

SUMMARY
T cell (TC) activation requires the coordinated signaling of the T cell receptor (TCR) and co-receptor molecules, allowing TCs to respond to lower degrees of TCR occupancy. Co-receptor molecules set the threshold for TC activation by controlling different regulatory signaling loops. The Cbl family members prevent undesired activation of TCs by regulating TCR signals. In this report we show that TC pre-stimulation by the CD43 co-receptor molecule before TCR engagement inhibits TCR-dependent c-Cbl tyrosine phosphorylation, c-Cbl interaction with the adapter molecule Crk-L and promotes Cbl-b degradation in a PKCθ–dependent manner. Consequently, the prolonged tyrosine phosphorylation and delayed degradation of ZAP-70 and of the ζ chain lead to enhanced MAPK activation and robust TC response. These data indicates that CD43-mediated signals lower the threshold for TC activation by restricting the c-Cbl and Cbl-b inhibitory effects on TCR signaling. In addition to the strength and duration of intracellular signals, our data underscore temporality with which certain molecules are engaged as yet another mechanism to fine tune TC signal quality, and ultimately immune function.
Keywords: T cell activation, CD43, Cbl, PKCθ
INTRODUCTION
In addition to the signals induced by the specific interaction of the TCR with peptides presented by MHC molecules on the antigen-presenting cell (APC), TC activation requires amplification signals provided by the interaction of other TC surface molecules, known as co-receptors, with their ligands on APCs (1). Co-stimulatory receptors regulate the threshold for TC activation and thus determine immunity or tolerance to self-molecules (2, 3)
CD43 is an abundant transmembrane glycoprotein with an elongated extracellular domain and a highly conserved intracytoplasmic tail (4, 5). Multiple ligands have been described for CD43: ICAM-1, MHC-I, galectin-1, seroalbumin, Siglec-1, cell surface nucleolin and E-selectin. In addition to interact with cytoskeletal elements and to participate in directing cell migration to inflammation sites, CD43 modulates cell–cell contacts and activation, regulating cell number and homeostasis (6). Interestingly, the complex glycosylation pattern of CD43 functions also as bait for multiple pathogens: HIV, Influenza virus, M. tuberculosis, T. cruzi, suggesting that CD43 can function as a pathogen recognition receptor as well (6). In T lymphocytes, co-ligation of CD43 with the TCR enhances TC proliferation above levels observed when cross-linking the TCR alone, in normal as well as in CD28−/− mice (7). CD43 modulates TCR signaling and immune response (8) by activating specific signaling pathways through its intracytoplasmic domain (9). Consistent with this, we have shown that by preventing the SHP-1-LCK interaction, CD43 prolongs ζ chain and ZAP-70 phosphorylation, and significantly augments ERK activation both in terms of intensity and duration, ultimately promoting TC activation and proliferation (10, 11).
Cbl family members play an important role in setting the threshold for signaling in B and T cells, negatively regulating the development of autoimmunity (12, 13). In Jurkat cells, c-Cbl over-expression inhibits the Ras-dependent activation of the MAP kinase pathway and AP-1 transcriptional activity (14). Accordingly, upon TCR engagement (15), mouse c-Cbl−/− TCs show enhanced ZAP-70 and MAPK kinase activity. Differential phosphorylation patterns of c-Cbl have been found to regulate its functions, modifying its capacity to interact with different signaling molecules (16, 17). Unlike TCR signaling (18), CD43-specific signals do not lead to c-Cbl tyrosine phosphorylation. Instead, by inducing its phosphorylation on serine residues (19), CD43 signals prevent the TCR-mediated c-Cbl tyrosine phosphorylation, leading us to hypothesize that CD43 generates a signaling cascade that antagonizes the c-Cbl-dependent inhibitory function on TCR signaling, lowering the threshold for TCR activation.
Herein, we present evidence supporting this hypothesis. When low numbers of TCR molecules where engaged, the co-stimulatory signals provided by CD43 led to enhanced ERK activation, IL-2 production and proliferation. Consistent with this, CD43 signals targeted the negative effect that both c-Cbl and Cbl-b exert on the TCR signaling cascade. Engaging CD43 before the TCR on human TCs prevented TCR-induced c-Cbl tyrosine phosphorylation, lowered the c-Cbl association with the adapter molecule Crk-L and reduced Cbl-b protein levels, while it prolonged ZAP-70 and ζ chain phosphorylation, resulting in enhanced ERK activation, all this in a PKCθ-dependent manner. These data, in combination with previous reports from our laboratory (10, 20, 21) evidence that CD43-mediated signals fine-tune TC activation through several molecular mechanisms.
MATERIALS AND METHODS
Antibodies
L10 and S7 and S11 recognize human (22) and murine CD43 (23) respectively. The anti-phosphotyrosine, anti-c-Cbl, Cbl-b, p-ERK, ERK, ubiquitin, IκB, c-Rel, NFAT and p65 antibodies were from Santa Cruz Biotechnology. The anti-p-c-Jun and anti-phosphorylated c-Cbl (p-c-Cbl Y743) antibodies were from Cell Signaling.
Cell Culture
Jurkat cells stably expressing histidine-tagged wild type PKCθ or the dominant negative PKCθ mutant (dnPKCθ) were obtained by transfecting the corresponding expression vectors (kindly provided by Dr. A. Altman) with the Amaxa transfection system and selection with geneticin (800 μg/ml) containing medium. The IL-2 producer murine TC hybridoma (By155.16) expressing the wild type human CD43, or a mutant lacking the intracytoplasmic domain (CD43ΔIC) or the CD4 molecules has been described previously (9). Peripheral blood TCs were isolated as described (11).
T cell activation, immunoprecipitation and immunoblotting
2 ×107 purified TCs or Jurkat cells were incubated in 0.5 ml of cold RPMI for 15 min at 4°C in the absence or presence of L10 mAb (4 μg/ml of purified IgG or otherwise indicated). Pre-activation was achieved by incubating the cells with RaMIG (4 μg/ml) for the indicated time at 37°C, following which cells were washed with cold RPMI and resuspended in 0.5 ml of cold RPMI. Cells were further stimulated with or without 4 μg/ml of the anti-CD3 mAb OKT3 and cross-linked with 4 μg/ml of RaMIG at 37°C for 5 min. When activated with TPA [Tetradecanoylphorbol acetate] (20ng/ml), cells were incubated for 10 min at 37°C. Non-activated TCs went through all incubation and centrifugation steps. When used, the PKC (RO31822; 10 μM), PLCγ (U73122; 2 μM) or MEK (PD98059; 50 μM) inhibitors were added as previously described (20). Cells lysates were subjected to imunoprecipitation and immunobloting as previously described (20). Densitometric analysis was performed with the help of an Alpha Innotech image analyzer, using the Fluorchem 8800 software from Alpha Innotech.
In vitro kinase assay
PKCθ was immunoprecipitated from stimulated or control cell extracts and the in vitro kinase assay was performed as described (20).
Murine T cell hybridoma activation assays
By155.16 hybridoma cells expressing wild type human CD43 or CD43ΔIC (8) were stimulated by cross-linking the TCR and the CD43 molecule. Cells (2.5 × 104/well) were incubated with sub-optimal doses of the anti-Vβ8 mAb F23.1 (0 to 10 ng/ml, final concentration) and with saturating amounts of the anti-CD43 mAb L10 (500 ng/ml, final concentration). Antibodies were cross-linked on the cell surface with M280 beads (Dynal) coated with goat anti-mouse IgG at a bead to cell ratio of 10:1. After 24 hours at 37°C in 5% CO2, supernatants were assayed for IL-2 content by their ability to support the proliferation of the IL-2 dependent murine TC line CTLL-20 as described (24).
Murine lymphocyte proliferation assays
Females B10.BR mice, age 6 to 8 weeks, were maintained in our animal facility in accordance with the Institute’s guidelines. Thymus and spleen cells were cultured at 2×105 thymocytes/well or 5×104 splenocytes/well for 4 days at 37°C in 5% CO2. Before plating, 96-microwell plates were coated overnight at 37°C with RaMIG (5 μg/ml of PBS), blocked for one hour with 1% gelatin and finally incubated for four hours at 37°C with varying concentrations of the anti-TCR Vβ8 mAb F23.1 and S7 or S11 culture supernatant. The anti-H-2k mAb 11.4.2 (1 μg/ml) was used as a control antibody for co-stimulation experiments. Excess of mAb was removed and IL-2 was added to the plates at the onset of the experiment at 50 IU/ml for thymocyte experiments and at 10 IU/ml for spleenocytes. Proliferation was measured by 3H-thymidine incorporation (1 μCi/well) during the final 18 hours of culture.
Human T cell proliferation assays
Purified human peripheral TCs (4×104/well) were stimulated with the indicated amounts of the OKT3 mAb and saturating amounts of the anti-CD43 mAb L10 (500 ng/ml). M280 beads coated with goat anti-mouse IgG were added to the wells to cross-link the antibodies on the cell surface at a 10:1 bead to cell ratio. Plates were incubated for 96 hours at 37°C in 5% CO2, 12 hours before harvesting, 3H-thymidine was added to the cultures.
RESULTS
CD43 lowers the threshold for TCR activation
We have shown that CD43 signals promote ERK activation and recruit the AP-1, NFκB and NFAT transcription factors (25). Accordingly, engaging CD43 prior to TCR activation on hTCs results in stronger ERK activation (supporting Fig. 1A and 1B) and enhanced nuclear localization of NFkB and NFAT (supporting Fig. 2A) as well as in higher IL-2 levels compared to cells stimulated through the TCR alone (supporting Fig. 2B and 2C), suggesting that CD43 lowers the threshold for TC activation. In order to address this possibility, we took advantage of the fact the TC activation is proportional to the number of TCR molecules engaged (26, 27). Clones of the By155.16 murine TC hybridoma expressing equivalent amounts (data not shown) of TCR-CD3 and human wild type CD43 or a mutant form of CD43 lacking its cytoplasmic domain (CD43ΔIC) (9) were activated with increasing amounts of anti-TCR mAb in the absence or presence of CD43 co-stimulation and their ability to produce IL-2 was determined. Engagement of CD43 in combination with sub-optimal doses of anti-TCR mAb on the hybridoma cells expressing wild type CD43 resulted in enhanced IL-2 production (Fig. 1A, upper panel). This effect was specific since an irrelevant antibody (anti-CD4 mAb, Leu3a) had no effect on the TCR-mediated IL-2 production (Fig. 1A, lower panel). Similarly, By155.16 hybridoma cells expressing human CD4 instead of human CD43 failed to produce IL-2 in response to sub-optimal anti-TCR mAb and anti-CD43 mAb, but did in response to CD4 and TCR ligation (supporting Fig. 3). Moreover, hybridoma cells expressing CD43ΔIC failed to produce higher amounts of IL-2 in response to co-stimulation through TCR and CD43 cross-linking than TCR-only stimulated cells (Fig. 1A upper panel). This effect could not be attributed to an intrinsic defect of the CD43+ΔIC cells to produce IL-2, since both By155.16CD43+ and CD43ΔIC cells responded with equivalent amounts of IL-2 when stimulated with saturating quantities of anti-TCR mAb (CD43+: 146 U/IL-2; CD43ΔIC: 135 U/IL-2). Altogether, these results show that when signals of the CD43 co-receptor molecule complement those of the TCR, IL-2 production augments synergistically, and that the cytoplasmic domain of CD43 is required to generate co-stimulatory signals that lower the threshold for TCR activation.
Fig. 1. CD43 signaling lowers the threshold for TCR activation and enhances TCR-induced IL-2 production and cell proliferation.

A) By155.16 CD43+, ΔCD43+ or CD4+ hybridoma cells expressing equivalent amounts of the TCR-CD3 complex and wild type (Wt) or truncated (ΔIC) human CD43 or CD4 molecules were stimulated with the indicated amount of anti-Vβ8 F23.1 in the presence or absence of saturating amounts of anti-human CD43 L10 or anti-human CD4 Leu3a. Cells were cultured for 24 h and supernatant was collected for IL-2 determination. B) Human TCs were stimulated with 0.1μg/ml of OKT3 mAb alone or in the presence of 500 ng/ml of L10 mAb; proliferation assay was performed as described in methods. C) Human TCs were non-pre-stimulated or pre-stimulated with the L10 mAb as described, followed by TCR engagement with suboptimal concentrations of the OKT3 mAb. Total cell extracts were prepared and levels of active (pERK) or total ERK were determined by immunoblot analysis. D) Thymocytes (upper panel) and spleen cells (bottom panel) from B10.BR mice were cultured in 96-well plates pre-coated with the indicated amounts of anti-TCR (anti-Vβ8) and with 0.5 μg/ml of anti-mCD43 (S7 or S11) mAbs. Proliferation was assessed by [3H]-TdR incorporation during the last 18 h of a 96 h culture assay. Results are representative of three independent experiments.
To further corroborate that CD43-mediated signals regulate the threshold for TC activation, we tested whether CD43 engagement promoted hTCs proliferation when low numbers of TCR molecules are engaged. The proliferation of normal purified TCs in response to a sub-optimal dose of OKT3 mAb (0.1μg/ml) was dramatically augmented when saturating amounts of the anti-CD43 mAb L10 were added (Fig. 1B). Consistent with these results, the low levels of ERK activation resulting from engaging the TCR with suboptimal doses of OKT3 were substantially enhanced by CD43 co-ligation (Fig. 1C).
In addition, we assessed the ability of the CD43 molecule to enhance TCR-mediated proliferation of murine thymocytes and peripheral TCs. Cross-linking the CD43 molecule enhanced TCR-induced thymocyte (Fig. 1D, upper panel) and splenic TCs (Fig. 1D, lower panel) proliferation. In contrast, cross-linking class I molecules with the TCR using the 11.4.2 mAb and F23.1 mAb did not increase cell proliferation above that induced by anti-TCR mAb alone (data not shown). These results show that CD43-dependent signals lower the threshold for TCR activation, enhancing TCR-induced cellular responses both, in human and murine TCs.
CD43 pre-stimulation prevents TCR-induced c-Cbl tyrosine phosphorylation and promotes ERK activation through a PKCθ-dependent mechanism
c-Cbl tyrosine phosphorylation targets signaling molecules associated to c-Cbl to degradation, thus attenuating signal propagation and TC activation (28). To test whether the strong ERK activation we observed when pre-stimulating TCs through CD43 (supporting Fig. 1A and 1B) resulted from preventing the TCR-dependent c-Cbl-tyrosine phosphorylation, c-Cbl was immunoprecipitated from CD43-pre-stimulated or TCR-only activated hTC. As previously reported (17), c-Cbl-tyrosine phosphorylation augmented in response to TCR engagement (Fig. 2A, upper panel). However, when cells were pre-stimulated through CD43, c-Cbl-tyrosine phosphorylation levels were comparable to background levels (Fig. 2A, upper panel). Pre-stimulation through CD43 prevented only partially the TCR-dependent c-Cbl/Crk-L interaction, but enhanced considerably the levels of phosphorylated ERK, compared to solely TCR-activated cells (Fig. 2A, lower panel). As expected (15, 16), addition of TPA before TCR engagement considerably reduced c-Cbl tyrosine phosphorylation as well as Cbl/Crk-L interaction, and increased the levels of phosphorylated ERK (Fig. 2A).
Fig. 2. Loss of the TCR-induced c-Cbl tyrosine phosphorylation and ERK activation resulting of CD43-pre-stimulation is mediated through PKCθ.

A) Human TCs (2×107) were stimulated as described in methods. c-Cbl was immunoprecipitated from non pre-stimulated (−), CD43-pre-stimulated (L10) or TPA pre-stimulated cells (TPA) that were left untreated (−) or treated with the OKT3 mAb (+). Immunoprecipitates were resolved by SDS-PAGE, transferred to nitrocellulose and blotted with anti-p-tyrosine (p-Y-Cbl), Crk-L (Crk-L), or c-Cbl (Cbl) antibodies (upper panel). Levels of phoshorylated ERK (p-ERK) and ERK were determined by inmmunoblotting using total cell lysates (2×106 ceqs) from the same experiment (lower panel). B) c-Cbl was immunoprecipitated from non pre-stimulated (−) or CD43-pre-stimulated cells in the absence or presence of 10 μM RO318220. Immunoprecipitates were resolved by SDS-PAGE, transferred to nitrocellulose and blotted with anti-p-tyrosine (p-Y-Cbl), or c-Cbl (Cbl) antibodies. C) TCs were stimulated as indicated and ERK activation was evaluated using anti-p-ERK and ERK antibodies (upper panel). The intensity of the bands was determined by densitometry; the ratio of p-ERK/ERK is shown (lower panel). D) TCs were stimulated as indicated, PKCθ was immunoprecipitated from total cell lysates and subjected to in vitro kinase assay in the presence of 32P-γATP and 1 μg of myelin basic protein as exogenous substrate. Proteins were resolved by SDS-PAGE, and transferred to nitrocellulose. Membranes were exposed to X-ray films, and blotted with anti-PKCθ antibodies. PKCθ activity was determined as the ratio of 32P-MBP/MBP/amount of PKC in the precipitate. Data shown is representative of three independent experiments.
Serine phosphorylation of c-Cbl by PKCθ prevents its tyrosine phosphorylation and thus its interaction with molecules such as Crk-L and PI3K (16). The fact that CD43 engagement promotes PKCθ kinase activity (20) and that the CD43-dependent phosphorylation of c-Cbl on serine residues is a PKC-dependent event (19) lead us to test whether a CD43-dependent activation of PKC would prevent the TCR-induced c-Cbl tyrosine phosphorylation and ERK activation. Consistent with our hypothesis, blocking PKC activity with RO-31-8220, a general PKC inhibitor, restored the TCR dependent c-Cbl-tyrosine phosphorylation (Fig. 2B), and reduced the ERK phosphorylation resulting from pre-stimulating TCs through CD43 before TCR engagement (Fig. 2C). The effect of RO-31-8220 on ERK activation was as efficient as inhibiting the ERK upstream activator MEK with the inhibitor PD98059 (Fig. 2C). In agreement with our previous report (20), CD43 ligation induced PKCθ activity (3.4 fold above basal levels) (Fig. 2D). Although no further kinase activity was detected when ligating CD43 before TCR engagement (3.7 fold), this PKCθ activity as well as that derived from TPA-stimulated TCs decreased in the presence of the PKC inhibitor RO-31-8220 (Fig. 2D). Under our experimental conditions, engaging the TCR alone did not result in PKCθ activation (Fig. 2D).
To corroborate that PKCθ activation is required for CD43 to prevent the TCR-induced c-Cbl-tyrosine phosphorylation, we generated Jurkat cell lines expressing either the wild type PKCθ or dnPKCθ. In cells expressing wild type PKCθ, a marginal but reproducible reduction in TCR-induced c-Cbl tyrosine phosphorylation was observed when cells were pre-stimulated through CD43 (Fig. 3). In contrast in cells expressing dnPKCθ, CD43 signals failed to reduce the levels of tyrosine-phosphorylated c-Cbl (Fig. 3) as well as to augment the activation of ZAP-70 and ERK resulting from engaging CD43 prior to the TCR (Fig 3). As previously published (19), activation through CD43 alone did not result in c-Cbl tyrosine phosphorylation (Fig. 3). Altogether, these results indicate that the CD43-dependent activation of PKCθ is required to prevent the TCR-induced c-Cbl-tyrosine phosphorylation, ultimately augmenting ZAP-70 and ERK activation.
Fig. 3. The CD43-dependent PKCθ kinase activity blocks the c-Cbl inhibitory effect on ERK and ZAP-70 activation.
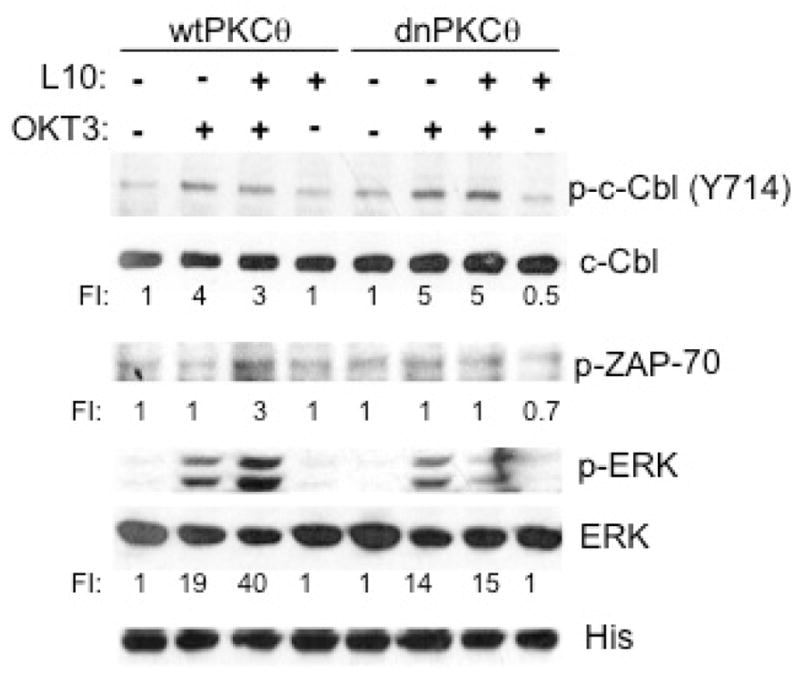
1×106 Jurkat cells stably expressing wild type PKCθ (wtPKCθ) or a dominant negative PKCθ mutant (dnPKCθ) were non treated (−) or stimulated with 4 μg/ml L10 mAb (L10), with 4 μg/ml OKT3 mAb (OKT3) or with both mAbs as described under material and methods. Cell extracts were resolved by SDS-PAGE and transferred to nitrocellulose. The levels of phosphorylated c-Cbl (p-c-Cbl Y743), ZAP-70 (p-ZAP-70) and ERK (p-ERK) as well as those of total c-Cbl, ERK and PKCθ wt or dn (His) were evaluated in the same extracts. Data shown is representative of two independent experiments.
CD43 co-stimulatory signals prevent the c-Cbl and Cbl-b inhibitory effects on TC activation
TCR activation leads also to ZAP-70-c-Cbl interaction, and down-modulation of the ZAP-70 kinase activity through ubiquitination and degradation (29–31), thus switching off its own signaling cascade. In order to evaluate if CD43 co-stimulatory signals prevented the c-Cbl-mediated ZAP-70 inhibition, we assessed the levels of ZAP-70 associated to c-Cbl immunoprecipitates following TC activation. As reported, TCR engagement resulted in a transient association between c-Cbl and ZAP-70 that was no longer detectable after 5 min activation (Fig. 4A). In contrast, and consistent with our previous data (10), the CD43-induced c-Cbl-ZAP-70 interaction was clearly detectable after 5 min stimulation (Fig. 4A), suggesting that the extended c-Cbl-ZAP-70 interaction we detected in CD43-TCR co-stimulated cells reflected the inability of c-Cbl to direct ZAP-70 to degradation.
Fig. 4. CD43 co-stimulatory signals prevent c-Cbl-mediated ZAP-70 and TCR ζ chain degradation.
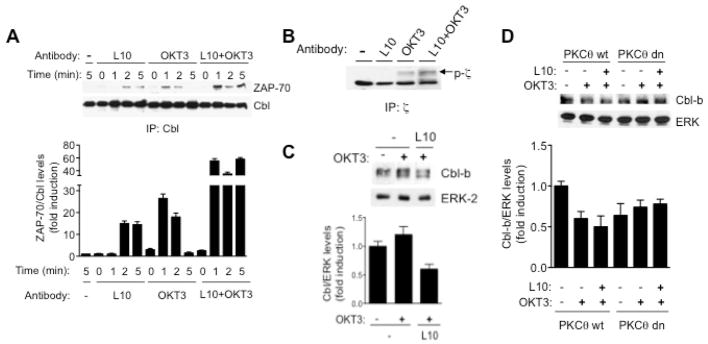
A) 2×107 Jurkat cells were non treated (−) or stimulated with 4 μg/ml L10 mAb (L10), with 4 μg/ml OKT3 mAb (OKT3) or with both mAbs (L10+OKT3), for the indicated period of time. Total cell extracts were prepared and c-Cbl was immunoprecipitated, proteins were resolved by SDS-PAGE, transferred to nitrocellulose and blotted with anti-ZAP-70 or anti-c-Cbl antibodies. B) 2×107 Jurkat cells were non stimulated (−), or stimulated with 4 μg/ml L10 mAb (L10), with 4 μg/ml OKT3 mAb (OKT3) or both mAbs (L10+OKT3) for 5 min. ζ chain immunoprecipitates were resolved by SDS-PAGE, transferred to nitrocellulose and blotted with anti-phosphotyrosine antibodies (p-ζ). C) 1×107 TCs were not pre-stimulated (−) or pre-stimulated with 4 mg/ml of the L10 mAb for 10 min before TCR engagement (L10). After 5 min of TCR stimulation, total cell extracts were prepared and 2×106 cellular equivalents were separated by SDS-PAGE and levels of Cbl-b were determined by blotting with anti-Cbl-b antibodies. ERK-2 levels are shown as loading control. D) 1×106 Jurkat cells stably expressing either wild type (PKCθ Wt) or a dominant negative PKCθ mutant (PKCθ dn) were non treated (−) or stimulated with 4 μg/ml OKT3 mAb (OKT3) alone or pre-stimulated with 4 μg/ml L10 mAb (L10) for 10 min before addition of the OKT3 antibody as described under material and methods. Proteins from total cell extracts were resolved by SDS-PAGE and transferred to nitrocellulose. The levels Cbl-b and ERK were evaluated. Similar results were obtained in three independent experiments.
ZAP-70 binds tyrosine phsophorylated ζ-chain and c-Cbl, acting as an adaptor molecule in the c-Cbl-dependent ubiquitination and degradation of the ζ chain that follows TCR activation (32). Since c-Cbl-ZAP-70 complexes were readily detectable in cells co-stimulated for 5 min (Fig. 4A), we assessed whether CD43 signals prevented the TCR-induced degradation of the tyrosine-phosphorylated ζ chain. Ligating CD43 before TCR engagement resulted in higher levels of tyrosine phosphorylated ζ chain, compared with TCs stimulated through the TCR only or with non-stimulated cells, where no detectable levels of tyrosine-phosphorylated ζ chain were found (Fig. 4B), suggesting that ubiqutination and degradation of the ζ chain were delayed when CD43-dependent signals preceded those of the TCR.
Cbl-b also regulates the threshold for TCR activation by negatively controlling Vav activity (13, 33). However, co-stimulation reduces Cbl-b protein levels by a mechanism involving its ubiquitination and degradation (3). Consistent with the fact that Vav plays a positive role in the CD43 signaling pathway (34), reduced Cbl-b protein levels were observed in hTCs pre-stimulated with CD43 as compared with cells stimulated through the TCR only (Fig. 4C). In agreement with previous results showing that Cbl-b phosphorylation by PKCθ targets it for ubiquitination and degradation (35), activation through the TCR alone was sufficient to reduce Cbl-b levels in Jurkat cells expressing wild type PKCθ (Fig. 4D), although co-stimulation with CD43 further diminished Cbl-b levels (Fig. 4D). In contrast, neither TCR nor CD43 engagement reduced Cbl-b levels in Jurkat cells expressing dnPKCθ (Fig. 4D). Together, these data suggest that the CD43 co-stimulatory signals leading to elevated levels of tyrosine-phosphorylated ζ chain and ZAP-70 and enhanced ERK activation depend on PKCθ to block c-Cbl ubiquitin ligase functions and promote Cbl-b ubiquitination and degradation.
DISCUSSION
The threshold for TCR activation is regulated by positive feedback loops triggered upon inhibition of negative regulatory molecules (36) and is critical to prevent anergy and autoimmunity (12, 37). The ubiquitin ligases of the Cbl family set the threshold for TC activation, negatively regulating ERK phosphorylation and signaling (14, 32). If only the TCR is engaged, c-Cbl and Cbl-b are tyrosine phosphorylated, and interact with downstream effector molecules such as PI3K, Vav, Crk-L, ZAP-70 and Syk, tagging some of them for degradation by ubiquitination (32, 38, 39), terminating TCR signaling. A weak ERK activation resulting from engaging low numbers of TCR molecules or from the lack of co-stimulation is not sufficient to overcome the threshold for TC activation, and leads to anergy (40). Co-receptor molecules reduce the threshold for TCR activation by blocking the negative role of members of the Cbl family on the TCR signaling pathway (2, 41), thus favoring TC activation and a functional immune response even when low numbers of TCR are being compromised.
Here we show that engaging the CD43 co-receptor molecule augmented the TCR-mediated ERK activation and proliferation, regardless of the number of TCR molecules engaged. This was dependent on the signaling cascade generated by the cytoplasmic domain of CD43 and did not result of the mere aggregation of CD43 molecules on the cell surface, as cells expressing the truncated version of CD43 produced similar amounts of IL-2 than cells stimulated only through the TCR. The fact that IL-2 levels were not enhanced in hybridoma cells expressing the human CD4 molecule when incubated with the anti-CD43 mAb L10 demonstrates that the effect detected in the CD43 expressing cells was not the result of a non-specific interaction of the L10 mAb with the murine CD43 molecule or any other cell surface protein. Co-stimulation with distinct commercially available anti-CD43 monoclonal antibodies resulted also in enhanced T cell proliferation, ruling out the possibility that the co-stimulatory effect observed in human T lymphocytes using the L10 mAb results from a non-physiological effect of this antibody (data not shown).
Co-receptor molecules lower the threshold for TC activation by targeting c-Cbl and Cbl-b functions through a mechanism involving PKC-mediated phosphorylation on serine residues (16, 17), favoring sustained TCR signaling and thus, TC activation. In agreement with this and with our previous results showing that CD43 signaling pathway leads to the phosphorylation of c-Cbl on serine residues through a PKC-dependent process (19), TCs activated through CD43 before TCR engagement exhibited low levels of tyrosine phosphorylated c-Cbl as well as of c-Cbl-Crk-L complexes, but enhanced ERK activation. This effect was dependent on PKCθ, as incubating TCs with the PKC inhibitor RO-31-8220 or dnPKCθ expression in Jurkat cells abrogated the synergic increase in ERK activity and restored c-Cbl tyrosine phosphorylation in response to TCR activation. Overall, these results indicate that the PKCθ activity resulting of CD43 signals preclude the negative regulatory effect of c-Cbl on the ERK pathway by preventing c-Cbl phosphorylation on tyrosine residues, therefore canceling critical interactions and the subsequent degradation of signaling molecules.
In addition, and consistent with the ubiquination and degradation of Zap-70 bound to the PTB domain of c-Cbl, and subsequent TCR ζ-chain degradation (32), we found that when co-stimulated through CD43 and the TCR, TCs exhibited higher levels of c-Cbl-associated ZAP-70 as well as of tyrosine-phosphorylated ζ-chain than cells stimulated through the TCR only. This data suggests that the prolonged activation we reported for these molecules in TCs pre-activated through CD43 prior to TCR engagement results from inhibiting the SHP-1-LCK negative regulatory loop (10) and from the inability of c-Cbl to promote their degradation. Whether the phosphorylated ζ chain and ZAP-70 we detected up to two hours after co-stimulating the cells (10) are associated to serine-phosphorylated c-Cbl, and whether SHP-1 is involved in the regulation of the tyrosine phosphoryation of c-Cbl remain to be investigated.
The critical events leading to TC proliferation, the expression of IL-2 and of its high affinity receptor (42) all depend on ERK activation. ERK plays also a pivotal role in the CD43 co-stimulatory functions: the induction of AP-1 and NFκB in response to CD43 ligation requires ERK activity (20) and, preventing ERK activity reduces IL-2 production and TC proliferation in response to CD43 and TCR engagement (10). The fact that the PKC inhibitor RO-31-8220 blocked ERK activation, and the downstream events leading to IL-2 production in hTCs and that dnPKCθ inhibited the CD43-dependent ERK activation suggests that, by inducing PKCθ activity, CD43 prevents the negative regulatory functions of c-Cbl on the ERK pathway as well as that of Cbl-b on NFkB activation (43) and Jun phosphorylation (supporting Fig 2A) (44), leading to enhanced IL-2 production and TC proliferation both, in human and murine TCs.
Our results indicate also that, similar to CD28 (35), CD43 signaling leading to Cbl-b degradation involves Cbl-b phosphorylation by PKCθ. The fact that both, CD43 and CD28 mediate c-Cbl inactivation and Cbl-b ubiquitination and degradation is in agreement with data showing that CD43 and CD28 complement each other during TC activation in the CD28−/− and CD43−/− mice, respectively (7, 45). In T cells, both CD28 and CD43 activate similar signaling pathways that regulate cell cycle progression, apoptosis, and Il-2 transcription. However, how these two molecules together regulate T cell activation when they are both present is not known.
All together, these results indicate that the PKCθ activity resulting of CD43 signals preclude the negative regulatory effect of c-Cbl on the ERK pathway by preventing c-Cbl phosphorylation on tyrosine residues, therefore canceling critical interactions and the subsequent degradation of signaling molecules. Moreover, data shown here provide evidence that besides the establishment of a positive feed back loop that ultimately relies on the duration and intensity of ERK activation (10), signaling through CD43 before TCR engagement also lowers the threshold of TC activation by negatively modulating c-Cbl and Cbl-b. This suggests that when CD43 interacts with its counter-receptor(s) on the APC, a TC may require engaging less TCR molecules in order to sustain a productive cellular response. In addition to the strength and duration of intracellular signals, our data underscore temporality with which certain molecules are engaged as yet another mechanism to fine tune TC signal quality, and ultimately immune function.
Supplementary Material
Acknowledgments
We thank Dr. Leonor Pérez Martinez for critical reading of the manuscript and O. López for technical help. We also thank the blood bank of the Hospital Regional del IMSS in Cuernavaca for leukocyte concentrates. This work was partially funded by grants from the Consejo Nacional de Ciencia y Tecnología (CONACyT), and the Dirección General de Apoyo al Personal Académico (DGAPA)/UNAM, México.
Abbreviations used are
- TCR
T cell receptor
- TC
T cell
- TCs
T cells
- hTC
human T cell
- APC
antigen presenting cell
- RaMIG
rabbit anti-mouse IgG
- mAb
monoclonal antibody
- MAPK
mitogen activated protein kinase
- ERK
extracellular signal-regulated kinase
- dnPKCθ
dominant negative PKCθ mutant
Footnotes
CONFLICT OF INTEREST
The authors declare they have no financial or commercial conflict of interest.
References
- 1.Lanzavecchia A, Sallusto F. From synapses to immunological memory: the role of sustained T cell stimulation. Curr Opin Immunol. 2000;12:92–98. doi: 10.1016/s0952-7915(99)00056-4. [DOI] [PubMed] [Google Scholar]
- 2.Li D, Gal I, Vermes C, Alegre ML, Chong AS, Chen L, Shao Q, Adarichev V, Xu X, Koreny T, Mikecz K, Finnegan A, Glant TT, Zhang J. Cutting edge: Cbl-b: one of the key molecules tuning CD28- and CTLA-4-mediated T cell costimulation. J Immunol. 2004;173:7135–7139. doi: 10.4049/jimmunol.173.12.7135. [DOI] [PubMed] [Google Scholar]
- 3.Zhang J, Bardos T, Li D, Gal I, Vermes C, Xu J, Mikecz K, Finnegan A, Lipkowitz S, Glant TT. Cutting edge: regulation of T cell activation threshold by CD28 costimulation through targeting Cbl-b for ubiquitination. J Immunol. 2002;169:2236–2240. doi: 10.4049/jimmunol.169.5.2236. [DOI] [PubMed] [Google Scholar]
- 4.Cyster J, Somoza C, Killeen N, Williams AF. Protein sequence and gene structure for mouse leukosialin (CD43), a T lymphocyte mucin without introns in the coding sequence. Eur J Immunol. 1990;20:875–881. doi: 10.1002/eji.1830200424. [DOI] [PubMed] [Google Scholar]
- 5.Shelley CS, Remold-O’Donnell E, Davis AE, 3rd, Bruns GA, Rosen FS, Carroll MC, Whitehead AS. Molecular characterization of sialophorin (CD43), the lymphocyte surface sialoglycoprotein defective in Wiskott-Aldrich syndrome. Proc Natl Acad Sci U S A. 1989;86:2819–2823. doi: 10.1073/pnas.86.8.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bravo-Adame ME, Sandoval-Hernandez MA, Migueles-Lozano OA, Rosenstein Y. In: CD43, in Encyclopedia of Signaling Molecules. Choi S, editor. Springer; (in press) [DOI] [Google Scholar]
- 7.Sperling AI, Green JM, Mosley RL, Smith PL, DiPaolo RJ, Klein JR, Bluestone JA, Thompson CB. CD43 is a murine T cell costimulatory receptor that functions independently of CD28. J Exp Med. 1995;182:139–146. doi: 10.1084/jem.182.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thurman EC, Walker J, Jayaraman S, Manjunath N, Ardman B, Green JM. Regulation of in vitro and in vivo T cell activation by CD43. Int Immunol. 1998;10:691–701. doi: 10.1093/intimm/10.5.691. [DOI] [PubMed] [Google Scholar]
- 9.Park JK, Rosenstein YJ, Remold-O’Donnell E, Bierer BE, Rosen FS, Burakoff SJ. Enhancement of T-cell activation by the CD43 molecule whose expression is defective in Wiskott-Aldrich syndrome. Nature. 1991;350:706–709. doi: 10.1038/350706a0. [DOI] [PubMed] [Google Scholar]
- 10.Fierro NA, Pedraza-Alva G, Rosenstein Y. TCR-dependent cell response is modulated by the timing of CD43 engagement. J Immunol. 2006;176:7346–7353. doi: 10.4049/jimmunol.176.12.7346. [DOI] [PubMed] [Google Scholar]
- 11.Pedraza-Alva G, Merida LB, Burakoff SJ, Rosenstein Y. CD43-specific activation of T cells induces association of CD43 to Fyn kinase. J Biol Chem. 1996;271:27564–27568. doi: 10.1074/jbc.271.44.27564. [DOI] [PubMed] [Google Scholar]
- 12.Bachmaier K, Krawczyk C, Kozieradzki I, Kong YY, Sasaki T, Oliveira-dos-Santos A, Mariathasan S, Bouchard D, Wakeham A, Itie A, Le J, Ohashi PS, Sarosi I, Nishina H, Lipkowitz S, Penninger JM. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature. 2000;403:211–216. doi: 10.1038/35003228. [DOI] [PubMed] [Google Scholar]
- 13.Chiang YJ, Kole HK, Brown K, Naramura M, Fukuhara S, Hu RJ, Jang IK, Gutkind JS, Shevach E, Gu H. Cbl-b regulates the CD28 dependence of T-cell activation. Nature. 2000;403:216–220. doi: 10.1038/35003235. [DOI] [PubMed] [Google Scholar]
- 14.Rellahan BL, Graham LJ, Stoica B, DeBell KE, Bonvini E. Cbl-mediated regulation of T cell receptor-induced AP1 activation. Implications for activation via the Ras signaling pathway. J Biol Chem. 1997;272:30806–30811. doi: 10.1074/jbc.272.49.30806. [DOI] [PubMed] [Google Scholar]
- 15.Murphy MA, Schnall RG, Venter DJ, Barnett L, Bertoncello I, Thien CB, Langdon WY, Bowtell DD. Tissue hyperplasia and enhanced T-cell signalling via ZAP-70 in c-Cbl-deficient mice. Mol Cell Biol. 1998;18:4872–4882. doi: 10.1128/mcb.18.8.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, Liu YC, Meller N, Giampa L, Elly C, Doyle M, Altman A. Protein kinase C activation inhibits tyrosine phosphorylation of Cbl and its recruitment of Src homology 2 domain-containing proteins. J Immunol. 1999;162:7095–7101. [PubMed] [Google Scholar]
- 17.Fernandez B, Czech MP, Meisner H. Role of protein kinase C in signal attenuation following T cell receptor engagement. J Biol Chem. 1999;274:20244–20250. doi: 10.1074/jbc.274.29.20244. [DOI] [PubMed] [Google Scholar]
- 18.Donovan JA, Wange RL, Langdon WY, Samelson LE. The protein product of the c-cbl protooncogene is the 120-kDa tyrosine-phosphorylated protein in Jurkat cells activated via the T cell antigen receptor. J Biol Chem. 1994;269:22921–22924. [PubMed] [Google Scholar]
- 19.Pedraza-Alva G, Sawasdikosol S, Liu YC, Merida LB, Cruz-Munoz ME, Oceguera-Yanez F, Burakoff SJ, Rosenstein Y. Regulation of Cbl molecular interactions by the co-receptor molecule CD43 in human T cells. J Biol Chem. 2001;276:729–737. doi: 10.1074/jbc.M008494200. [DOI] [PubMed] [Google Scholar]
- 20.del Rio R, Rincon M, Layseca-Espinosa E, Fierro NA, Rosenstein Y, Pedraza-Alva G. PKCtheta is required for the activation of human T lymphocytes induced by CD43 engagement. Biochem Biophys Res Commun. 2004;325:133–143. doi: 10.1016/j.bbrc.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 21.Cruz-Munoz ME, Salas-Vidal E, Salaiza-Suazo N, Becker I, Pedraza-Alva G, Rosenstein Y. The CD43 co receptor molecule recruits the zeta-chain as part of its signaling pathway. J Immunol. 2003;171:1901–1908. doi: 10.4049/jimmunol.171.4.1901. [DOI] [PubMed] [Google Scholar]
- 22.Remold-O’Donell E, Kennedy DM, Parkman R, Cairns L, Savage B, Rosen FS. Characterization of a human lymphocyte surface syaloglycoprotein that is defective in Wiskott -Aldrich syndrome. J Exp Med. 1984;159:1705–1723. doi: 10.1084/jem.159.6.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baecher-Allan CM, Kemp JD, Dorfman KS, Barth RK, Frelinger JG. Differential epitope expression of Ly-48 (mouse leukosialin) Immunogenetics. 1993;37:183–192. doi: 10.1007/BF00191883. [DOI] [PubMed] [Google Scholar]
- 24.Gillis S, Ferm MM, Ou W, Smith KA. T cell growth factor: parameters of production and a quantitative microassay for activity. J Immunol. 1978;120:2027–2032. [PubMed] [Google Scholar]
- 25.Santana MA, Pedraza-Alva G, Olivares-Zavaleta N, Madrid-Marina V, Horejsi V, Burakoff SJ, Rosenstein Y. CD43-mediated signals induce DNA binding activity of AP-1, NF-AT, and NFkappa B transcription factors in human T lymphocytes. J Biol Chem. 2000;275:31460–31468. doi: 10.1074/jbc.M005231200. [DOI] [PubMed] [Google Scholar]
- 26.Homer RJ, Mamalaki C, Kioussis D, Flavell RA. T cell unresponsiveness correlates with quantitative TCR levels in a transgenic model. Int Immunol. 1993;5:1495–1500. doi: 10.1093/intimm/5.12.1495. [DOI] [PubMed] [Google Scholar]
- 27.Feito MJ, Ballester S, Diez-Orejas R, Ojeda G, Criado G, Portoles P, Rojo JM. CD4 dependence of activation threshold and TCR signalling in mouse T lymphocytes. Scand J Immunol. 1997;45:166–174. doi: 10.1046/j.1365-3083.1997.d01-388.x. [DOI] [PubMed] [Google Scholar]
- 28.Huang F, Gu H. Negative regulation of lymphocyte development and function by the Cbl family of proteins. Immunol Rev. 2008;224:229–238. doi: 10.1111/j.1600-065X.2008.00655.x. [DOI] [PubMed] [Google Scholar]
- 29.Fournel M, Davidson D, Weil R, Veillette A. Association of tyrosine protein kinase Zap-70 with the protooncogene product p120c-cbl in T lymphocytes. J Exp Med. 1996;183:301–306. doi: 10.1084/jem.183.1.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meng W, Sawasdikosol S, Burakoff SJ, Eck MJ. Structure of the amino-terminal domain of Cbl complexed to its binding site on ZAP-70 kinase. Nature. 1999;398:84–90. doi: 10.1038/18050. [DOI] [PubMed] [Google Scholar]
- 31.Rao N, Lupher ML, Jr, Ota S, Reedquist KA, Druker BJ, Band H. The linker phosphorylation site Tyr292 mediates the negative regulatory effect of Cbl on ZAP-70 in T cells. J Immunol. 2000;164:4616–4626. doi: 10.4049/jimmunol.164.9.4616. [DOI] [PubMed] [Google Scholar]
- 32.Wang HY, Altman Y, Fang D, Elly C, Dai Y, Shao Y, Liu YC. Cbl promotes ubiquitination of the T cell receptor zeta through an adaptor function of Zap-70. J Biol Chem. 2001;276:26004–26011. doi: 10.1074/jbc.M010738200. [DOI] [PubMed] [Google Scholar]
- 33.Fang D, Wang HY, Fang N, Altman Y, Elly C, Liu YC. Cbl-b, a RING-type E3 ubiquitin ligase, targets phosphatidylinositol 3-kinase for ubiquitination in T cells. J Biol Chem. 2001;276:4872–4878. doi: 10.1074/jbc.M008901200. [DOI] [PubMed] [Google Scholar]
- 34.Pedraza-Alva G, Merida LB, Burakoff SJ, Rosenstein Y. T cell activation through the CD43 molecule leads to Vav tyrosine phosphorylation and mitogen-activated protein kinase pathway activation. J Biol Chem. 1998;273:14218–14224. doi: 10.1074/jbc.273.23.14218. [DOI] [PubMed] [Google Scholar]
- 35.Gruber T, Hermann-Kleiter N, Hinterleitner R, Fresser F, Schneider R, Gastl G, Penninger JM, Baier G. PKC-theta modulates the strength of T cell responses by targeting Cbl-b for ubiquitination and degradation. Sci Signal. 2009;2:ra30. doi: 10.1126/scisignal.2000046. [DOI] [PubMed] [Google Scholar]
- 36.Altan-Bonnet G, Germain RN. Modeling T cell antigen discrimination based on feedback control of digital ERK responses. PLoS Biol. 2005;3:e356. doi: 10.1371/journal.pbio.0030356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jeon MS, Atfield A, Venuprasad K, Krawczyk C, Sarao R, Elly C, Yang C, Arya S, Bachmaier K, Su L, Bouchard D, Jones R, Gronski M, Ohashi P, Wada T, Bloom D, Fathman CG, Liu YC, Penninger JM. Essential role of the E3 ubiquitin ligase Cbl-b in T cell anergy induction. Immunity. 2004;21:167–177. doi: 10.1016/j.immuni.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 38.Joazeiro CA, Wing SS, Huang H, Leverson JD, Hunter T, Liu YC. The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science. 1999;286:309–312. doi: 10.1126/science.286.5438.309. [DOI] [PubMed] [Google Scholar]
- 39.Ota S, Hazeki K, Rao N, Lupher ML, Jr, Andoniou CE, Druker B, Band H. The RING finger domain of Cbl is essential for negative regulation of the Syk tyrosine kinase. J Biol Chem. 2000;275:414–422. doi: 10.1074/jbc.275.1.414. [DOI] [PubMed] [Google Scholar]
- 40.Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;273:104–106. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]
- 41.Krawczyk CM, Jones RG, Atfield A, Bachmaier K, Arya S, Odermatt B, Ohashi PS, Penninger JM. Differential control of CD28-regulated in vivo immunity by the E3 ligase Cbl-b. J Immunol. 2005;174:1472–1478. doi: 10.4049/jimmunol.174.3.1472. [DOI] [PubMed] [Google Scholar]
- 42.Cantrell D. T cell antigen receptor signal transduction pathways. Annu Rev Immunol. 1996;14:259–274. doi: 10.1146/annurev.immunol.14.1.259. [DOI] [PubMed] [Google Scholar]
- 43.Qiao G, Li Z, Molinero L, Alegre ML, Ying H, Sun Z, Penninger JM, Zhang J. T-cell receptor-induced NF-kappaB activation is negatively regulated by E3 ubiquitin ligase Cbl-b. Mol Cell Biol. 2008;28:2470–2480. doi: 10.1128/MCB.01505-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bustelo XR, Crespo P, Lopez-Barahona M, Gutkind JS, Barbacid M. Cbl-b, a member of the Sli-1/c-Cbl protein family, inhibits Vav-mediated c-Jun N-terminal kinase activation. Oncogene. 1997;15:2511–2520. doi: 10.1038/sj.onc.1201430. [DOI] [PubMed] [Google Scholar]
- 45.Manjunath N, Correa M, Ardman M, Ardman B. Negative regulation of T-cell adhesion and activation by CD43. Nature. 1995;377:535–538. doi: 10.1038/377535a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.