

Abstract
X-linked lymphoproliferative (XLP) disease is a primary immunodeficiency syndrome associated with the inability to control Epstein-Barr virus (EBV), lymphoma, and hypogammaglobulinemia. XLP is caused by mutations in the SH2D1A gene, which encodes the SLAM-associated protein (SAP), or in the BIRC4 gene, which encodes the X-linked inhibitor of apoptosis protein (XIAP).
Here we report a patient with recurrent respiratory tract infections and early onset agammaglob-ulinemia who carried a unique disease-causing intronic loss-of-function mutation in SH2D1A. The intronic mutation affected SH2D1A gene transcription but not mRNA splicing, and led to markedly reduced level of SAP protein. Despite undetectable serum immunoglobulins, the patient's B cells replicated and differentiated into antibody producing cells normally in vitro.
Keywords: SAP, X-linked lymphoproliferative disease, agammaglobulinemia, EBV, B cells
1. Introduction
X-linked lymphoproliferative disease (XLP) is a primary immunodeficiency (PID) syndrome caused by either mutations in the SH2D1A gene, encoding for the SLAM-associated protein (SAP), or in the gene BIRC4 encoding the signalling molecule X-linked inhibitor of apoptosis (XIAP) with some but not complete clinical overlap [1,2]. In the majority of patients, infection with Epstein-Barr virus (EBV) leads to lymphoproliferation, hypogammaglobulinemia and macro-phage activation syndrome.
Mice deficient in SAP recapitulate the human XLP phenotype [3]. Murine and human SAP deficient T cells and NK cells fail to mediate virus-specific cytotoxicity [3–6]. A recent report demonstrated that human SAP deficient, EBV-specific T cells have a disadvantage in vivo compared to their SAP-competent counterparts, while no in vivo disadvantage was found for influenza-specific T cells, potentially explaining the selective susceptibility to EBV infection in this disease [7]. Hypogammaglobulinemia is observed in many patients with SAP deficiency [2]; yet the underlying mechanism is not fully understood. Hypogammaglobulinemia may develop also in the absence of EBV infection [8]. SAP deficient mice develop normal B cell responses to T cell-independent antigens, while T helper cell-dependent antibody responses are defective [9]. In agreement with the SAP-deficient murine model, defective T cell help with resultant reduced formation of memory B cells has been described in affected humans [10,11]. Here we report a patient with early onset agammaglobulinemia due to a disease-causing intronic SH2D1A mutation that results in severely diminished SAP expression.
2. Materials and methods
2.1. Monoclonal antibodies and reagents
The following monoclonal antibodies (mAbs) were used: anti-CD19 PE (BD Biosciences, San Jose, CA, USA); anti-CD27 APC (BD Biosciences, San Jose, CA, USA); anti-CD38 PE-Cy7 (BD Biosciences, San Jose, CA, USA); anti-IgD FITC (BD Biosciences, San Jose, CA, USA); anti-IgM PE (BD Biosciences, San Jose, CA, USA); anti-CD24 APC (Biolegend, San Diego, CA, USA).
2.2. B cell phenotyping and isolation
Peripheral blood mononuclear cells (PBMC) were isolated from anticoagulated peripheral blood by Ficoll (Histopaque 1077, Sigma-Aldrich, St. Louis, MO, USA) gradient centrifugation. Frequencies of B cell subpopulations were calculated based on flow-cytometric analysis (Flowjo, Tree Star INC., Ashland, OR, USA).
2.3. In vitro activation of naive B cells and B-cell lines
PBMC were labeled with 5 μM CFSE and cultured in 96-well round-bottomed plates at a concentration of 3×105/200 μL in absence or presence of CD40L/IL-21 (provided by Dr. Eibel, used as described in [12]) or CpG 2006 (Invivogen, San Diego, CA, USA, 1.25 μM final concentration). After 4 days, CFSE dilution was determined by flow cytometry. Secretion of IgM was measured in the culture supernatants using standard ELISA technology (Zeptometrix, Buffalo, NY, USA).
2.4. Generation of T cell blasts
PBMC were activated with Phytohaemagglutinin (PHA, Enzo life sciences, Ann Arbor, MI, USA, 100 ng/ml) and interleukin-2 (IL-2, Roche, Basel, Switzerland, 100 ng/ml) and T cell blasts expanded by adding fresh IL-2 every 3-4 days.
2.5. Genetic analysis
Genomic DNA was isolated from whole blood and the SH2D1A gene was amplified using the following primers: Exon 1 Forward, 5′-TTTGCACATCTGGCTGAACT-3′; Exon 1 Reverse, 5′-GCCCATGTCCACCGTATCA-3′; Exon 2 Forward, 5′-GTG TCCTACTATATGGACATT-3′; Exon 2 Reverse, 5′-CAAATACG TCCTTGCACCCC-3′; Exon 3 Forward, 5′-TTTGTATCATTAT GAGATACGTA-3′; Exon 3 Reverse, 5′-CTGAGCTTCCAAAC CCTGTC-3′; Exon 4 Forward, 5′-TTATAAGTTTGAGTTAATC TGT-3′; and Exon 4 Reverse, 5′-CATTTGTAGCTCACCGAAC TG-3′. PCR products were analyzed after cycle sequencing (BigDye Teminator, Applied Biosystems) using an ABI3130 Genetic analyzer (Applied Biosystems).
RNA was isolated from T cell blasts using Trizol according to standard protocols. Reverse transcription was performed with qScript cDNA SuperMix (QuantaBioSciences, Inc., Gaithesburg, MD, USA) using 1 microgram of RNA. SH2D1A cDNA (ENST00000371139) was amplified using primers 1 F and 4R and then analyzed after cycle sequencing. For real-time PCR analysis, TaqMan primers and probes spanning exon 1-2 of the SH2D1A gene were used and the level of expression was normalized using GAPDH as a reference (Hs00158978_m1 and Hs99999905_m1, Life Technologies, Grand Island, NY, USA).
2.6. Western blot
PHA blasts from the patient and a healthy control were lysed at 106 cells per 20 μl lysis buffer containing 1%Triton-X100, 100 mM NaCl and 50 mM EDTA pH 7.5. Lysates were cleared by centrifugation then separated by SDS/PAGE gel and transferred to nitrocellulose membranes. SAP was detected using mouse anti-human SAP mAb clone 10C4.2 (gift from Dr. Terhorst) that recognizes a peptide located at the C-terminal end of SAP protein (aa 112-123). Actin was detected with a mAb (Millipore, Billerica, MA, USA) and used as a loading control.
2.7. NKT cell analysis
Peripheral blood lymphocytes were used for NKT cell staining. Hematopoietic cells were identified by gating on cells stained with PE-Texas red conjugated anti-human CD45 mAb (Life Technologies, Grand Island, NY, USA). Alexafluor780 conjugated CD3 and PE conjugated CD1d tetramers loaded with PBS57 (NIH, NIAID MHC tetramer core facility, Atlanta, GA, USA) were used to identify NKT cells. Unloaded CD1d tetramers were used as a negative control. The difference in the proportion of PBS57-loaded minus unloaded tetramer positive CD3+ T cells indicated the frequency of NKT cells.
3. Results
The index patient is a 2-year-old Caucasian boy born to non-consanguineous parents. He was healthy until 8 months of age, when he developed recurrent suppurative otitis media and bacterial conjunctivitis. An initial screening laboratory evaluation at 11 months revealed normal serum immunoglob-ulin levels (Table 1). Subsequently, he developed an episode of bronchopneumonia due to RSV infection, associated with transient neutropenia and hepatosplenomegaly. EBV PCR was negative at that time as was PCR for CMV and HIV. He was subsequently hospitalized several times for ill-defined febrile illnesses, empirically treated with antibiotics. Serum immunoglobulins were re-measured at 13 months and revealed undetectable serum IgG, IgA, and IgM (Table 1). Intravenous immunoglobulin replacement (IVIG) was started at 13 months of age. After initiation of treatment with IVIG, IgG trough levels were in the desired range, whereas serum IgM and IgA stayed below detection limits (Table 1). Peripheral B cell numbers were below the normal range for age at 13 months of age (CD19+ cell count: 278/μl) but normalized when retested at 17 months of age and remained in the normal range. X-linked agammaglobulinemia (XLA) was formally ruled out by BTK gene mutation analysis and testing for BTK protein expression by intracellular flow-cytometric analysis of monocytes and B cells. The patient never developed peripheral lymphadenopathy. As part of a pre-bone marrow transplant evaluation at 3 years of age was found to have developed EBV viremia (16,380 copies/ml), which remained clinically silent and resolved after B cell depleting therapy with anti-CD20 monoclonal antibody (Rituximab).
Table 1.
Clinical Immunology phenotype Immunoglobulins and T and B cell lymphocytes were analyzed at the indicated age. Normal values are indicated in brackets.
| 11 months | 13 months | 17 months | 24 months | |
|---|---|---|---|---|
| IgG, mg/dL | 598 (300-1500) | <100 (300-1500) | 773* (400-1300) | 1000* (400-1300) |
| IgM, mg/dL | 74 (25-115) | <5 (25-115) | <5 (30-120) | <5 (30-120) |
| IgA, mg/dL | 26 (16-100) | <5 (16-100) | <7 (20-230) | <7 (20-230) |
| IgE, IU/ml | ND | ND | <1 (0-30) | <1 (0-30) |
| CD3+CD4+, cells/μL | ND | 1863 (1300-3400) | 1716 (1300-3400) | 1494 (700-2200) |
| CD3+CD8+, cells/μL | ND | 1906 (500-1700) | 1966 (620-2000) | 1605 (490-1300) |
| CD19+, cells/μL | ND | 278 (720-2600) | 1733 (720-2600) | 1004 (390-1400) |
| CD19+IgD-CD27+(% of CD19+ cells) | ND | ND | 0.1 (3.9-13.6) | 0.1 (4.7-21.2) |
| CD3-CD16+/CD56+, cells/μL | ND | 790 (180-920) | 275 (180-920) | 201 (130-720) |
Values in parentheses represent reference ranges;
Abnormal values are bolded;
ND=Not done;
on IVIG replacement therapy.
Analysis of blood derived B cells showed a relative increase of CD24+CD38+ transitional (immature) B cells and a decrease of IgD-CD27+ memory B cells (Figs. 1a and b). This phenotype may be partly reflecting the young age of the patient. In vitro activation of blood derived B cells by the toll like receptor (TLR) 9 agonist CpG or CD40L+IL-21 demonstrated a normal capacity of the patient's B cells to proliferate and to secrete IgM (Figs. 1c and d). Since the patient had reduced serum immunoglobulin levels in combination with normal B cell numbers and normal in vitro B cell function as reported in patients with XLP due to SAP deficiency [10], we evaluated him for XLP. NKT cells are known to be severely reduced in XLP patients [13]. The frequency of NKT cells among peripheral blood CD3+ T cells was assessed by CD1 tetramer staining. NKT cells could not be detected in the patient, but were readily detected in a healthy control at a frequency of 0.017% of CD45 hematopoietic cells (Supplementary Figure). We next sequenced the SH2D1A gene in the patient. An intronic guanine to cytidine (g>c) substitution – at position +5 in intron 1 (137+5 g>c) – was identified (Fig. 2a). To determine the impact of the mutation on SAP expression, we analysed the SH2D1A mRNA transcript derived from T cell blasts of the patient and a healthy control. SH2D1A mRNA in the patient was of normal length and sequence (data not shown), but its expression was reduced 10-fold compared to the control as assessed by quantitative PCR (Fig. 2b). Reduced SH2D1A mRNA levels were associated with reduced expression of a normal-sized SAP protein as assessed by Western blot of T cell blast lysates (Fig. 2c). Flow-cytometric analysis of in vitro activated peripheral blood derived T cells revealed that SAP expression was virtually abrogated in the patient (Fig. 2d). The mother of the patient was identified as a carrier of the SH2D1A 137+5 g>c mutation; the father had a wild-type SH2D1A gene (data not shown).
Figure 1.
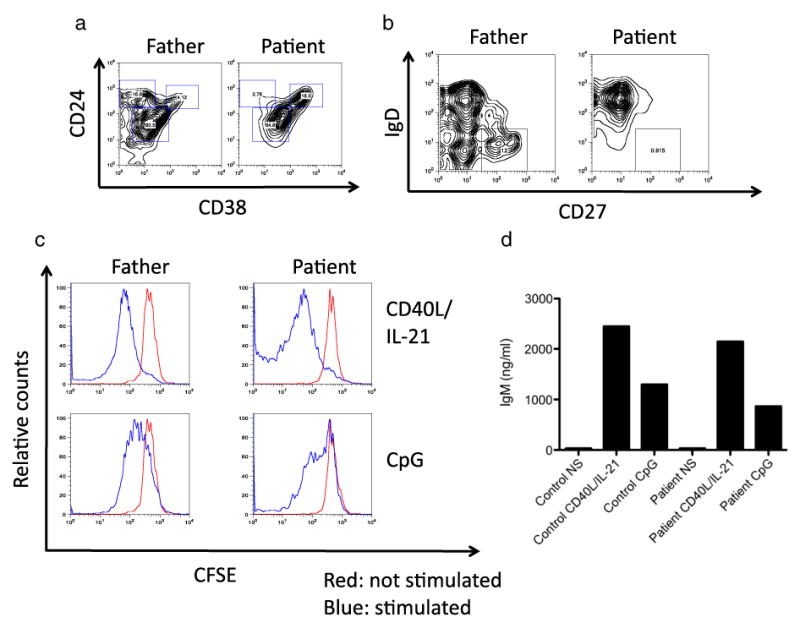
Immunological characterization of a patient with SAP-mutation associated agammaglobulinemia. (a+b) PBMC derived CD19+ B cells were stained with antibodies specific for CD24 and CD38, and relative frequencies of transitional B cells (CD24hiCD38hi), marginal zone like B cells (CD24hiCD38lo), naïve mature B cells (CD24intCD38int) and memory B cells (IgD-CD27+) were assessed by flow cytometry. (c+d) PBMC were labeled with CFSE and stimulated with CpG or CD40L/IL-21. After 4 days of stimulation, proliferation – as indicated by CFSE dilution– of CD19+ B cells was assessed by flow cytometry (c). Secretion of IgM was measured in the culture supernatants by ELISA (d).
Figure 2.
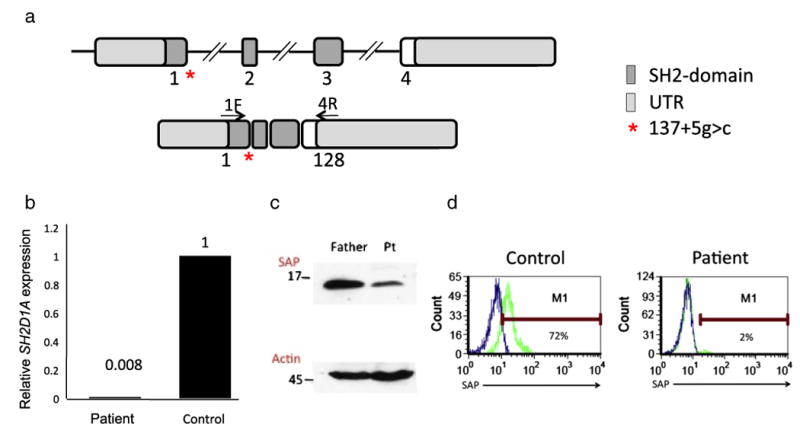
Genetic characterization of a patient with SAP-mutation associated agammaglobulinemia. (a-c) PHA T cell blasts derived from the patient or, as a control, from the father were analyzed. (a) Sequencing analysis of the novel intronic (137+5 g>c) mutation in the SH2D1A gene (b); quantitative PCR demonstrates very low levels of SH2D1A mRNA expression in the patient (c); SAP expression was assessed by western blot analysis. Expression of actin served as an internal loading control. (d) Activated peripheral blood derived CD8+ T cells were stained for SAP expression by flow-cytometry.
4. Discussion
The immunologic phenotype of our patient is characterized by agammaglobulinemia with normal B cell number and normal B cell function in vitro, but almost absent switched memory B cells (Table 1 and Fig. 1b). The patient was found to have a 137 (+5) g>c mutation in intron 1 of the SH2D1A gene that resulted in severely diminished SH2D1A mRNA and SAP protein expression.
Previous work in mouse models has demonstrated that SAP expressed by T cells plays a critical role in promoting normal germinal center formation and T cell dependent antibody production [14]; in particular SAP was important for sustained interactions between follicular T helper (Tfh) cells and B cells. SAP may also influence B cell function intrinsically [15,16]. B cells from SAP deficient patients mature normally, but the transition to memory B cells is abrogated [10]. This could be a result of a block in germinal center formation as found in murine models of SAP deficiency [3]. Thus, it has been hypothesized that SAP deficiency may lead to impaired T cell help to B cells. Tangye et al. [10] have found reduced generation of IL-10 producing T helper cells in SAP deficient patients and suggested that it may contribute to their hypogammaglobulinemia. However, IL-10 deficiency in humans is associated with hypergammaglobulinemia [17]. Tfh cell derived IL-21 was relatively normal in the absence of SAP [18]. Differential expression of some co-stimulating molecules involved in T dependent B cell responses, such as ICOS, has been reported but may not fully explain the hypogammaglobulinemia observed in vivo [11].
As in our patient, hypogammaglobulinemia may occur in SAP deficient patients in the absence of EBV infection, suggesting that B cell dysregulation can occur independently of chronic viral infections [8,19]. Interestingly, none of 13 male XLP patients tested in that study had reduced serum IgM prior to EBV infection. In contrast, 8/13 male XLP patients studied by Grierson et al. showed increased IgM serum concentrations. The complete absence of serum IgM in our patient cannot be explained by absent T cell derived B cell help but raises the possibility that T cells or other cells may inhibit T cell independent B cell responses in vivo through mechanisms yet to be defined. The residual expression of SAP protein in the cells of our patient may relate to the development of agammaglobulinemia, as distinct phenotypes have been described for hypomorphic mutations in other primary immunodeficiency genes [20]. Our study does not completely rule out that mutations in PID-genes other than SH2D1A might be involved in the observed agammaglobulinemia. However, flow cytometric analysis of the patient's peripheral blood revealed normal expression of BTK, CD19, CD21, CD40, BAFFR and MHC II on B cells, with no increase of CD21lo B cells as found in a subgroup of CVID patients [21] (data not shown). Furthermore, the total absence of NKT cells in the patient is highly consistent with SAP deficiency [13].
The murine and human SAP gene and mutations thereof have been analyzed in detail [22–24]. A 137 (+5) g>t variation of the SH2D1A gene, similar to the 137 (+5) g>c mutation described here, was reported recently [2], but has not been further characterized. The mutation identified in our patient is unique as it leads to drastic reduction in the level of wild-type SH2D1A mRNA and SAP protein. The replacement of G residues at position +5 of donor splice-sites is predicted to reduce the stability of base-pairing of the splice site with the complementary region of U1 snRNA, and hence to significantly decrease the efficiency of splicing [25]. The present case highlights the need to consider mutations in the non-coding regions of the SH2D1A gene and to measure SAP protein expression in patients with agammaglobulinemia and normal B cell numbers.
Supplementary Material
Acknowledgments
LDN, RSG and SG were supported by the following PPG grant from the NIH: 5P01AI076210-04. LDN was also supported by The Manton Foundation. RSG was additionally supported by the Dubai-Harvard Foundation of Medical Research, the Jeffrey Modell Foundation, and a NIH grant on Combined SNP Analysis and Whole Genome Sequencing to discover immunodeficiency genes, 5R03AI094017-02.MR was supported by a grant from the Swiss National Science Foundation (SNSF/SSMBS) grant: PASMP3-127678. CH was supported by a grant from the Swiss National Science Foundation (SNSF): 31003A_135677/1. SG was supported by the Fondazione Nocivelli.
Abbreviations
- XLP
X-linked lymphoproliferative disease
- SAP
SLAM-associated protein
- EBV
Epstein Barr Virus
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.clim.2012.11.007.
Conflicts of Interest: The authors have no conflict of interest to declare.
References
- 1.Pachlopnik Schmid J, et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency) Blood. 2011;117:1522–1529. doi: 10.1182/blood-2010-07-298372. [DOI] [PubMed] [Google Scholar]
- 2.Filipovich AH, Zhang K, Snow AL, Marsh RA. X-linked lymphoproliferative syndromes: brothers or distant cousins? Blood. 2010;116:3398–3408. doi: 10.1182/blood-2010-03-275909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crotty S, McCausland MM, Aubert RD, Wherry EJ, Ahmed R. Hypogammaglobulinemia and exacerbated CD8 T-cell-mediated immunopathology in SAP-deficient mice with chronic LCMV infection mimics human XLP disease. Blood. 2006;108:3085–3093. doi: 10.1182/blood-2006-04-018929. [DOI] [PubMed] [Google Scholar]
- 4.Bloch-Queyrat C, et al. Regulation of natural cytotoxicity by the adaptor SAP and the Src-related kinase Fyn. J Exp Med. 2005;202:181–192. doi: 10.1084/jem.20050449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hislop AD, et al. Impaired Epstein-Barr virus-specific CD8+ T-cell function in X-linked lymphoproliferative disease is restricted to SLAM family-positive B-cell targets. Blood. 2010;116:3249–3257. doi: 10.1182/blood-2009-09-238832. [DOI] [PubMed] [Google Scholar]
- 6.Sharifi R, et al. SAP mediates specific cytotoxic T-cell functions in X-linked lymphoproliferative disease. Blood. 2004;103:3821–3827. doi: 10.1182/blood-2003-09-3359. [DOI] [PubMed] [Google Scholar]
- 7.Palendira U, et al. Molecular pathogenesis of EBV susceptibility in XLP as revealed by analysis of female carriers with heterozygous expression of SAP. PLoS Biol. 2011;9:e1001187. doi: 10.1371/journal.pbio.1001187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grierson HL, Skare J, Hawk J, Pauza M, Purtilo DT. Immunoglobulin class and subclass deficiencies prior to Epstein-Barr virus infection in males with X-linked lymphopro-liferative disease. Am J Med Genet. 1991;40:294–297. doi: 10.1002/ajmg.1320400309. [DOI] [PubMed] [Google Scholar]
- 9.Hron JD, Caplan L, Gerth AJ, Schwartzberg PL, Peng SL. SH2D1A regulates T-dependent humoral autoimmunity. J Exp Med. 2004;200:261–266. doi: 10.1084/jem.20040526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma CS, et al. Impaired humoral immunity in X-linked lymphoproliferative disease is associated with defective IL-10 production by CD4+ T cells. J Clin Invest. 2005;115:1049–1059. doi: 10.1172/JCI23139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma CS, Deenick EK. The role of SAP and SLAM family molecules in the humoral immune response. Ann N Y Acad Sci. 2011;1217:32–44. doi: 10.1111/j.1749-6632.2010.05824.x. [DOI] [PubMed] [Google Scholar]
- 12.Warnatz K, et al. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc Natl Acad Sci U S A. 2009;106:13945–13950. doi: 10.1073/pnas.0903543106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nichols KE, et al. Regulation of NKT cell development by SAP, the protein defective in XLP. Nat Med. 2005;11:340–345. doi: 10.1038/nm1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Veillette A, et al. SAP expression in T cells, not in B cells, is required for humoral immunity. Proc Natl Acad Sci U S A. 2008;105:1273–1278. doi: 10.1073/pnas.0710698105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Al-Alem U, et al. Impaired Ig class switch in mice deficient for the X-linked lymphoproliferative disease gene Sap. Blood. 2005;106:2069–2075. doi: 10.1182/blood-2004-07-2731. [DOI] [PubMed] [Google Scholar]
- 16.Ostrakhovitch EA, Wang Y, Li SS. SAP binds to CD22 and regulates B cell inhibitory signaling and calcium flux. Cell Signal. 2009;21:540–550. doi: 10.1016/j.cellsig.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 17.Glocker EO, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–2045. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deenick EK, et al. Follicular helper T cell differentiation requires continuous antigen presentation that is independent of unique B cell signaling. Immunity. 2010;33:241–253. doi: 10.1016/j.immuni.2010.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sumegi J, et al. Correlation of mutations of the SH2D1A gene and epstein-barr virus infection with clinical phenotype and outcome in X-linked lymphoproliferative disease. Blood. 2000;96:3118–3125. [PubMed] [Google Scholar]
- 20.Niehues T, Perez-Becker R, Schuetz C. More than just SCID–the phenotypic range of combined immunodeficiencies associated with mutations in the recombinase activating genes (RAG) 1 and 2. Clin Immunol. 2010;135:183–192. doi: 10.1016/j.clim.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 21.Wehr C, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2008;111:77–85. doi: 10.1182/blood-2007-06-091744. [DOI] [PubMed] [Google Scholar]
- 22.Wu C, et al. Genomic organization and characterization of mouse SAP, the gene that is altered in X-linked lymphoproliferative disease. Immunogenetics. 2000;51:805–815. doi: 10.1007/s002510000215. [DOI] [PubMed] [Google Scholar]
- 23.Coffey AJ, et al. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat Genet. 1998;20:129–135. doi: 10.1038/2424. [DOI] [PubMed] [Google Scholar]
- 24.Brandau O, et al. Epstein-Barr virus-negative boys with non-Hodgkin lymphoma are mutated in the SH2D1A gene, as are patients with X-linked lymphoproliferative disease (XLP) Hum Mol Genet. 1999;8:2407–2413. doi: 10.1093/hmg/8.13.2407. [DOI] [PubMed] [Google Scholar]
- 25.Cooper DN, Krawczak M. Single base-pair substitutions in human gene mRNA splice junctions and their phenotypic consequences. In: Cooper DN, Krawczak M, editors. Human Gene Mutation. Bios Scientific Publishers; 1993. pp. 239–260. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.