatment increased the detergent solubility of the raft-associated protein CD44 and decreased its association with the cytoskeleton upon EGF stimulation (Figure 1D).17 Together these data confirm that EGF-mediated activation of EGFR results in its redistribution to the detergent-insoluble fraction and are consistent with EGFR being tethered to the actin cytoskeleton upon activation.
Biochemical Isolation and Purification of the Tethered Membrane Proteome
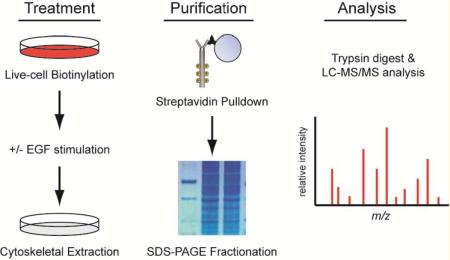
Regulated tethering of EGFR in HK2 cells provided us with a useful framework to validate a novel strategy for isolating and characterizing the tethered membrane proteome (Figure 2A). In this strategy, we combined the biochemical fractionation method described above with cell surface biotinylation, which was performed with sulfo-NHS-SS-biotin, a cell nonpermeable cross-linking reagent that adds a biotin tag to primary amines. This reagent can be applied to live cells in phosphate-buffered saline (PBS) to label the lysine residues of extracellular proteins. When labeling is performed at temperatures lower than 4 °C, vesicular trafficking to the cell surface is inhibited, thus ensuring that a stable population of biotinylated receptors is present when cells are experimentally stimulated. After stimulation and fractionation, the membrane protein component of the detergent-insoluble cytoskeletal fraction can be isolated by affinity purification over streptavidin beads and eluted by reduction of the biotin cross-link with dithiothreitol (DTT).
Figure 2.

Proteomic workflow and experimental validation. (A) Flow diagram of the methods used to isolate and purify the fraction of membrane proteins associated with the cytoskeleton. See the Experimental Section for detailed procedures. (B) Cytoskeletal protein lysates isolated by the methods shown in A were analyzed be immunoblot before (input) and after purification with streptavidin beads (pulldown). Purification enriches for membrane proteins (EGFR and CD44) over intracellular cytoskeletal elements (tubulin and actin). (C) Annotated mass spectra for EGFR peptide (TIQEVAGYVIALNTVER) representative of the detection metrics for the peptides identified in this study. A minimum of two unique peptides with ≥50% probability (Prophet score) were used to identify each protein. (D) Annotated mass spectra for CD44 peptide (LVINSGNGAVEDR). (E) Quantification of relative EGFR (44/102 peptides) and CD44 (50/56 peptides) abundance in the cytoskeletal fraction of HK2 cells treated without (−) and with (+) EGF stimulation for 5 min using the label-free spectral counting method.
We validated the experimental scheme presented above by labeling HK2 cells with sulfo-NHS-SS-biotin for 30 min on ice and then allowing a 5 min recovery in prewarmed culture media in the presence or absence of EGF (100 ng/mL). Cells were then extracted with ice-cold detergent (TritonX-100) to remove the soluble/untethered protein fraction. The remaining cytoskeletal fraction was scraped into a second buffer containing a cocktail of detergents (NP40, deoxycholate and Brij-35) and homogenized by shearing the cytoskeletal fraction through a 25 gauge needle. Remaining debris was removed by centrifugation, and the resulting protein lysate was purified over streptavidin beads to isolate all membrane proteins associated with the cytoskeleton. Immunoblot analysis of the input and purified fractions clearly demonstrate enrichment of EGFR in the cytoskeletal fraction after EGF stimulation (Figure 2B). As controls for the reaction, we also probed lysates for CD44. As expected, CD44 was isolated with the purified membrane protein fraction, and was found to be associated with the cytoskeletal fraction independent of EGF stimulation (Figure 2B). Immunblot analysis of the streptavidin-purified fraction also reveals that even highly abundant cytoplasmic proteins from the cytoskeletal fraction, such as tubulin and actin, are largely excluded by purification of biotinylated membrane proteins away from the rest of the cytoskeletal fraction (Figure 2B).
Quantitative Analysis of Tethered Membrane Proteome Composition
To identify the composition of the tethered membrane proteome, we performed label-free tandem mass spectrometry (MS/MS) on the streptavidin-purified fractions from HK2 cells treated in the presence or absence of EGF for 5 min. Because detection of unique peptides using label-free methods of quantification such as spectral counting can be confounded by large differences in protein abundance, we first separated individual samples by one-dimensional polyacrylamide gel electrophoresis and fractionated them into 20 separate gel slices by protein mass. Individual gel fractions were trypsin digested and separated by liquid chromatography interfaced directly with the mass spectrometer. The 15 most abundant ions for each peptide were selected for MS/MS analysis. Spectra for each peptide were matched to the Swissprot human database using a local copy of Mascot and parsed into the Scaffold software package to filter and simply identified hits into a nonredundant list of proteins for each sample.
Quantitative analysis of the MS/MS data was performed by analyzing spectral counts (SpC) of peptides mapped to unique proteins within the Swissprot database. Previous studies have shown that this method can accurately discriminate differences of abundance as low as 1.4-fold with 95% confidence providing that at least one of the two samples displays ≥4 SpC.13 In our analysis of the data, we filtered out all proteins that produced a SpC <4 for either sample (± EGF) to ensure maximum confidence in protein identification (Supporting Information Tables S1 and S2). We then determined the SpC ratio of stimulated-to-unstimulated cells to distinguish positively identified proteins whose association with the cytoskeleton changed upon stimulation of cells with EGF. To ensure that these quantitative predictions from the MS/MS data accurately represent protein abundance, we compared the spectral count ratios of EGFR and CD44 in EGF-stimulated versus -unstimulated cells (Figure 2C–E). As previously demonstrated by immunoblot (Figure 2B), the amount of EGFR in the tethered membrane proteome increases upon EGF stimulation (44/101 SpC), whereas the amount of CD44 stays relatively constant under the same treatment conditions (50/56 SpC, Figure 2E).
Activation of EGFR by its ligand promotes its intrinsic tyrosine kinase activity and induces a variety of downstream signaling pathways associated with proliferation, survival, and migration. In addition, EGF signaling also engages the cellular machinery required for internalization and trafficking of activated receptors. Because the cytoskeleton is intimately involved in signaling and receptor-mediated endocytosis, we asked whether stimulation of HK2 cells with EGF altered the association of proteins beside EGFR into the cytoskeletal fraction. Analysis of normalized spectral count ratios between stimulated and unstimulated cells revealed that 12.2% (61/499) of proteins detected by MS/MS display a 2-fold or greater change in abundance in the purified cytoskeletal fraction (Tables 1 and 2, Supporting Information Table S1). This list revealed the differential redistribution of several different protein classes expected to function downstream of EGFR including those involved in cell signaling, cytoskeletal regulation, migration, and proteolysis. Interestingly, many of the proteins identified in this analysis are functionally associated with RNA binding/processing and translation (Table 2). While neither of these cellular functions is obviously associated with cytoskeletal regulation or EGFR signaling, they have been connected to cellular spreading and adhesion, which are modulated by EGF in HK2 cells.18 In addition, one of the RNA processing factors we found to be differentially tethered in response to EGF, small nuclear ribonucleoprotein F (SNRPF), has been previously shown to bind directly to the EGFR effector Grb2, which was also differentially tethered in our study (Figure 3A).19
Table 1.
Proteins with Incresed Cytoskeletal Association upon EGF Stimulationa
| unique peptide |
coverage (%) |
spectral counts |
quantitative value |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cell signaling | accession numberb | gene | (−) EGF | (+) EGF | (−) EGF | (+) EGF | (−) EGF | (+) EGF | (−) EGF | (+) EGF | fold Δ | log Δ |
| Peptidyl-prolyl cis–trans isomerase B | P23284 | PPIB | 5 | 8 | 22.0 | 44.0 | 5 | 16 | 4.2 | 25.9 | 6.13 | 2.61 |
| Cysteine and glycine-rich protein 2 | Q16527 | CSRP2 | 5 | 8 | 32.0 | 45.0 | 8 | 16 | 6.3 | 24.8 | 3.90 | 1.96 |
| CD70 antigen | P32970 | CD70 | 5 | 5 | 28.0 | 38.0 | 16 | 19 | 3.6 | 12.5 | 3.48 | 1.80 |
| Gremlin-1 | O60565 | GREM1 | 7 | 8 | 37.0 | 49.0 | 29 | 39 | 21.6 | 66.6 | 3.09 | 1.63 |
| Peroxiredoxin-1 | Q06830 | PRDX1 | 12 | 15 | 59.0 | 56.0 | 35 | 56 | 31.1 | 80.5 | 2.59 | 1.37 |
| Guanine nucleotide-binding protein G(i) subunit alpha-2 | P04899 | GNAI2 | 8 | 13 | 26.0 | 43.0 | 13 | 22 | 7.1 | 17.6 | 2.48 | 1.31 |
| 14-3-3 protein epsilon | P62258 | YWHAE | 4 | 6 | 16.0 | 27.0 | 5 | 9 | 5.3 | 12.5 | 2.38 | 1.25 |
| Epidermal growth factor receptor | P00533 | EGFR | 30 | 42 | 31.0 | 37.0 | 44 | 101 | 26.7 | 61.4 | 2.30 | 1.20 |
| Prohibitin-2 | Q99623 | PHB2 | 11 | 13 | 33.0 | 40.0 | 18 | 27 | 12.6 | 28.7 | 2.28 | 1.19 |
| Cytoplasmic FMR1-interacting protein 1 | Q7L576 | CYFP1 | 7 | 13 | 6.1 | 12.0 | 10 | 14 | 4.0 | 8.9 | 2.20 | 1.14 |
| Peroxiredoxin-2 | P32119 | PRDX2 | 6 | 6 | 28.0 | 30.0 | 8 | 9 | 6.3 | 13.6 | 2.14 | 1.10 |
| Receptor-type tyrosine-protein phosphatase S | Q13332 | PTPRS | 10 | 14 | 6.6 | 9.6 | 12 | 18 | 5.2 | 10.8 | 2.08 | 1.06 |
| Vesicular Trafficking | ||||||||||||
| Ras-related protein Rab-10 | P61026 | RAB10 | 4 | 5 | 22.0 | 26.0 | 6 | 14 | 4.2 | 19.1 | 4.52 | 2.18 |
| Dynein light chain 1 | P63167 | DYNLL1 | 6 | 5 | 65.0 | 64.0 | 12 | 20 | 22.3 | 54.4 | 2.44 | 1.29 |
| Coatomer subunit alpha | P53621 | COPA | 11 | 20 | 9.8 | 18.0 | 13 | 21 | 6.5 | 13.3 | 2.05 | 1.04 |
| ECM and Cytoskeletal Regulation | ||||||||||||
| Urokinase-type plasminogen activator | P00749 | PLAU | 6 | 10 | 13.0 | 19.0 | 11 | 16 | 2.2 | 6.1 | 2.76 | 1.47 |
| Protein S100-A9 | P06702 | S100A9 | 4 | 7 | 45.0 | 53.0 | 12 | 25 | 22.3 | 53.0 | 2.38 | 1.25 |
| Prolactin-inducible protein | P12273 | PIP | 5 | 6 | 44.0 | 44.0 | 17 | 30 | 9.9 | 23.1 | 2.33 | 1.22 |
| Collagen alpha-1(XVIII) chain | P39060 | COL18A1 | 6 | 9 | 5.8 | 7.7 | 15 | 23 | 3.7 | 8.2 | 2.21 | 1.15 |
| Membrane Transport | ||||||||||||
| Voltage-dependent anion-selective channel protein 3 | Q9Y277 | VDAC3 | 4 | 9 | 16.0 | 44.0 | 8 | 14 | 4.5 | 15.3 | 3.40 | 1.77 |
| Voltage-dependent anion-selective channel protein 2 | P45880 | VDAC2 | 12 | 12 | 52.0 | 56.0 | 21 | 32 | 15.3 | 32.3 | 2.12 | 1.08 |
Proteins that were identified with confidence of >99% and were scored with ≥4 spectral counts (SpC) in both unstimulated and stimulated cells were sorted by the SpC ratio in stimulated/unstimulated cells. Refer to Table S1 (Supporting Information) for a complete list of proteins identified in this study.
Accession numbers were compiled from the SwissProt database.
Table 2.
Proteins with Decresed Cytoskeletal Association upon EGF Stimulationa
| unique peptide |
coverage (%) |
spectral counts |
quantitative value |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cell signaling | accession numberb | gene | (−) EGF | (+) EGF | (−) EGF | (+) EGF | (−) EGF | (+) EGF | (−) EGF | (+) EGF | fold Δ | log Δ |
| Protein RRP5 homologue | Q14690 | PDCD11 | 11 | 5 | 6.40 | 2.70 | 11 | 5 | 6.5 | 3.2 | 0.49 | −1.04 |
| Cyclin-dependent kinase inhibitor 2A, isoform 4 | Q8N726 | CDKN2A | 6 | 5 | 43 | 28 | 13 | 7 | 27.3 | 9.7 | 0.35 | −1.50 |
| ECM and Cytoskeletal Regulation | ||||||||||||
| Desmoglein-1 | Q02413 | DSG1 | 13 | 13 | 16 | 11 | 29 | 18 | 12.6 | 6.3 | 0.50 | −1.00 |
| Plectin | Q15149 | PLEC | 65 | 21 | 14 | 3.50 | 84 | 27 | 48.7 | 18.0 | 0.37 | −1.44 |
| Filaggrin-2 | Q5D862 | FLG2 | 12 | 9 | 6.70 | 5.20 | 28 | 29 | 23.7 | 7.0 | 0.30 | −1.76 |
| Hornerin | Q86YZ3 | HRNR | 9 | 5 | 5.70 | 2.60 | 52 | 22 | 22.5 | 3.0 | 0.13 | −2.92 |
| Transcription and Chromatin Binding | ||||||||||||
| FACT complex subunit SPT16 | Q9Y5B9 | SSRP1 | 25 | 13 | 31 | 16 | 33 | 15 | 18.1 | 8.9 | 0.49 | −1.02 |
| DNA topoisomerase 2-alpha | P11388 | TOP2A | 11 | 7 | 11 | 8.40 | 13 | 7 | 14.4 | 7.0 | 0.49 | −1.03 |
| High mobility group protein HMGI-C | P52926 | HMGA2 | 5 | 4 | 50 | 32 | 13 | 7 | 17.9 | 7.7 | 0.43 | −1.21 |
| Chromodomain-helicase-DNA-binding protein 4 | Q14839 | CHD4 | 37 | 18 | 23 | 11 | 46 | 21 | 27.5 | 11.5 | 0.42 | −1.26 |
| THO complex subunit 4 | Q86V81 | ALYREF | 7 | 4 | 47 | 27 | 21 | 5 | 23.5 | 6.9 | 0.30 | −1.76 |
| DNA Replication | ||||||||||||
| Replication factor C subunit 1 | P35251 | RFC1 | 10 | 5 | 12 | 5.50 | 10 | 7 | 6.5 | 3.2 | 0.49 | −1.03 |
| Replication factor C subunit 2 | P35250 | RFC2 | 6 | 4 | 23 | 18 | 11 | 4 | 9.3 | 3.4 | 0.36 | −1.47 |
| RNA Binding/Processing | ||||||||||||
| Plasminogen activator inhibitor 1 RNA-binding protein | Q8NC51 | SERBP1 | 12 | 7 | 33 | 21 | 13 | 7 | 7.5 | 3.8 | 0.50 | −1.00 |
| Nucleolar protein 6 | Q9H6R4 | UTP6 | 11 | 5 | 11 | 6.20 | 11 | 5 | 6.3 | 3.0 | 0.47 | −1.08 |
| Protein C22orf28 | Q9Y3I0 | C22orf28 | 8 | 6 | 21 | 13 | 13 | 6 | 6.9 | 2.7 | 0.39 | −1.37 |
| U4/U6 small nuclear ribonucleoprotein Prp3 | O43395 | PRPF3 | 11 | 4 | 22 | 6.10 | 13 | 4 | 6.7 | 2.3 | 0.35 | −1.51 |
| Nucleolar complex protein 3 homologue | Q8WTT2 | NOC3L | 13 | 7 | 19 | 5.40 | 16 | 9 | 8.3 | 2.9 | 0.35 | −1.51 |
| Small nuclear ribonucleoprotein F | P62306 | SNRPF | 4 | 4 | 49 | 49 | 38 | 24 | 151.2 | 49.7 | 0.33 | −1.60 |
| U1 small nuclear ribonucleoprotein 70 kDa | P08621 | SNRNP70 | 11 | 4 | 28 | 12 | 17 | 6 | 9.0 | 2.7 | 0.30 | −1.73 |
Proteins that were identified with confidence of >99% and were scored with ≥4 spectral counts (SpC) in both unstimulated and stimulated cells were sorted by the SpC ratio in stimulated/unstimulated cells. Refer to Table S1 (Supporting Information) for a complete list of proteins identified in this study.
Accession numbers were compiled from the SwissProt database.
Figure 3.

EGF signaling promotes association of Grb2, 14-3-3 epsilon and PTPRS with the cytoskeleton. (A) HK2 cells were plated at fixed density, serum-starved and then stimulated for the indicated time with 100 ng/mL recombinant human EGF. Cells were then biochemically fractionated, and lysates were analyzed by immunoblot for the indicated proteins. The antibody to PTPRS detects endogenous full-length (FL) protein, the processed phosphatase subunit (P-sub), and a further proteolytic C-terminal fragment (CTF). (B) HK2 cells were transfected with pCDNA6.2-PTPRS, which expresses full-length PTPRS under control of the CMV promoter. After 48 hours, the cells were treated with 100 ng/mL EGF for the indicated time and fractionated to isolate the soluble and cytoskeletal fractions of protein lysate. Lysates were separated by SDS-PAGE, transferred to nitrocellulose and probed with primary antibodies to the indicated proteins. (C–F) HK2 cells infected with adenoviruses expressing PTPRS for 24 hours were serum-starved overnight prior to treatment +/− 100 ng/mL EGF for 5 minutes. After stimulation the cells were either fixed directly (C,E) or extracted with buffer containing 1% TritonX-100 prior to fixation (D,F). Cells were then labeled with antibodies for PTPRS (green) and clathrin (red), and co-stained with Hoescht stain (blue) to detect nuclei. Images were captured using an epifluorescent inverted microscope at 60× magnification. Scale bars indicate 20 microns. 2× magnifications of the indicated cells (arrows) are shown in the upper right inset of each merged image to demonstrate separation or colocalization of the PTPRS and clathrin signals.
EGF Signaling Promotes Cytoskeletal Association and Processing of PTPRS
To ensure that the findings from our MS/MS analysis accurately represent redistribution of proteins to the cytoskeleton, we performed immunoblot analysis on three of the differentially detected proteins in the soluble and cytoskeletal fractions of EGF-stimulated HK2 cells. Consistent with the MS/MS quantification, we found that the EGFR-associated adaptor proteins Grb2 and 14-3-3 epsilon increase in the cytoskeletal fraction of EGF-stimulated cells along with EGFR (Figure 3A). We also observed that the type-S receptor phosphotyrosine phosphatase (PTPRS) increased in association with the cytoskeleton upon EGFR activation, which is intriguing since a recent study has implicated this protein as a tumor suppressor and negative regulator of EGFR.20 The temporal increase in association of PTPRS with the cytoskeleton also correlates with its proteolytic processing, as full-length receptor is increased in the cytoskeletal fraction at 5 min after EGF stimulation, whereas the processed intracellular domain is increased at 15 min after EGF stimulation (Figure 3A). By 60 min poststimulation, levels of both isoforms return to basal levels of abundance within the cytoskeletal fraction. Similar results are achieved with overexpression of full-length recombinant PTPRS in HK2 cells (Figure 3B).
Consistent with previous data, we observed that overexpression of PTPRS in HK2 cells using an adenoviral vector dampened basal EGFR phosphorylation and weakly antagonized EGF-induced activation of EGFR (Supporting Information Figure S3A).20 Together with the coordinate increase in cytoskeletal association, these data suggest that PTPRS may localize to the same detergent-insoluble endocytic compartment that EGFR resides in upon EGF stimulation. To test this hypothesis, we immunofluorescently labeled HK2 cells overexpressing PTPRS with antibodies to PTPRS and the endosomal marker clathrin, which dynamically associates with the cytoskeleton during vesicular trafficking through the endosomal system via an assortment of different adaptor proteins.5a In unstimulated HK2 cells, PTPRS primarily localizes to punctate spots that are evenly distributed throughout the cell and do not colocalize with clathrin (Figure 3C,D). Extraction of unstimulated cells with detergent does not dramatically alter this pattern of staining, suggesting that PTPRS constitutively associates with the cytoskeleton but resides in a nonclathrin coated vesicular compartment in unstimulated HK2 cells (Figure 3D). Upon stimulation of cells with EGF, PTPRS puncta rapidly condense to the perinuclear region and colocalize with clathrin-coated vesicles, consistent with endosomal relocalization (Figure 3E,F). As predicted, both PTPRS and clathrin reside in the detergent-insoluble fraction after EGF stimulation, indicating that both proteins remain associated with the cytoskeleton after EGF stimulation (Figure 3F). We suggest that the relative increase of PTPRS in the cytoskeletal fraction predicted by mass spectrometry and confirmed by immunoblot results from increased flux through the endocytic system induced by EGF. Whether PTPRS is passively or actively recruited into clathrin-coated vesicles in response to EGF awaits further investigation.
CONCLUSIONS
Tethering of membrane proteins to the cortical actin cytoskeleton is a dynamic process that plays an important role in many different cellular functions including endocytosis, vesicular trafficking, migration, and signaling. The regulation of all these processes can be coordinated by extracellular signals, which leads to increased endocytosis and turnover of activated receptors and changes in cellular behavior, such as migration, in response to that signal. The binding of EGF to its receptor is a prototypical example of how a single extracelluar signal can orchestrate rapid changes in cellular behavior concomitant with alterations in endocytosis, vesicular trafficking, and cytoskeletal reorganization.
We used EGF signaling as a tool to demonstrate that tethering of membrane proteins to the actin cytoskeleton rapidly changes in response to a well-defined stimulus. Because activated EGFR has previously been shown to redistribute to a detergent-insoluble compartment in association with the tethering proteins EBP50 and ezrin, we used changes in the biochemical fractionation of EGFR as a positive control for changes in tethering.7a,21 We then combined biochemical fractionation of the cytoskeletal proteome with membrane protein biotinylation to specifically purify proteins that are physically associated with the cytoskeleton, either by direct tethering or indirect association with biotinylated membrane proteins. Quantitative MS/MS analysis of this purified fraction revealed the composition of the tethered membrane proteome, or “membrane tetherome”, in HK2 cells before and after EGF stimulus.
While the results of our analysis clearly demonstrate that this purification scheme enriches for transmembrane proteins such as EGFR and CD44, the purified fraction also contained proteins that are indirectly associated with the cytoskeletal fraction by binding to the membrane proteins themselves. These interactors can largely be removed by adding SDS to the dissociation buffer used to solubilize the cytoskeletal fraction or by heat denaturation of this fraction prior to streptavidin purification (Supporting Information Figure S2). In this study, we chose not to include this step to allow for detection of important signaling intermediates, such as Grb2 and 14-3-3 epsilon, which redistribute to the cytoskeletal fraction with EGFR. For applications where the presence of indirect interactors is not desired, however, these proteins can easily be removed by chemical or physical means.
Several of the changes in protein tethering we observed are clearly related to changes in signal transduction and cellular behavior elicited by EGF. In HK2 cells, treatment with EGF results in a rapid induction of cellular migration and a more delayed increase in cellular proliferation (data not shown). Since we measured the difference in membrane tetherome composition after only 5 min of EGF stimulation, we anticipated that most of the differentially tethered proteins would be ones that mediate EGFR signaling, membrane trafficking/endocytosis, and cytoskeletal reorganization. Consistent with this hypothesis, we found that many of the proteins that showed a differential abundance of 2-fold or greater in the cytoskeletal fraction after EGF stimulation had been previously associated with these processes.
Interestingly, two large classes of proteins that are not usually associated with EGF-mediated signaling or the cytoskeleton were also highly represented in our analysis. These two classes of proteins are collectively involved in the binding, processing and translation of various types of RNA in the cytoplasm. Importantly, most of these proteins show a loss of cytoskeletal association upon EGF stimulation, suggesting that cellular centers of RNA biosynthesis and protein translation may be stably associated with the actin cytoskeleton under homeostatic growth conditions but are actively released from microfilaments when the cell receives promigratory signals from its environment. One of the RNA processing factors we found to be differentially tethered in response to EGF, small nuclear ribonucleoprotein F (SNRPF), has also been previously shown to bind directly to the EGFR effector Grb2, suggesting a possible mechanistic connection between EGF signaling and RNA processing in the cytoplasm.19 While further studies are required to further test this hypothesis, our data are strikingly similar to a previous study that identified a large collection of RNA binding and processing proteins physically associated with focal adhesions during the processes of cellular adhesion and spreading.18 As these cellular processes are directly involved in migration, it is possible that signals that regulate migration also control the association of RNA processing centers with the cytoskeleton.
Under normal homeostatic conditions, it is important for cells to restrict the aberrant activation of signaling receptors by incidental contact with the plasma membrane. In the case of receptor tyrosine kinases (RTKs) such as EGFR, this is achieved by a variety of means including the presence of membrane-associated tyrosine phosphatases that rapidly dephosphorylate low levels of activated receptors. This balance is frequently interrupted in cancer by mutation or loss of these phosphatases.20,22 Loss of PTPRS, one of the cytoskeletal interactors identified in our study, has recently been associated with head and neck cancers in which EGFR is aberrantly activated. While it has been proposed that PTPRS directly dephosphorylates EGFR in normal epithelial cells, the mechanism by which PTPRS associates with EGFR has not been clearly elucidated. Our findings suggest that PTPRS normally resides in a vesicular compartment that can be rapidly mobilized to the endosome upon EGFR activation.23 Trafficking of these vesicles is apparently associated with increased cytoskeletal tethering of PTPRS, which explains why this protein was identified in our proteomic analysis. We propose that this phenomenon underlies that ability of PTPRS to localize with and dephosphorylate activated EGFR, though confirmation of this hypothesis awaits further studies.
Supplementary Material
ACKNOWLEDGMENTS
We would like to acknowledge the personnel from MS Bioworks (Ann Arbor, MI) for their exceptional work with the mass spectrometry and data analysis involved in this project. We particularly thank Dr. Michael Ford for his intellectual contributions and help in preparing technical aspects of this manuscript. This work was supported by the Department of Defense Prostate Cancer Research Program of the Office of Congressionally Directed Medical Research Programs PC081089 to JPM. JPM is also supported by Award Number R01CA138651 from the National Cancer Institute.
Footnotes
Supporting Information Figures S1–S3 and Tables S1–S3M. The full list of proteins with 4 or more spectral counts identified by LC–MS/MS, along with quantitative information, can be found in Table S1. CAMthiopropanyol modified peptides indicative of extracellular biotinylation sites are listed in Table S2. Complete HPLC gradient conditions used are listed in Table S3. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Morone N, Nakada C, Umemura Y, Usukura J, Kusumi A. Three-dimensional molecular architecture of the plasma-membrane-associated cytoskeleton as reconstructed by freeze-etch electron tomography. Methods Cell Biol. 2008;88:207–36. doi: 10.1016/S0091-679X(08)00412-3. [DOI] [PubMed] [Google Scholar]; (b) Golan DE, Veatch W. Lateral mobility of band 3 in the human erythrocyte membrane studied by fluorescence photobleaching recovery: evidence for control by cytoskeletal interactions. Proc. Natl. Acad. Sci. U. S. A. 1980;77(5):2537–41. doi: 10.1073/pnas.77.5.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Svetina S, Bozic B, Derganc J, Zeks B. Mechanical and functional aspects of membrane skeletons. Cell. Mol. Biol. Lett. 2001;6(3):677–90. [PubMed] [Google Scholar]
- (3).(a) Edidin M, Zagyansky Y, Lardner TJ. Measurement of membrane protein lateral diffusion in single cells. Science. 1976;191(4226):466–8. doi: 10.1126/science.1246629. [DOI] [PubMed] [Google Scholar]; (b) Leitner DM, Brown FL, Wilson KR. Regulation of protein mobility in cell membranes: a dynamic corral model. Biophys. J. 2000;78(1):125–35. doi: 10.1016/S0006-3495(00)76579-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ritchie K, Iino R, Fujiwara T, Murase K, Kusumi A. The fence and picket structure of the plasma membrane of live cells as revealed by single molecule techniques (Review) Mol. Membr. Biol. 2003;20(1):13–8. doi: 10.1080/0968768021000055698. [DOI] [PubMed] [Google Scholar]
- (4).(a) Sheetz MP. Integral membrane protein interaction with Triton cytoskeletons of erythrocytes. Biochim. Biophys. Acta. 1979;557(1):122–34. doi: 10.1016/0005-2736(79)90095-6. [DOI] [PubMed] [Google Scholar]; (b) Mangeat P, Burridge K. Actin-membrane interaction in fibroblasts: what proteins are involved in this association? J. Cell Biol. 1984;99(1 Pt 2):95s–103s. doi: 10.1083/jcb.99.1.95s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Schafer DA. Coupling actin dynamics and membrane dynamics during endocytosis. Curr. Opin. Cell Biol. 2002;14(1):76–81. doi: 10.1016/s0955-0674(01)00297-6. [DOI] [PubMed] [Google Scholar]; (b) Cavey M, Lecuit T. Molecular bases of cell-cell junctions stability and dynamics. Cold Spring Harbor Perspect. Biol. 2009;1(5):a002998. doi: 10.1101/cshperspect.a002998. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chichili GR, Rodgers W. Cytoskeleton-membrane interactions in membrane raft structure. Cell. Mol. Life Sci. 2009;66(14):2319–28. doi: 10.1007/s00018-009-0022-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Gautreau A, Louvard D, Arpin M. ERM proteins and NF2 tumor suppressor: the Yin and Yang of cortical actin organization and cell growth signaling. Curr. Opin. Cell Biol. 2002;14(1):104–9. doi: 10.1016/s0955-0674(01)00300-3. [DOI] [PubMed] [Google Scholar]; (b) Fehon RG, McClatchey AI, Bretscher A. Organizing the cell cortex: the role of ERM proteins. Nat. Rev. Mol. Cell Biol. 2010;11(4):276–87. doi: 10.1038/nrm2866. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bretscher A, Chambers D, Nguyen R, Reczek D. ERM-Merlin and EBP50 protein families in plasma membrane organization and function. Annu. Rev. Cell Dev. Biol. 2000;16:113–43. doi: 10.1146/annurev.cellbio.16.1.113. [DOI] [PubMed] [Google Scholar]
- (7).(a) Chirivino D, Del Maestro L, Formstecher E, Hupe P, Raposo G, Louvard D, Arpin M. The ERM proteins interact with the HOPS complex to regulate the maturation of endosomes. Mol. Biol. Cell. 2011;22(3):375–85. doi: 10.1091/mbc.E10-09-0796. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Claperon A, Guedj N, Mergey M, Vignjevic D, Desbois-Mouthon C, Boissan M, Saubamea B, Paradis V, Housset C, Fouassier L. Loss of EBP50 stimulates EGFR activity to induce EMT phenotypic features in biliary cancer cells. Oncogene. 2012;31(11):1376–88. doi: 10.1038/onc.2011.334. [DOI] [PubMed] [Google Scholar]
- (8).(a) Cole BK, Curto M, Chan AW, McClatchey AI. Localization to the cortical cytoskeleton is necessary for Nf2/merlin-dependent epidermal growth factor receptor silencing. Mol. Cell. Biol. 2008;28(4):1274–84. doi: 10.1128/MCB.01139-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Curto M, Cole BK, Lallemand D, Liu CH, McClatchey AI. Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J. Cell Biol. 2007;177(5):893–903. doi: 10.1083/jcb.200703010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Foster LJ, De Hoog CL, Mann M. Unbiased quantitative proteomics of lipid rafts reveals high specificity for signaling factors. Proc. Natl. Acad. Sci. U. S. A. 2003;100(10):5813–8. doi: 10.1073/pnas.0631608100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shin BK, Wang H, Yim AM, Le Naour F, Brichory F, Jang JH, Zhao R, Puravs E, Tra J, Michael CW, Misek DE, Hanash SM. Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J. Biol. Chem. 2003;278(9):7607–16. doi: 10.1074/jbc.M210455200. [DOI] [PubMed] [Google Scholar]; (c) Zhang M, Liu J, Cheng A, Deyoung SM, Saltiel AR. Identification of CAP as a costameric protein that interacts with filamin C. Mol. Biol. Cell. 2007;18(12):4731–40. doi: 10.1091/mbc.E07-06-0628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Weekes MP, Antrobus R, Lill JR, Duncan LM, Hor S, Lehner PJ. Comparative analysis of techniques to purify plasma membrane proteins. J. Biomol. Tech. 2010;21(3):108–15. [PMC free article] [PubMed] [Google Scholar]
- (11).Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002;74(20):5383–92. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- (12).Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003;75(17):4646–58. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- (13).(a) Hoehenwarter W, Wienkoop S. Spectral counting robust on high mass accuracy mass spectrometers. Rapid Commun. Mass Spectrom. 2010;24(24):3609–14. doi: 10.1002/rcm.4818. [DOI] [PubMed] [Google Scholar]; (b) Liu H, Sadygov RG, Yates JR., 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 2004;76(14):4193–201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- (14).Sorkin A, Goh LK. Endocytosis and intracellular trafficking of ErbBs. Exp. Cell Res. 2009;315(4):683–96. doi: 10.1016/j.yexcr.2008.07.029. [DOI] [PubMed] [Google Scholar]
- (15).London E, Brown DA. Insolubility of lipids in triton X-100: physical origin and relationship to sphingolipid/cholesterol membrane domains (rafts) Biochim. Biophys. Acta. 2000;1508(1–2):182–95. doi: 10.1016/s0304-4157(00)00007-1. [DOI] [PubMed] [Google Scholar]
- (16).Oliferenko S, Paiha K, Harder T, Gerke V, Schwarzler C, Schwarz H, Beug H, Gunthert U, Huber LA. Analysis of CD44-containing lipid rafts: Recruitment of annexin II and stabilization by the actin cytoskeleton. J. Cell Biol. 1999;146(4):843–54. doi: 10.1083/jcb.146.4.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 2003;4(1):33–45. doi: 10.1038/nrm1004. [DOI] [PubMed] [Google Scholar]
- (18).de Hoog CL, Foster LJ, Mann M. RNA and RNA binding proteins participate in early stages of cell spreading through spreading initiation centers. Cell. 2004;117(5):649–62. doi: 10.1016/s0092-8674(04)00456-8. [DOI] [PubMed] [Google Scholar]
- (19).Hino N, Oyama M, Sato A, Mukai T, Iraha F, Hayashi A, Kozuka-Hata H, Yamamoto T, Yokoyama S, Sakamoto K. Genetic incorporation of a photo-crosslinkable amino acid reveals novel protein complexes with GRB2 in mammalian cells. J. Mol. Biol. 2011;406(2):343–53. doi: 10.1016/j.jmb.2010.12.022. [DOI] [PubMed] [Google Scholar]
- (20).Morris LG, Taylor BS, Bivona TG, Gong Y, Eng S, Brennan CW, Kaufman A, Kastenhuber ER, Banuchi VE, Singh B, Heguy A, Viale A, Mellinghoff IK, Huse J, Ganly I, Chan TA. Genomic dissection of the epidermal growth factor receptor (EGFR)/PI3K pathway reveals frequent deletion of the EGFR phosphatase PTPRS in head and neck cancers. Proc. Natl. Acad. Sci. U. S. A. 2011;108(47):19024–9. doi: 10.1073/pnas.1111963108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Balbis A, Parmar A, Wang Y, Baquiran G, Posner BI. Compartmentalization of signaling-competent epidermal growth factor receptors in endosomes. Endocrinology. 2007;148(6):2944–54. doi: 10.1210/en.2006-1674. [DOI] [PubMed] [Google Scholar]
- (22).(a) Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 2008;27(2):179–92. doi: 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]; (b) Veeriah S, Brennan C, Meng S, Singh B, Fagin JA, Solit DB, Paty PB, Rohle D, Vivanco I, Chmielecki J, Pao W, Ladanyi M, Gerald WL, Liau L, Cloughesy TC, Mischel PS, Sander C, Taylor B, Schultz N, Major J, Heguy A, Fang F, Mellinghoff IK, Chan TA. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc. Natl. Acad. Sci. U. S. A. 2009;106(23):9435–40. doi: 10.1073/pnas.0900571106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Martin KR, Xu Y, Looyenga BD, Davis RJ, Wu CL, Tremblay ML, Xu HE, MacKeigan JP. Identification of PTPsigma as an autophagic phosphatase. J. Cell Sci. 2011;124(Pt 5):812–9. doi: 10.1242/jcs.080341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.