

Abstract
Neuropathic pain is a refractory disease characterized by maladaptive changes in gene transcription and translation within the sensory pathway. Long noncoding RNAs (lncRNAs) are emerging as new players in gene regulation, but how lncRNAs operate in the development of neuropathic pain is unclear. Here we identify a conserved lncRNA for Kcna2 (named Kcna2 antisense RNA) in first-order sensory neurons of rat dorsal root ganglion (DRG). Peripheral nerve injury increases Kcna2 antisense RNA expression in injured DRG through activation of myeloid zinc finger protein 1, a transcription factor that binds to Kcna2 antisense RNA gene promoter. Mimicking this increase downregulates Kcna2, reduces total Kv current, increases excitability in DRG neurons, and produces neuropathic pain symptoms. Blocking this increase reverses nerve injury-induced downregulation of DRG Kcna2 and attenuates development and maintenance of neuropathic pain. These findings suggest native Kcna2 antisense RNA as a new therapeutic target for the treatment of neuropathic pain.
Neuropathic pain is a major public health problem. Current treatment for this disorder has had limited success owing to our incomplete understanding of the mechanisms that underlie the induction of neuropathic pain1. Nerve injury-induced neuropathic pain is thought to be induced by abnormal spontaneous activity that arises in neuromas and first-order sensory neurons of dorsal root ganglion (DRG)2,3. The abnormal excitability may result from maladaptive changes in gene transcription and translation of receptors, enzymes, and voltage-dependent ion channels in the DRG4. Voltage-dependent potassium channels govern cell excitability. Peripheral nerve injury downregulates expression of Kv mRNA and protein in the DRG5–10, a phenomenon that may contribute to induction of neuropathic pain11–14. However, the molecular mechanisms that underlie this downregulation are still unknown.
Recent studies suggest that the mechanism for gene regulation involves widespread non-coding RNAs (ncRNAs), including long ncRNAs (lncRNAs)15–17. The study of lncRNAs is still in its infancy18,19. A few lncRNAs have been identified in mammalian cells and implicated in gene-regulatory roles such as transcription and translation18,19. Their expression is associated with some physiological and pathological processes, but how they are causally linked to disease development is elusive18,19. Here, we report a new native lncRNA that is expressed in mammalian DRG neurons. Because most of its sequence is complementary to Kcna2 RNA, we named it Kcna2 antisense RNA. We found that Kcna2 antisense RNA may act as a biologically active regulator and participate in induction and maintenance of neuropathic pain by specifically silencing Kcna2 expression in the DRG.
RESULTS
Identification of natural Kcna2 antisense RNA in DRG neurons
To detect Kcna2 antisense RNA, we first searched a database using the complete published Kcna2 cDNA sequence. Although many of the expressed sequence tags reflected portions of Kcna2 transcript, a few were in the antisense direction. Using strand-specific primers for reverse transcription we identified Kcna2 antisense transcript in the DRGs of rat, mouse, monkey, and human (Fig. 1a), although the sequences were not completely identical among species. This transcript was also detected in spinal cord, various brain regions, and other body organs of rats (Supplemental Fig. 1a). Using rapid amplification of cDNA ends for directional sequencing of 5′ and 3′ ends, we identified a 2.52-kb Kcna2 antisense RNA in rat DRG (Supplemental Fig. 1b). Most of its sequence overlapped that of Kcna2 RNA, including the coding sequence, the 3′ untranslated region, and part of the 5′ untranslated region, but Kcna2 antisense RNA had unique regions at the 5′ and 3′ ends (Fig. 1b). It had no apparent open reading frame (Supplemental Fig. 1b), indicating that Kcna2 antisense RNA is non-coding RNA.
Figure 1.
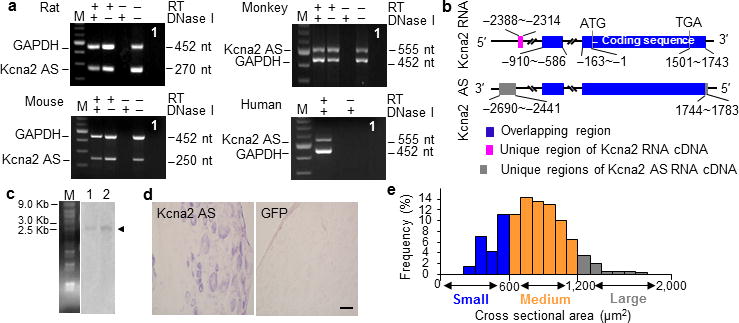
Identification and expression of Kcna2 antisense RNA in naïve dorsal root ganglion. (a) Native Kcna2 antisense transcripts were detected in the DRGs of rat, mouse, monkey, and human by using reverse transcription (RT)-PCR with strand-specific primers. To exclude genomic DNA contamination, the extracted RNA samples were pretreated with excess DNase I. In addition, GAPDH was used as a control. Without RT primers, neither GAPDH nor Kcna2 antisense PCR products were detected in the DNase I-treated samples, indicating absence of genomic DNA. n = 3 repeated experiments/specie. The existence of native Kcna2 antisense RNA in the tissues was further confirmed by using specific intron-spanning primers. Lane 1: H2O. M: 100-bp ladder. (b) Schematic diagrams show the extent of sequence overlap (blue-highlighted boxes) between the cDNAs of Kcna2 RNA and Kcna2 antisense RNA. (c) Northern blot expression analysis of Kcna2 antisense RNA in the DRG (lane 1) and spinal cord (lane 2) of rats (arrowhead). n = 3 repeated experiments. M: RNA marker. (d) In situ hybridization histochemistry shows the distribution of Kcna2 antisense RNA in rat DRG. GFP, which is not expressed in mammalian cells, was used as a negative control. n = 5 rats. Scale bar: 40 μm. (e) Histogram shows the distribution of Kcna2 antisense RNA-positive somata in normal rat DRG.
The Kcna2 antisense RNA was further confirmed at the expected size by Northern blot analysis of RNA from adult rat DRG and spinal cord, although the signals were weak (Fig. 1c). In situ hybridization histochemistry showed that Kcna2 antisense RNA was expressed weakly in DRG neurons (n=5 rats; Fig. 1d). Approximately 21.5% of DRG neurons (228/1060) were labeled. Most were medium-sized (69%; 25–35 μm in diameter), although some were small (24%; <25 μm in diameter) and a few large (7%; >35 μm in diameter) (Fig. 1e). Approximately 60.6% of Kcna2 antisense RNA-labeled neurons were positive for neurofilament-200 (NF-200), 18.1% for P2X3, 15.3% for isolectin B4, and 28.7% for calcitonin gene-related peptide (CGRP) (Fig. 2). Although the distribution pattern of Kcna2 antisense RNA partially overlaps of Kcna2 protein in DRG (Fig. 1e and 3a, b), most Kcna2 antisense-labeled neurons express a low level of Kcna2 protein (Fig. 3c).
Figure 2.
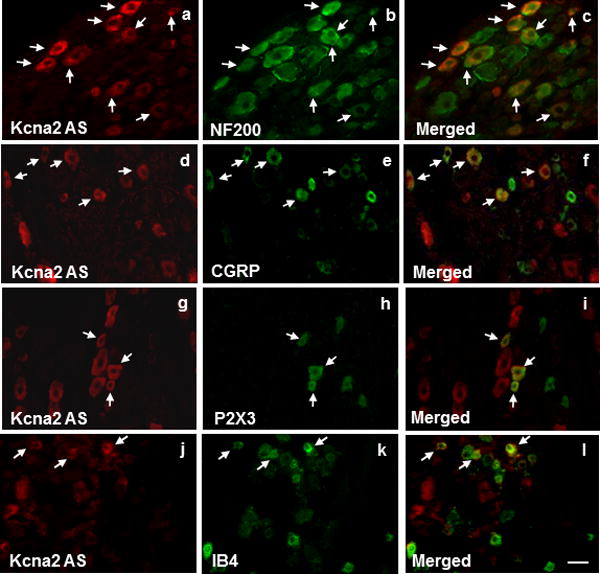
Subpopulation distribution of Kcna2 antisense RNA-containing neurons in dorsal root ganglion of naïve rats. Neurons were double labeled with Kcna2 antisense RNA and neurofilament-200 (NF200; a–c), calcitonin gene-related peptide (CGRP; d–f), P2X3 (g–i), or isolectin B4 (IB4; j–l). Arrows: double-labeled neurons. n = 5 rats. Scale bar: 40 μm.
Figure 3.
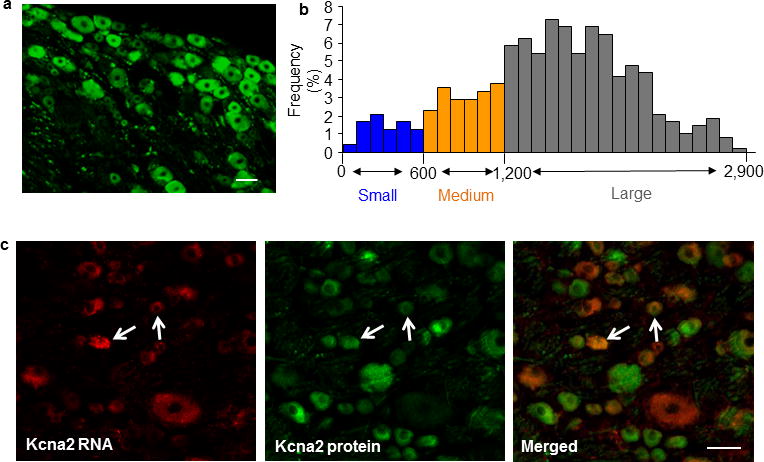
Distribution of Kcna2 protein and double labeling of Kcna2 antisense RNA with Kcna2 protein in normal rat DRG. (a) A representative example showing the distribution of Kcna2-positive neurons. Approximately 70.1% (855/1220) of DRG neurons are positive for Kcna2. (b) Histogram showed the distribution of Kcna2-positive somata (large: 72.6%; medium: 18.6%; small: 8.6%). (c) Representative examples showing that most Kcna2 antisense RNA-labeled neurons in the DRG express a low level of Kcna2 protein, although a few display high-density Kcna2 protein staining (arrows). n = 5 rats. Scale bars: 50 μm.
DRG Kcna2 antisense RNA expression after peripheral nerve injury
Next, we examined whether expression of DRG Kcna2 antisense RNA is altered in rat after peripheral nerve injury. Consistent with previous studies5–10, unilateral L5 spinal nerve ligation (SNL), but not sham surgery, time-dependently downregulated the expression of Kcna2 mRNA (n=12 rats/group/time point; P<0.01; Fig. 4a) and protein (n=12 rats/group/time point, P<0.05 or 0.01; Fig. 4b) in the ipsilateral L5 DRG. Interestingly, the level of Kcna2 antisense RNA was time-dependently increased in the ipsilateral L5 DRG after SNL (n=12 rats/group/time point, P<0.05 or 0.01; Fig. 4c). Neither SNL nor sham surgery changed the expression of Kcna2 mRNA, Kcna2 protein, or Kcna2 antisense RNA in the ipsilateral L4 DRG (n=12 rats/group/time point, P>0.05; Fig. 4a, c) or L5 spinal cord (n=4 rats/group/time point, P>0.05; Supplemental Fig. 2a–c). Furthermore, the staining density and number of Kcna2 antisense RNA-labeled neurons in the ipsilateral L5 DRG were higher than those in the contralateral L5 DRG on days 3, 7, and 14 post-SNL (n=5 rats/time point; P<0.05 or 0.01; Fig. 4d, e). These changes occurred predominantly in large DRG neurons (Fig. 1e,4f). Similar results were observed following sciatic nerve axotomy. On day 7 post-axotomy, the ratio of ipsilateral to contralateral Kcna2 antisense RNA was increased by 2.2-fold in the injured L5 DRG compared to that in the sham groups, whereas the corresponding ratio for Kcna2 mRNA was decreased by 75% (n=12/group, P<0.01; Fig. 4g). Additionally, Kcna2 protein in the ipsilateral L5 DRG was reduced by 51.8% compared to that in the contralateral L5 DRG from the sham groups (n=12/group, P<0.05; Supplemental Fig. 2d).
Figure 4.
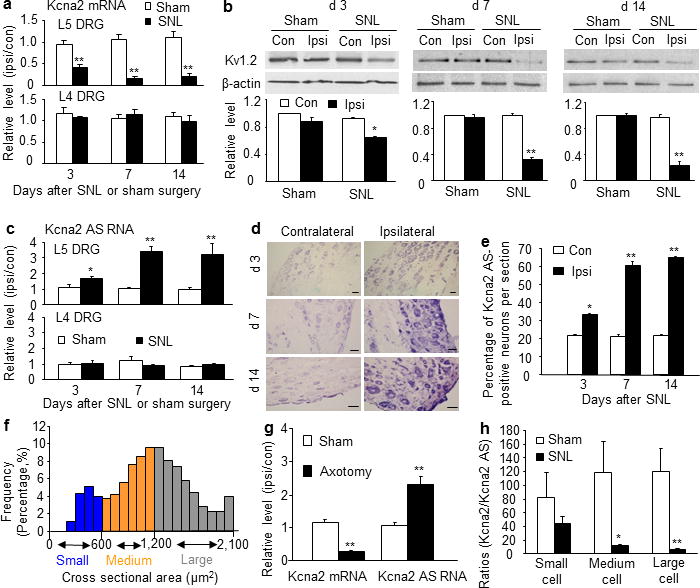
Expressional changes of DRG Kcna2 antisense RNA and Kcna2 after peripheral nerve injury. (a) Kcna2 mRNA expression in L4/5 DRGs after SNL or sham surgery. Ipsi: ipsilateral. Con: contralateral. n=12 rats/group/time point. F=60.05. **P<0.01 vs the sham group at the corresponding time point. (b) Kcna2 protein expression in L5 DRG after SNL or sham surgery. n=12 rats/group/time point. F=6.90 for day 3, 74.11 for day 7, and 351.39 for day 14. *P<0.05, **P<0.01 vs the contralateral side of the sham group at the corresponding time point. (c) Kcna2 antisense RNA expression in L4/5 DRGs after SNL or sham surgery. n=12 rats/group/time point. F=35.51. *P<0.05, **P<0.01 vs the sham group at the corresponding time point. (d. e) Kcna2 antisense RNA-positive neurons in L5 DRGs after SNL. n=5 rats/time point. F=358.18. *P<0.05, **P<0.01 vs the corresponding contralateral side. Scale bars: 40 μm. (f) Histogram showing that 46.4% of Kcna2 antisense RNA-positive neurons are large, 39.1% medium, and 14.5% small in the ipsilateral L5 DRG on day 14 post-SNL. (g) Expression of Kcna2 antisense RNA and Kcna2 mRNA in L4/5 DRGs on day 7 after axotomy or sham surgery. n=12 rats/group. t = −14.19 for antisense RNA and 7.55 for mRNA. **P<0.01 vs the corresponding sham group. (h) The ratios of Kcna2 mRNA to Kcna2 antisense RNA in individual DRG neurons on day 7 after SNL or sham surgery. n=15 neurons/cell type/group. t = 1.01 for small cells, 3.35 for medium cells, and 4.48 for large cells. *P<0.05, **P<0.01 vs the corresponding sham group.
We further examined the opposing SNL-induced changes in Kcna2 antisense RNA and Kcna2 mRNA in individual DRG neurons. Kcna2-to-Kcna2 antisense RNA ratios were approximately 82, 118, and 121 in small, medium, and large DRG neurons, respectively, from sham rats (n=15 neurons/cell type/group, P<0.05 or 0.01; Fig. 4h). These ratios decreased, particularly in medium and large DRG neurons, 7 days after SNL (Fig. 4h). Taken together, these results demonstrate that Kcna2 antisense RNA can be induced in the injured DRG after peripheral nerve injury.
MZF1 promotes Kcna2 antisense RNA gene activity after SNL
How is DRG Kcna2 antisense RNA upregulated after nerve injury? Using the online software TFSEARCH, we found a consensus binding motif (−161/agtgggga/−154) for transcriptional activator myeloid zinc finger protein 1 (MZF1) in the promoter region of Kcna2 antisense RNA gene20,21. An electrophoretic mobility shift assay demonstrated binding of MZF1 to this motif in the DRG (Fig. 5a). A chromatin immunoprecipitation assay revealed that a fragment of the Kcna2 antisense RNA promoter that includes the binding motif could be amplified from the complex immunoprecipitated with MZF1 antibody in nuclear fractions from DRGs in sham rats (Fig. 5b). This amplification did not occur with normal serum (Fig. 5b) or after pre-absorption of MZF1 antibody (data not shown), indicating that the binding of MZF1 to Kcna2 antisense RNA promoter is specific and selective. SNL increased the binding of MZF1 to the Kcna2 antisense gene promoter, as demonstrated by a 4.12-fold increase in band density in the ipsilateral L5 DRG from SNL rats compared to that from sham rats on day 14 (n=6 rats/group, P<0.05). This increase may result from SNL-induced time-dependent upregulation of MZF1 in the ipsilateral L5 DRG (n=9 rats/time point/group, P<0.05; Fig. 5c, d). As expected, neither sham nor SNL surgery altered basal binding activity or MZF1 expression in the contralateral L5 DRG and ipsilateral L4 DRG (data not shown). Moreover, MZF1 mRNA co-expressed with Kcna2 antisense RNA in the DRG neurons (Supplementary Fig. 3a). These in vivo findings suggest that peripheral nerve injury increases DRG MZF1 expression, allowing the binding of more MZF1 to the promoter region of Kcna2 antisense gene in the injured DRG neurons.
Figure 5.
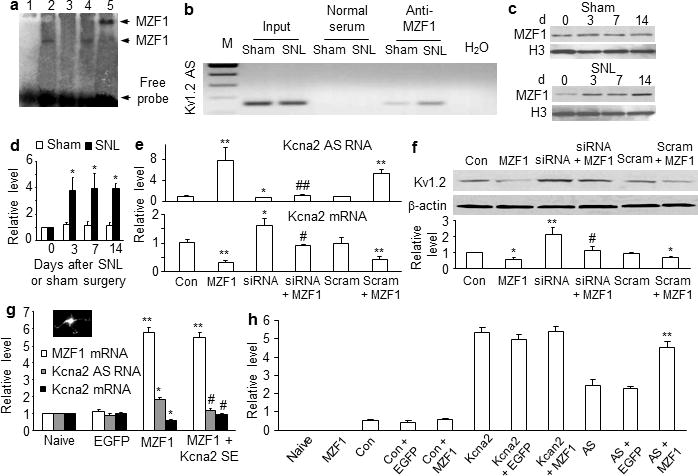
MZF1 mediates nerve injury-induced upregulation of DRG Kcna2 antisense RNA. (a) The specific-binding activity of MZF1 to the Kcna2 antisense promoter. Labeled probe alone (lane 1) or plus nuclear extract (lane 2), nuclear extract and 50-fold unlabeled probe (lane 3), nuclear extract and 50-fold unlabeled mutant probe (lane 4), or nuclear extract and rabbit anti-MZF1 antibody (lane 5). n=3 repeats. (b) Kcna2 antisense promoter fragments immunoprecipitated by rabbit anti-MZF1 antibody in the ipsilateral L5 DRGs on day 14 after SNL or sham surgery. Input: total purified fragments. M: ladder marker. (c, d) MZF1expression in the ipsilateral L5 DRGs after SNL or sham surgery. n=9 rats/time point/group. F=14.13. *P<0.05 vs the corresponding naive group (0 d). (e, f) The levels of Kcna2 antisense RNA, Kcna2 mRNA, and Kcna2 protein in HEK-293T cells transfected as shown. Control: EGFP; siRNA: MZF1 siRNA; Scram: scrambled MZF1 siRNA. n=5 repeats/treatment. F=8.53 for antisense RNA, 12.92 for mRNA, and 7.93 for protein. *P<0.05, **P<0.01 vs control EGFP. #P<0.05, ##P<0.01 vs MZF1 alone. (g) The levels of MZF1 mRNA, Kcna2 antisense RNA, and Kcna2 mRNA in rat DRG cultured neurons transduced as shown. Inset: an AAV5-EGFP-labeled neuron. n=3 repeats/treatment. F=168.61 for MZF1 mRNA, 30.84 for Kcna2 antisense RNA, and 17.79 for Kcna2 mRNA. *P<0.05, **P<0.01 vs the corresponding naïve. #P<0.05 vs the corresponding AAV5-MZF1 alone. (h) Activity levels of Kcna2 gene promoter and Kcna2 antisense gene promoter in HEK-293T cells transfected as shown. Con: control vector (pGL3-Basic). n=3 repeats/treatment. F=82.09. **P<0.01 vs pGL3-Kcna2 antisense vector alone.
To further examine whether MZF1 directly regulates Kcna2 antisense RNA expression, we overexpressed full-length MZF1 in cultured HEK-293T cells (Supplementary Fig. 3b. c) that express endogenous Kcna2 antisense RNA, Kcna2, and other Kv channels. MZF1 overexpression significantly increased Kcna2 antisense RNA and correspondingly decreased Kcna2 mRNA and protein (n=5 repeats/treatment, P<0.05 or 0.01; Fig. 5e, f). These responses were abolished in cells co-transfected with full-length MZF1 vector and MZF1-specific siRNA (but not scrambled MZF1 siRNA) (Fig. 5e, f and Supplementary Fig. 3d), indicating that upregulation of Kcna2 antisense RNA was specific in response to MZF1. MZF1 siRNA transfection alone also reduced basal Kcna2 antisense RNA expression and increased basal expression of Kcna2 mRNA and protein (Fig. 5e, f). MZF1-triggered upregulation of Kcna2 antisense RNA and downregulation of Kcna2 mRNA were confirmed in cultured DRG neurons that were transduced with recombinant adeno-associated virus 5 (AAV5) that expressed full-length MZF1 (n=3 repeats/treatment, P<0.05 or 0.01; Fig. 5g).
A software prediction showed that the promoter region of Kcna2 RNA does not contain a consensus MZF1-binding motif. MZF1 does not enhance the activity of Kcna2 gene promoter but dramatically activates the Kcna2 antisense gene promoter (n=3 repeats/treatment, P<0.01; Fig. 5h). In naïve rats, Kcna2 RNA promoter fragments are not amplified from the DRG nuclear complex immunoprecipitated by MZF1 antibody (data not shown). MZF1-triggered downregulation of Kcna2 does not occur likely by direct binding of MZF1 to the Kcna2 gene promoter. To examine whether Kcna2 antisense RNA mediates this effect, we cloned an AAV5 vector that expresses a Kcna2 sense (SE) RNA fragment (−311 to +40). This fragment can significantly block Kcna2 antisense RNA expression (n=4 repeats/treatment, P<0.01; Fig. 6a, b) but does not alter basal expression of Kcna2 mRNA or protein or produce truncated Kcna2 protein in cultured HEK-293T cells or DRG neurons (Fig. 6a, b). We found that Kcna2 SE RNA fragment blocked MZF1-induced increase in Kcna2 antisense RNA and reversed MZF1-induced reduction in Kcna2 mRNA in DRG neurons (Fig. 5g). Thus, MZF1-induced Kcna2 downregulation may be attributable to MZF1-triggered Kcna2 antisense gene expression.
Figure 6.
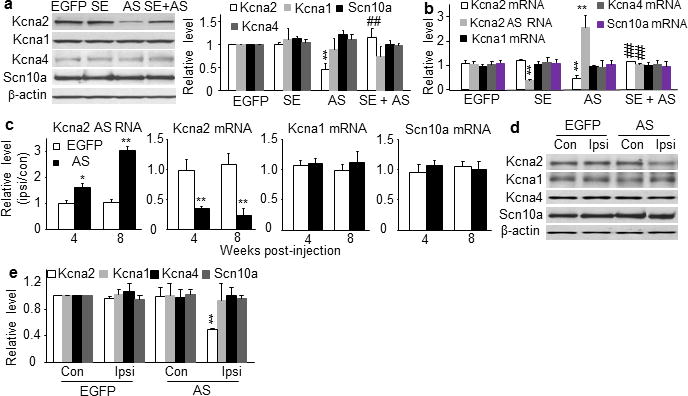
Kcna2 antisense RNA specifically targets Kcna2. (a) Left: Representative Western blots showing the amounts of Kcna2, Kcna1, Kcna4, and Scn10a protein in HEK-293T cells transfected with control EGFP vector, Kcna2 SE vector, Kcna2 antisense vector, or Kcna2 SE + Kcna2 antisense. Right: Statistical summary of the densitometric analysis. n=4 repeats/treatment., F=21.37 for Kcna2, 0.65 for Kcna1, 0.61 for Kcna4, and 0.45 for Scn10a. **P<0.01 vs the corresponding EGFP groups. ##P<0.01 vs the corresponding Kcna2 antisense vector alone. (b) Levels of Kcna2 antisense RNA and mRNA of various ion channels in rat DRG cultured neurons transduced with AAV5-EGFP, AAV5-Kcna2 SE, AAV5-Kcna2 antisense, or AAV5-Kcna2 SE + AAV5-Kcna2 antisense. n=3 repeats/treatment. F=10.06 for Kcna2 mRNA, 11.90 for antisense RNA, 0.24 for Kcna1 mRNA, 0.65 for Kcna4 mRNA, and 0.87 for Scn10a mRNA. **P<0.01 vs AAV5-EGFP alone. ##P<0.01 vs the corresponding AAV5-Kcna2 antisense alone. (c) Levels of Kcna2 antisense RNA and mRNA of various ion channels in the ipsilateral (ipsi) and contralateral (con) L4/5 DRGs 4 and 8 weeks after injection with AAV5-EGFP or AAV5-Kcna2 antisense. n=12 rats/treatment. F=15.91 for antisense RNA, 20.45 for Kcna2 mRNA, 0.39 for Kcna1 mRNA, and 0.56 for Scn10a mRNA. *P<0.05, **P<0.01 vs the corresponding EGFP-treated group. (d) Representative Western blots of ipsilateral and contralateral L4/5 DRGs 8 weeks after injection with AAV5-EGFP or AAV5-Kcna2 antisense. (e) Statistical summary of the densitometric analysis. n=10 rats/group. F=15.51 for Kcna2, 0.35 for Kcna1, 0.78 for Kcna4 and 0.48 for Scn10a. **P<0.01 vs corresponding contralateral side of the AAV5-EGFP-treated group.
DRG Kcna2 antisense RNA leads to neuropathic pain symptoms
We next investigated whether mimicking nerve injury-induced upregulation of DRG Kcna2 antisense RNA alters DRG Kcna2 expression and function, DRG neuronal excitability, and nociceptive thresholds. To this end, we transfected Kcna2 antisense RNA proviral vector or control EGFP vector into cultured HEK-293T cells and transduced AAV5 that expressed Kcna2 antisense RNA (AAV5-Kcna2 antisense) or EGFP (AAV5-EGFP) into cultured DRG neurons. Kcna2 antisense RNA dramatically decreased Kcna2 mRNA and protein expression, but not Kcna1, Kcna4, or Na1.8expression (Fig. 6a, b). Then we injected AAV5-Kcna2 antisense or AAV5-EGFP unilaterally into L4/5 DRGs. Four weeks after injection, EGFP-labeled AAV5 was limited to the ipsilateral L4/5 DRG neurons and their fibers and terminals (Supplementary Fig. 4a–g). Approximately 87.12% of labeled cells were positive for NF-200, 4.21% for substance P, 6.32% for CGRP, and 10.01% for P2X3 (Supplementary Fig. 4h), a distribution similar to that of Kcna2 antisense RNA-labeled neurons in the injured DRG post-SNL (Fig. 4f). Expression of the Kcna2 antisense RNA was significantly increased in the L4/5 DRGs at 4 weeks, reached a peak at 8 weeks, and remained high for at least 12 weeks after viral injection (n=12 rats/group, P<0.05 or 0.01; Fig. 6c). In contrast, the expression of Kcna2 mRNA (n=12 rats/group) and protein (n=10 rats/group) was significantly and temporally reduced in the ipsilateral L4/5 DRGs (P<0.01; Fig. 6c–e). The amounts of mRNA and protein of Kcna1, Kcna4, and Scn10a were unaffected (P>0.05; Fig. 6c–e). These results indicate that Kcna2 antisense RNA specifically and selectively targets Kcna2.
Using a voltage-clamp technique, we recorded Kcna2-related current in neurons freshly dissociated from the injected L4/5 DRGs 8–12 weeks post-injection. To increase the recording efficiency, we injected AAV5-EGFP alone (control group) or a mixed viral solution of AAV5-Kcna2 antisense plus AAV5-EGFP (Kcna2 antisense-treated group) and recorded only green DRG neurons (Fig. 7a). In the Kcna2 antisense-treated group, total Kv current density was significantly reduced in large- and medium-diameter neurons (Fig. 7a, b and Supplemental Fig. 5a–c). To verify whether this reduction is due to Kcna2 downregulation, we utilized bath application of 100 nM maurotoxin (MTX), a selective Kcna2 current inhibitor22–24. MTX produced greater reductions in total Kv current in large (n=14/group) and medium (Control: n=17; antisense: n=15) neurons from the control group than in those from the Kcna2 antisense-treated group at depolarized voltages (P<0.05 or 0.01; Fig. 7a, b and Supplemental Fig. 5a–c). When tested at +50 mV, large and medium neurons in the control group retained 81.7±1.7% and 85.1±2.2% of current, respectively, after MTX treatment, but large and medium neurons from the Kcna2 antisense-treated group retained 92.3±0.9% and 94.9±1.6% of current, respectively. In small DRG neurons, the current reduction by MTX was less prominent, but the difference between control and Kcna2 antisense-treated groups was still statistically significant (n=11 neurons/group, P<0.05; Supplemental Fig. 5d–f). These data indicate that Kcna2 antisense RNA reduces total Kv current densities in large and medium DRG neurons and decreases Kcna2-related current in all neurons.
Figure 7.
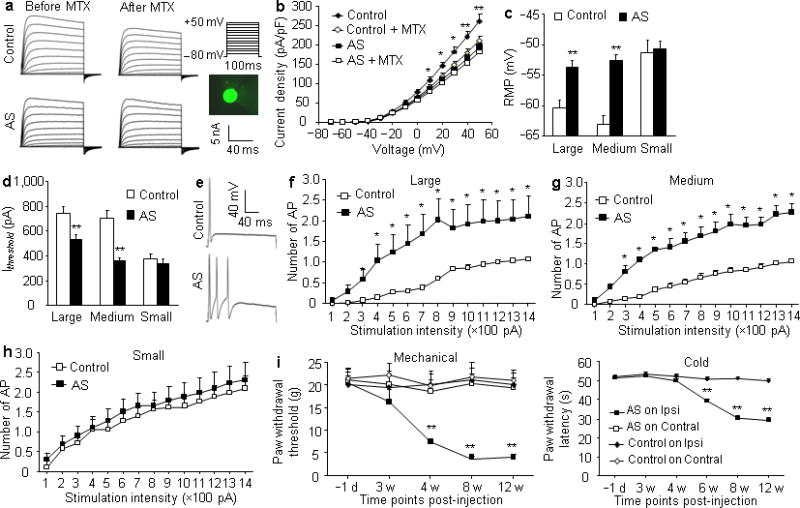
Kcna2 antisense RNA overexpression in DRG reduces total Kv current, increases excitability in large and medium DRG neurons, and produces neuropathic pain symptoms. (a) Representative traces of total Kv current in large DRG neurons from control and antisense-injected rats before or after bath perfusion of 100 nM maurotoxin (MTX). Inset: a green-labeled recording neuron. (b) I–V curve for control and antisense-treated large DRG neurons before or after 100 nM MTX treatment. n=14 cells/group. F=139.21, *P<0.05, **P<0.01 vs the antisense group. (c, d) Resting membrane potential (RMP, c; t = −4.21, for large, −7.04 for medium, and −0.32 for small) and current threshold for pulses (Ithreshold, d; t = 3.22 for large, 6.28 for medium, and 0.73 for small). n=33 large, 42 medium, and 30 small cells from the control group (12 rats). n=43 large, 70 medium, and 32 small cells from the antisense group (14 rats). **P<0.01 vs the corresponding control group. (e) Representative traces of the evoked action potential (AP) in DRG neurons. (f–h) Numbers of evoked APs from control and antisense-injected rats after application of different currents. Numbers of the recorded cells are the same as in c. F = 18.45 for large, 20.65 for medium, and 0.67 for small cells. *P<0.05 vs the same stimulation intensity in the control group. (i) Ipsilateral (Ipsi) and contralateral (Contral) paw withdrawal responses to mechanical (F=38.31) and cold (F=65.77) stimuli from control and antisense-injected rats. n=14 rats/group.. **P<0.01 vs control on the ipsilateral side at the corresponding time points.
To assess whether Kcna2 antisense RNA modulates DRG neuronal excitability, we carried out whole-cell current-clamp recording 8–12 weeks post-injection. Compared to the control group, Kcna2 antisense RNA treatment significantly increased resting membrane potentials by 6.74 mV and 10.52 mV in large (Control: n=33; antisense: n=43) and medium (Control: n=42; antisense: n=70) neurons, respectively (P<0.01; Fig. 7c), and reduced current thresholds by 217 pA and 344 pA, respectively (P<0.01; Fig. 7d). The average number of action potentials evoked by stimulation of ≥300 pA in the Kcna2 antisense-treated group was greater than the average number evoked by the corresponding stimulation intensity in the control group in large and medium neurons (P<0.05; Fig. 7e–g). No such changes were observed in small DRG neurons (Control: n=17; antisense: n=14; P>0.05; Fig. 7c, d, h). There were no apparent differences between the two groups in membrane input resistances or other action potential parameters, such as amplitude, threshold, duration, overshoot, and afterhyperpolarization amplitude (Supplemental Table 1). Application of MTX into DRG neurons produced similar effects (Supplemental Fig. 6 and Table 1). Our findings indicate that Kcna2 knockdown or current inhibition increases DRG neuronal excitability.
Lastly, we examined whether rats that received L4/5 DRG injections of Kcna2 antisense RNA displayed behavioral changes in nociceptive thresholds. Injection of AAV5-Kcna2 antisense, but not of AAV5-EGFP, produced mechanical and cold hypersensitivities as demonstrated by ipsilateral decreases in paw withdrawal threshold and paw withdrawal latency, respectively (n=14 rats/group, P<0.01; Fig. 7j). These hypersensitivities developed by 4 to 6 weeks, reached a peak at 8 weeks, and were maintained for at least 12 weeks (Fig. 7j). Neither AAV5-Kcna2 antisense nor AAV5-EGFP affected locomotor functions (Data not shown). These findings suggest that Kcna2 antisense RNA-triggered DRG Kcna2 downregulation induces mechanical and cold hypersensitivities, two major clinical symptoms of neuropathic pain.
Blocking DRG Kcna2 antisense RNA attenuates neuropathic pain
Finally, we inquired whether blocking nerve injury-induced upregulation of DRG Kcna2 antisense RNA would affect reductions in DRG Kcna2 expression and nociceptive thresholds after nerve injury. Consistent with our in vitro work (Fig. 6a, b), in vivo DRG injection of AAV5-Kcna2 SE, but not AAV5-EGFP, significantly blocked upregulation of Kcna2 antisense RNA and downregulation of Kcna2 mRNA and protein in the injured DRGs after SNL or chronic constriction injury (CCI) (n=12/group, P<0.05 or 0.01; Fig. 8a, b). These effects occurred at 4 weeks and were maintained for at least 12 weeks after viral injection. Injection of AAV5-Kcna2 SE alone did not alter basal expression of Kcna2 mRNA and protein or Kcna2 antisense RNA in the ipsilateral L5 DRG of sham-operated rats (Fig. 8a, b). To examine the role of Kcna2 antisense RNA in neuropathic pain induction, we subjected rats to SNL 4 weeks after DRG viral injection, as our pilot work showed that the level of Kcna2 SE RNA fragment was too low to block SNL-induced Kcna2 antisense RNA expression before that time. SNL produced mechanical, cold, and thermal hypersensitivities on the ipsilateral side in the EGFP-injected group (n=8 rats/group; Fig. 8c). However, hypersensitivity was attenuated in the Kcna2 SE-injected rats (P<0.05 or 0.01; Fig. 8c). Paw withdrawal threshold to mechanical stimulation and paw withdrawal latency to cold and thermal stimuli were higher in the Kcna2 SE-injected rats than in the EGFP-injected group from day 3 to day 14 post-SNL (Fig. 8c). Similar effects of AAV5-Kcna2 SE on neuropathic pain development were also observed in the CCI model (Fig. 8d). To further investigate the role of Kcna2 antisense RNA in neuropathic pain maintenance, we subjected rats to SNL 2 weeks after DRG viral injection. As shown in Fig. 8e (n=8 rats/groups), mechanical, cold, and thermal hypersensitivities were completely developed in both the Kcna2 SE-injected and EGFP-injected rats on day 7 post-SNL. These hypersensitivities were markedly attenuated on days 14, 21, and 28 post-SNL in the Kcna2 SE-injected rats (P<0.05 or 0.01; Fig. 8e). Neither AAV5-Kcna2 SE nor AAV5-EGFP affected paw withdrawal threshold or latency on the contralateral side (Fig. 8c, d, e), affected locomotor function (Data not shown), or altered basal responses to mechanical or cold stimuli in sham-operated rats (data not shown). Our findings indicate that Kcna2 antisense RNA contributes to neuropathic pain development and maintenance and that blocking its expression may have clinical applications in neuropathic pain treatment.
Figure 8.
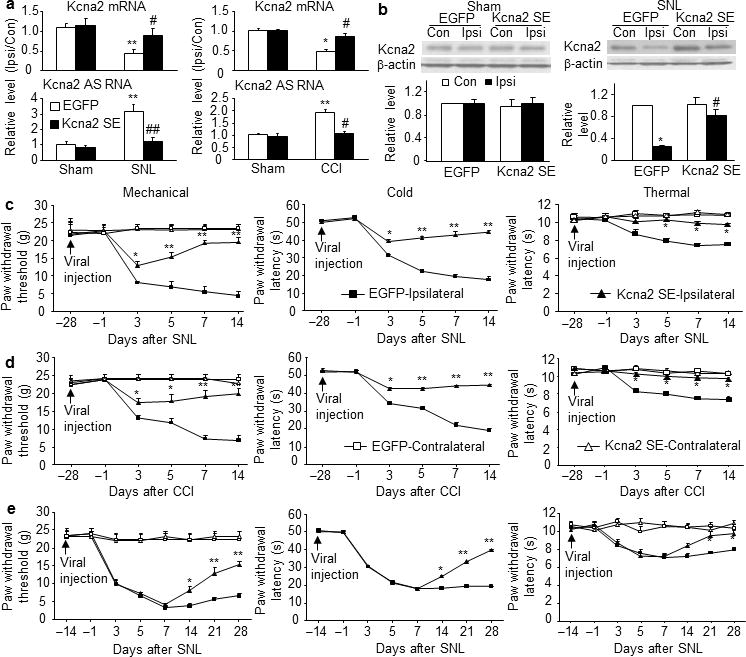
Blocking nerve injury-induced upregulation of DRG Kcna2 antisense RNA attenuates neuropathic pain. (a) Kcna2 antisense RNA and Kcna2 mRNA expression in the ipsilateral (Ipsi) and contralateral (Con) L5 DRGs on day 14 after SNL (F=41.03 for antisense RNA and 10.26 for mRNA), CCI, (F=35.91 for antisense RNA and 8.73 for mRNA) or sham surgery in the EGFP-treated and Kcna2 SE-treated groups. n=12 rats/group. **P <0.01 vs the EGFP-treated group after sham surgery. #P<0.05, ##P <0.01 vs the corresponding EGFP-treated group after SNL or CCI. (b) Kcna2 protein expression in the ipsilateral and contralateral L5 DRGs on day 14 after sham surgery or SNL in the EGFP-treated and Kcna2 SE-treated groups. n=8 rats/group. F=9.26 in SNL and 0.53 in sham. *P<0.05 vs corresponding contralateral side of the EGFP-treated group. #P<0.05 vs the corresponding ipsilateral side of the EGFP-treated group. (c, d) Effect of Kcna2 SE on the development of SNL- or CCI-induced pain hypersensitivities. Paw withdrawal responses at the times shown before and after SNL (F=23.25 for mechanical, 545.13 for cold, and 15.31 for thermal) or CCI (F=22.51 for mechanical, 267.42 for cold, and 12.45 for thermal). n=8 rats/group. *P<0.05, **P<0.01 vs the ipsilateral side of the EGFP-treated group at the corresponding time point. (e) Effect of Kcna2 SE on the maintenance of SNL-induced pain hypersensitivities. Paw withdrawal responses at the times shown before and after SNL (F=22.66 for mechanical, 104.16 for cold, and 7.64 for thermal). n=8 rats/group. *P<0.05, **P<0.01 vs the ipsilateral side of the EGFP-treated group at the corresponding time point.
DISCUSSION
Long ncRNAs were recently shown to occur naturally in mammals18,19. They can be transcribed in cis from the opposing DNA strands of the RNA genes at the same genomic locus, or in trans from a locus different from that of the RNA genes25. Rat Kcna2 antisense RNA is more than 2.5 kb and complementary to most of the Kcna2 RNA sequence, strongly suggesting that Kcna2 antisense RNA is a cis-encoded lncRNA. Interestingly, the Kcna2 antisense gene exhibits the same splicing patterns as the Kcna2 RNA gene. Because the splice junctions of the Kcna2 RNA gene are canonical (that is, they follow the GT-AG rule), splicing mechanisms of the Kcna2 antisense gene are unusual and merit additional investigation.
Expression of native Kcna2 antisense RNA, like that of mRNA, can be regulated via transcriptional activation. Nerve injury-induced upregulation of Kcna2 antisense RNA was triggered through DRG MZF1 activation. Whether other transcription factors also trigger activation of Kcna2 antisense gene transcription is unknown. Additionally, the increase in antisense RNA might be caused by increases in RNA stability and/or other epigenetic modification. These possibilities cannot be excluded and will be addressed in our future studies.
Kcna2 antisense RNA functions as a biologically active regulator of Kcna2 mRNA in primary afferent neurons. Normally, Kcna2 antisense RNA is expressed at a low level in a few (mostly medium-sized) DRG neurons, whereas Kcna2 protein is highly expressed in most medium- or large-sized DRG neurons9. Interestingly, injury to the peripheral nerve not only increased Kcna2 antisense RNA expression but also altered its subpopulation distribution pattern to large- and medium-sized neurons in the injured DRG. Conversely, Kcna2 mRNA and protein were correspondingly downregulated in these neurons5–10. This downregulation is likely caused by the increase in Kcna2 antisense RNA, as overexpression of Kcna2 antisense RNA in cultured HEK-293T cells or DRG neurons selectively and specifically inhibits Kcna2 mRNA and protein expression. This effect may be related to the extensive overlap of their complementary regions, including the transcription and translation initiation sites. DRG Kv1 subunits are functional heteromultimers9,26–28. The expression of other Kv1 subunits is unaffected, likely because they lack complementary sequences and the inhibitory effect of Kcna2 antisense RNA occurs before the formation of heteromultimers. However, the fact that Kcna2 downregulation markedly reduced total Kv current density in large and medium DRG neurons indicates that Kcna2 is a key subunit in determining Kv channel function in these neurons. Minimal reduction was observed in small DRG neurons, possibly because Kcna2 is poorly expressed in those neurons9.
We found that selective reduction of Kcna2 expression in DRG by Kcna2 antisense RNA decreased total Kv current, depolarized the resting membrane potential, decreased current threshold for activation of action potentials, and increased the number of action potentials in large and medium DRG neurons. Depolarization of DRG neuronal resting membrane potential by DRG Kcna2 downregulation was also reported previously29,30. Kcna2 antisense RNA did not affect action potential threshold or amplitude in DRG neurons, as these two parameters may be determined predominantly by Na+ channels. Kcna2 knockdown by Kcna2 antisense RNA produced a modest, but insignificant, increase in DRG neuronal membrane input resistances, an observation that is consistent with the fact that membrane input resistance also depends on other Kv members (e.g., Kcna1, Kcna4), hyperpolarization-activated cyclic nucleotide-gated channels31,32, and chloride channels33 expressed on DRG neuronal membrane. In addition, the depolarized resting membrane potential by itself may increase resting potassium conductance31,32, which may counteract Kcna2 deficiency-induced increase in membrane input resistance. The increase in membrane input resistance caused by blocking Kv current in DRG neurons was observed only in the absence of a significant resting membrane potential depolarization34. The fact membrane input resistance is unchanged but that resting membrane potentials are markedly depolarized in DRG neurons has been reported after peripheral nerve injury35.
Nerve injury-induced increases in spontaneous ectopic activity, which have been found primarily in injured myelinated afferents and the corresponding large and medium DRG neuronal bodies36,37, are considered to play a leading role in the genesis of neuropathic pain1,3. Peripheral nerve injury increased Kcna2 antisense RNA mainly in medium and large DRG neurons. Kcna2 antisense RNA-induced depolarization of the resting membrane potential of DRG neurons may render those neurons more prone to hyperexcitability. Indeed, animals that overexpressed Kcna2 antisense RNA exhibited significant hypersensitivities to mechanical and noxious cold stimuli. Substance P and CGRP in the injured myelinated fibers and in large and medium DRG neurons are markedly increased as early as 2 days after nerve injury3,38. It is very likely that the increase in excitability of large and medium DRG neurons drives the release of these neurotransmitters from their primary afferent terminals and leads to spinal central sensitization, which contributes to the development and maintenance of neuropathic pain. This conclusion is supported by the fact that blocking SNL-evoked upregulation of Kcna2 antisense RNA reversed the reduction in DRG Kcna2 and attenuated induction and maintenance of nerve injury-induced mechanical and cold hypersensitivities. It is still a puzzle how blocking SNL-induced downregulation of DRG Kcna2 almost abolishes SNL-induced pain hypersensitivity at the late time points. We think that blocking DRG Kcna2 downregulation causes persistent reduction in DRG excitability that may enhance the decrease in primary afferent transmitter release, resulting in attenuation of spinal central sensitization formation. Persistent reduction in DRG neuronal excitability may also block further SNL-induced changes in the expression of other DRG genes, including transcription factors that govern gene expression. This activity could create positive feedback to further reduce DRG excitability. These potential mechanisms remain to be confirmed. Taken together, our findings suggest that Kcna2 antisense RNA is an endogenous trigger in neuropathic pain development and maintenance. Regulation of Kcna2 channel expression may be a novel potential target for treating neuropathic pain.
In summary, identification of Kcna2 antisense RNA may point to new regulation of Kcna2 channel expression and neuronal excitability, a novel mechanism in neuropathic pain, and potential targets for the development of therapies of this disorder. Because Kcna2 antisense RNA and Kcna2 mRNA and protein are expressed broadly, they may be implicated in other pathological processes. In addition, demonstration of Kcna2 antisense RNA may challenge current molecular methodologies. For example, we cannot use sense probes usually designed as negative controls as they detect endogenous antisense RNAs in in situ hybridization or oligo-dT primers as they reverse both sense and antisense RNAs for RNA reverse transcription. Therefore, our findings not only provide conceptual advances regarding the development of neuropathic pain but also will affect the conduct of research in other fields.
ONLINE METHODS
Animals
Male Sprague-Dawley rats weighing 200–250 g were kept in a standard 12-h light/dark cycle, with water and food pellets available ad libitum. All procedures used were approved by the Animal Care and Use Committee at the Johns Hopkins University and consistent with the ethical guidelines of the National Institutes of Health and the International Association for the Study of Pain. All efforts were made to minimize animal suffering and to reduce the number of animals used. All of the experimenters were blind to treatment condition.
Nerve injury models
L5 spinal nerve ligation (SNL)39–41, chronic constriction injury (CCI)42, and sciatic nerve axotomy39 models of neuropathic pain were carried out as described previously. Sham groups underwent identical procedures but without transection of the respective nerve.
Behavioral analysis
Mechanical, cold, thermal, and locomotor behavioral tests were carried out. Each behavioral test was carried out at 1-h intervals. Paw withdrawal thresholds in response to mechanical stimuli were first measured with the up–down testing paradigm as described previously39. Paw withdrawal latencies to noxious cold (0°C) were then measured with a cold plate, the temperature of which was monitored continuously. Each animal was placed in a Plexiglas chamber on the cold plate, which was set at 0°C. The length of time between the placement of the hind paw on the plate and the animal jumping, with or without paw licking and flinching, was defined as the paw withdrawal latency. Each trial was repeated three times at 10-min intervals for the paw on the ipsilateral side. A cutoff time of 60 s was used to avoid tissue damage. Finally, paw withdrawal latencies to noxious heat were measured with Model 336 Analgesia Meter (IITC Inc./Life Science Instruments, Woodland Hills, CA, USA) as described previously41. Tests of locomotor function, including placing, grasping, and righting reflexes, were performed before and after viral injection according to previously described protocols39,40,43.
Cell line culture and transfection
HEK-293T cells were cultured in Dulbecco’s Modified Eagle’s Medium at 37°C in a humidified incubator with 5% CO2. The plasmids were transfected into the HEK-293T cells with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions.
Dorsal root ganglion (DRG) neuronal culture and AAV5 transduction
Adult male rats were euthanized with isoflurane. DRGs were collected in cold DH10 [90% DMEM/F-12 (Gibco, Grand Island, NY), 10% FBS (JR Scientific, Woodland, CA), 1% penicillin-streptomycin (Quality Biological, Gaithersburg, MD)] and then treated with enzyme solution [3.5 mg/ml dispase, 1.6 mg/ml collagenase type I in HBSS without Ca2+ and Mg2+ (Gibco)] at 37°C. After the centrifugation, dissociated cells were resuspended in DH10 and plated at a density of 1.5–4×105 cells on glass cover slips or in a 6-well plate coated with poly-L-lysine (0.5 mg/ml, Sigma, St. Louis, MO) and laminin (10 μg/ml, Invitrogen). The cells were incubated in 5% CO2 at 37°C. One day later, 1 μl of AAV5 virus (titer ≥ 1×1012) was added to each well. Cells were harvested 4 d later.
Reverse transcription (RT)-PCR, rapid amplification of cDNA ends (RACE), and quantitative RT-PCR
Total RNA was extracted by the Trizol method (Invitrogen) and treated with excess DNase I (New England Biolabs, Ipswich, MA). Highly purified, DNase-treated RNA samples from human DRG were purchased from Clontech Laboratories, Inc. (Mountain View, CA).. Using the Omniscript RT kit (QIAGEN, Valencia, CA) with strand-specific primers, we reverse transcribed single-stranded cDNA from RNA (1 μg). RT and PCR primers were determined from the UCSC genome database (Supplemental Table 2). Template (1 μl) was amplified by PCR with TaKaRa Taq™ DNA polymerase (Clontech Laboratories, Inc.) in 20 μl total reaction volume containing 0.5 μM of PCR primer. PCR amplification consisted of 30 s at 94°C, 20 s at 56°C, and 20 s at 72°C for 35 cycles.
RNA fragments amplified from the rat DRG were extended first by using RT-PCR with strand-specific primers and then by using a RACE Kit (2nd Generation, Roche Diagnostics, Indianapolis, IN). The 5′ RACE was used for amplification of the 5′-end of cDNA according to the manufacturer’s instructions. The 3′ RACE analysis was performed by ligating an adapter to the 3-hydroxyl group of RNA, followed by gene- and adapter-specific amplification. All primers are listed in Supplemental Table 2. PCR products from RT-PCR, 5′ RACE, and 3′ RACE were extracted, purified, and verified by automatic DNA sequencing. All sequences were analyzed and the full-length Kcna2 antisense RNA determined.
For quantitative real-time RT-PCR, three DRGs from 3 individual rats were pooled together to achieve enough RNA. cDNA was prepared as described above. Template (1 μl) was amplified by real-time PCR by using 1 μM of each probe and 0.5 μM of each primer listed in Supplemental Table 2. Each sample was run in quadruplicate in a 20-μl reaction with TaqMan Universal PCR master mix kit (Applied Biosystems, Grand Island, NY). Reactions were performed on 96-well plates in an ABI 7500 Fast real-time PCR system (Applied Biosystems). Ratios of ipsilateral-side mRNA levels to contralateral-side mRNA levels were calculated by using the ΔCt method (2−ΔΔCt) at a threshold of 0.02, as our pilot data indicated that the amplification reactions of the target genes and reference genes have similar PCR efficiency (Supplemental Fig. 7a). All data were normalized to GAPDH, which was demonstrated to be stable after SNL (Supplemental Fig. 7b–d).
For single-cell quantitative RT-PCR, freshly dissociated rat DRG neurons were first prepared as described below. Four hours after plating, small, medium, and large DRG neurons were randomly collected under an inverted microscope fit with a micromanipulator and microinjector. A single living neuron was selected with a glass micropipette, without contamination by other neurons, and placed in a PCR tube with 6 μl of cell lysis buffer (Signosis, Sunnyvale, CA) as described44. After centrifugation, the supernatants were collected. The remaining real-time RT-PCR procedure was carried out as described44 or according to the manufacturer’s instructions with the single-cell real-time RT-PCR assay kit (Signosis).
rAAV5 plasmid constructs and virus production
After RNA was extracted from the DRG, full-length Kcna2 antisense cDNA, full-length MZF1 cDNA, and Kcna2 sense (SE) cDNA fragment (–311 to +40) were amplified by nested RT-PCR (the primers in Supplemental Table 2). The restriction enzyme recognition sites were introduced at the 5′ and 3′ ends of the three fragments. The PCR products were cloned by using the pGEM®-T easy cloning kit (Invitrogen). The positive clones were identified by restriction enzyme analysis (BspEI/NotI) and clone sequencing.
The identified fragments were ligated into the BspEI/NotI sites of the proviral plasmids (University of North Carolina, Chapel Hill) to replace enhanced GFP (EGFP) and the S-D sequence. The resulting four vectors expressed EGFP, Kcna2 antisense RNA, Kcna2 SE RNA, and MZF1 under the control of the cytomegalovirus promoter. rAAV5 viral particles carrying the four cDNAs were produced at the University of North Carolina Vector Core.
Northern blotting
To prepare complementary RNA (cRNA) probes of rat Kcna2 antisense RNA, we constructed the pSC-A plasmid that contained a 0.946 kb DNA template, and identified the sequence using double-strand DNA sequencing. Plasmid construct was linearized by Acc65I and XhoI. Riboprobe was generated from in vitro transcription and labeled with 32P-dUTP.
Northern blot analysis was performed as described previously45. The extracted RNA (10 μg) was separated on a 1.5% agarose/formaldehyde gel, transferred to a BrightStar-plus positively charged nylon membrane, and cross-linked by using UV. After pre-hybridization, the membrane was hybridized overnight at 68°C with 32P-dUTP-labeled cRNA probes for Kcna2 antisense RNA. After the membrane was washed and dried, autoradiography was carried out.
In situ hybridization histochemistry (ISHH)
The ISHH was carried out as described previously with minor modification46,47. Two sets of 20-μm sections were collected from each DRG by grouping every third section. Kcna2 cRNA probe (0.268-kb fragment) and GFP cRNA probe (0.187-kb fragment) were prepared by in vitro transcription and labeled with digoxigenin-dUTP according to the manufacturer’s instructions (Roche Diagnostics, Indianapolis, IN). After treatment with proteinase K and pre-hybridization, the two sets of sections were hybridized with digoxigenin-dUTP-labeled cRNA probes for Kcna2 antisense RNA and GFP RNA for 18 h at 68°C. After being washed, the sections were incubated with anti-digoxigenin–conjugated alkaline phosphatase. The signals were developed with 5-Bromo-4-chloro-3′-indolyl phosphate p-toluidine salt and Nitro-blue tetrazolium chloride substrates. For the double labeling of ISHH and immunohistochemistry, the sections were treated as described above except that they were hybridized only with digoxigenin-dUTP–labeled cRNA probe for Kcna2 antisense RNA, and the fluorescent signals were developed with Fast Red.
Immunohistochemistry
After being blocked for 1 h at 37°C in PBS containing 10% goat serum and 0.3% Triton X-100, the sections were incubated with rabbit anti-NF200 (1:500, Sigma-Aldrich, St. Louis, MO)48, rabbit anti-P2X3 (1:500, Neuromics, Edina, MN)48, Biotinylated IB4 (1:100, Sigma)48, rabbit anti-CGRP (1:500, EMD, Billerica, MA)48, mouse anti-NeuN (1:600, EMD)48, mouse anti-GFAP (1:500, Sigma)48, or mouse anti-OX-42 (1:400, Sigma)48 overnight at 4°C. The sections were then incubated with goat anti-rabbit IgG conjugated with Cy2 (1:400, Jackson ImmunoResearch, West Grove, PA) or Cy3 (1:400, Jackson ImmunoResearch) or with FITC-labeled avidin D (1:200, Sigma) for 2 h at room temperature. Control experiments included substitution of normal mouse serum for the primary antiserum and omission of the primary antiserum. All immunofluorescence-labeled images were examined under a Nikon TE2000E fluorescence microscope (Nikon Co., Japan) and captured with a CCD spot camera. Single- and double-labeled neurons were counted by using stereological methods as described48.
Western blotting
For DRG, three DRGs from 3 individual rats were pooled together to achieve enough protein. The tissues were homogenized and the cultured cells ultrasonicated in chilled lysis buffer (50 mM Tris, 1 mM phenylmethylsulfonyl fluoride, 1 mM EDTA, 1 μM leupeptin). After the centrifugation at 4°C for 15 min at 1,000g, the supernatant was collected for cytosolic and membrane proteins and the pellet for nuclear proteins. After protein concentration was measured, the samples were heated at 99°C for 5 min and loaded onto a 4% stacking/7.5% separating SDS-polyacrylamide gel (Bio-Rad Laboratories, Hercules, CA). The proteins were then electrophoretically transferred onto a polyvinylidene difluoride membrane (Immobilon-P, Millipore, Billerica, MA). According to the targeted protein molecular weights, the membranes were cut into several small strips and then blocked with 3% nonfat milk in Tris-buffered saline containing 0.1% Tween-20 for 1 h. The following primary antibodies were used: mouse anti-Kcna1 (1:200, NeuroMab, Davis, CA)9, mouse anti-Kcna2 (1:200, NeuroMab)9, mouse anti-Kcna4 (1:1,000, Cell Signaling Technology, Danvers, MA)48, rabbit anti-PKCα (1:500, Santa Cruz Biotechnology, Santa Cruz, CA)40, mouse anti-Scn10a (1:1,000; NeuroMab)50, rabbit anti-MZF1 (1:200, provided by Dr. D.Y.H. Tuan, Medical College of Georgia)21, mouse anti-β-actin (1:2,000; Santa-Cruz Biotechnology), and rabbit anti-histone H3 (1:1,000, Cell Signaling Technology). The proteins were detected by horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary antibody and visualized by chemiluminescence regents (ECL; Amersham Pharmacia Biotech, Piscataway, NJ) and exposure to film. The intensity of blots was quantified with densitometry.
Electrophoretic mobility shift assay (EMSA)
32P-labeled double-stranded DNA probe was prepared by annealing synthetic oligonucleotide in H-Star polymerase PCR solution containing P32-dCTP, dATP, and dTTP at 56°C for 1 min and then at 72°C for 30 min. Unlabeled probe and unlabeled mutant probe were similarly prepared for use as competitors. Their oligonucleotide sequences are shown in Supplemental Table 2. All probes were purified in a G50 column (GE Healthcare, Silver Spring, MD). DRG nuclear extract (5 μg) was incubated with labeled probe (9 ng) alone or with 50× unlabeled probe or 50× unlabeled mutant probe at 25°C for 20 min. Labeled probe alone was used as a control. After incubation, the DNA-protein complexes were subjected to polyacrylamide gel electrophoresis. Autoradiography was carried out after the gel was dried. For supershift EMSA, the nuclear extracts were pre-incubated with rabbit MZF1 antibody (2 μg) at 25°C for 30 min prior to EMSA.
Chromatin immunoprecipitation (ChIP) assay
The homogenization solution from the DRG was cross-linked with 1% formaldehyde at 37°C for 5 min and the reaction terminated by the addition of 0.25 M glycine. After centrifugation, the pellet was collected, washed, and suspended in lysis buffer containing 0.1% SDS, 1% sodium deoxycholate, and 1% NP-40 in the presence of protease inhibitors. The suspension was sonicated with an ultrasonic cell disruptor (Misonix Inc., Farmingdale, NY) to shear chromatin and produce 0.2- to 1-kb DNA fragments. After the samples were pre-cleared with protein G agarose, they were immunoprecipitated with 5 μg of rabbit anti-MZF1 antibody, normal rabbit serum (5 μg), or rabbit anti-MZF1 antibody (5 μg) after pre-absorption with excess MZF-1 fusion protein (10 μg). Input (10% of the sample for immunoprecipitation) was used as a positive control. The fragment (156 nt) of Kcna2 antisense gene promoter containing the predicted MZF1 binding site was detected by PCR. All primers used are listed in Supplemental Table 2.
Luciferase assay
To construct the Kcna2 gene and Kcna2 antisense gene reporter plasmids, we amplified the 1,268-bp fragment from the Kcna2 gene promoter region and the 633-bp fragment from the Kcna2 antisense gene promoter (including MZF1-binding motif) by PCR from genomic DNA. The PCR products were subcloned into the SmaI and HindIII restriction sites of the pGL3-Basic vector (Promega, Madison, WI). The sequences of recombinant clones were verified by DNA sequencing. All primer sequences are shown in Supplemental Table 2.
HEK-293T cells were prepared as described above. After 24 h of culture, the cells were transfected with 40 ng of the pRL-TK plasmid (a normalizing control; Promega, Madison, WI) alone or plus 1 μg of the constructed plasmids using Lipofectamine 2000 (Invitrogen). After an additional 48 h of culture, the transfected cells were lysed with passive lysis buffer, and 40 μl of supernatant was assayed for luciferase activity with the Dual-Luciferase Reporter Assay System (Promega). The relative reporter activity was obtained by normalization of the firefly activity to renilla activity. Three independent transfection experiments were performed.
Whole-cell patch clamp recording
To record total potassium current in DRG neurons, we first prepared freshly dissociated rat DRG neurons as described above. Whole-cell patch clamp recording was carried out 4 to 24 h after plating. Cover slips were placed into the perfusion chamber (Warner Instruments, Hamden, CT). Only green-labeled neurons were recorded. The electrode resistances of micropipettes ranged from 2 to 4 MΩ. Cells were voltage-clamped with an Axopatch-700B amplifier (Molecular Devices, Sunnyvale, CA). The intracellular pipette solution contained (in mM): potassium gluconate 120, KCl 20, MgCl2 2, EGTA 10, HEPES 10, Mg-ATP 4 (pH = 7.3 with KOH, 310 mOsm). We minimized the Na+ and Ca2+ component in Kv current recording by using an extracellular solution composed of (in mM): choline-Cl 150, KCl 5, CdCl2 1, CaCl2 2, MgCl2 1, HEPES 10, glucose 10 (pH = 7.4 with Tris-base, 320 mOsm). Signals were filtered at 1 kHz and digitized by using a DigiData 1322A with pClamp 9.2 software (Molecular Devices). Series resistance was compensated by 60–80%. Cell membrane capacitances were acquired by reading the value for whole-cell capacitance compensation directly from the amplifier. An online P/4 leak subtraction was performed to eliminate leak current contribution. The data were stored on computer by a DigiData 1322A interface and were analyzed by the pCLAMP 9.2 software package (Molecular Devices).
To record the action potential, we switched the recording mode into current clamp. Cover slips were placed in the chamber and perfused with extracellular solution consisting of (in mM): NaCl 140, KCl 4, CaCl2 2, MgCl2 2, HEPES 10, and glucose 5, with pH adjusted to 7.38 by NaOH. The intracellular pipette solution contained (in mM): KCl 135, MgATP 3, Na2ATP 0.5, CaCl2 1.1, EGTA 2, and glucose 5; pH was adjusted to 7.38 with KOH and osmolarity adjusted to 300 mOsm with sucrose. The resting membrane potential was taken 3 min after a stable recording was first obtained. Depolarizing currents of 100–1400 pA (200-ms duration) were delivered in increments of 100 pA until an action potential (AP) was evoked. The injection current threshold was defined as the minimum current required to evoke the first AP. The membrane potential was held at the existing resting membrane potential during the current injection. The AP threshold was defined as the first point on the rapid rising phase of the spike at which the change in voltage exceeded 50 mV/ms. The AP amplitude was measured between the peak and the baseline. The membrane input resistance for each cell was obtained from the slope of a steady-state I–V plot in response to a series of hyperpolarizing currents, 200-ms duration delivered in steps of 100 pA from 200 pA to −2,000 pA. The afterhyperpolarization amplitude was measured between the maximum hyperpolarization and the final plateau voltage, and the AP overshoot was measured between the AP peak and 0 mV. The data were stored on computer by a DigiData 1322A interface and were analyzed by the pCLAMP 9.2 software package (Molecular Devices). All experiments were performed at room temperature (~25°C).
DRG microinjection
DRG microinjection was carried out as described51,52. Briefly, a midline incision was made in the lower lumbar back region, and the L5 vertebral body was exposed. After the lamina was removed and the DRG exposed, viral solution (2 μl) was injected into two sites in the L4 and L5 DRGs or into one site in the L5 DRG with a glass micropipette connected to a Hamilton syringe. The pipette was removed after 10 min. After injection, the skin incision closed with wound clips. The injected rats showed no signs of paresis or other abnormalities. The injected DRGs, stained with hematoxylin/eosin, retained their structural integrity and contained no visible leukocytes. The immune responses from viral injection were therefore minimal.
Statistical analysis
For in vitro experiments, the cells were evenly suspended and then randomly distributed in each well tested. For in vivo experiments, the animals were distributed into various treated groups randomly. All of the results are given as means ± SEM. Data distribution was assumed to be normal but this was not formally tested. The data were statistically analyzed with two-tailed, paired/unpaired Student’s t test and a one-way or two-way ANOVA. When ANOVA showed significant difference, pairwise comparisons between means were tested by the post hoc Tukey method (SigmaStat, San Jose, CA). No statistical methods were used to pre-determine sample sizes, but our sample sizes are similar to those reported previously in the fields.39–41 Significance was set at p < 0.05.
Supplementary Material
Supplementary Figure 1. Expression of Kcna2 antisense RNA in tissues and its whole sequence in rats.
Supplementary Figure 2. Expression of Kcna2 mRNA, Kcna2 protein, or Kcna2 antisense RNA in L4 DRG, L5 DRG, or L5 dorsal horn after peripheral nerve injury.
Supplementary Figure 3. MZF1 expression in Kcna2 antisense RNA-containing DRG neurons and MZF1 knockdown by its siRNA in HEK293T cells.
Supplementary Figure 4. EGFP expression is limited in L5 DRG and its fibers and terminals on the ipsilateral side 8 weeks after AAV5-EGFP injection into the left L5 DRG.
Supplementary Figure 5. Kcna2 antisense RNA overexpression in DRG reduces total Kv current density in medium, but not small, DRG neurons 8–12 weeks post-viral injection.
Supplementary Figure 6. Inhibition of Kcna2 function increases neuronal excitability in rat large and medium DRG neurons.
Supplementary Figure 7. SNL does not alter expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA in the injured DRG.
Supplementary Figure 8. Full-length pictures of the blots in the main figures
Supplementary Table 1. Effect of Kcna2 antisense RNA or MTX on membrane input resistance and other action potential parameters in the DRG
Supplementary Table 2. All primers and probes used
Acknowledgments
We thank Dr. R. Jude Samulski for providing the rAAV5 plasmid, Dr. Jiawei Zhou for providing the full-length EGFP-MZF1 plasmid, and Dr. Dorothy Y.H. Tuan for providing MZF1 antibody. This work was supported by NIH grants (NS054791 and GM087369) to X.D.; by NIH grants (MH084691 and GM078579) to M.L; by NIH grants (DA033390, NS072206) and the Rita Allen Foundation to Y.X.T. X.D. is an Early Career Scientist of the Howard Hughes Medical Institute. The authors thank Claire F. Levine, MS, for her editorial assistance.
Footnotes
AUTHOR CONTRIBUTIONS
Y.X.T. conceived the project and supervised most experiments. X.Z., Z.T., H.Z., F.E.A., J.Z., Y.J.G., H.C., M.L., X.D., and Y.X.T. designed the project. X.Z., F.E.A., J.Z., L.L, W.W., X.G., S.C.K., and V.T. performed molecular, biochemical, and behavioral experiments. Z.T. and X.D. performed current clamp experiments. H.Z. and M.L. carried out voltage clamp experiments. P.N.H. did microinjection. X.Z., Z.T., H.Z., F.E.A., J.Z. L.L., W.W., X.G., V.T., and Y.X.T. analyzed the data. Y.X.T. wrote the manuscript. All of the authors read and discussed the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Campbell JN, Meyer RA. Mechanisms of neuropathic pain. Neuron. 2006;52:77–92. doi: 10.1016/j.neuron.2006.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chung JM, Chung K. Importance of hyperexcitability of DRG neurons in neuropathic pain. Pain Pract. 2002;2:87–97. doi: 10.1046/j.1533-2500.2002.02011.x. [DOI] [PubMed] [Google Scholar]
- 3.Devor M. Ectopic discharge in Abeta afferents as a source of neuropathic pain. Exp Brain Res. 2009;196:115–128. doi: 10.1007/s00221-009-1724-6. [DOI] [PubMed] [Google Scholar]
- 4.Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. 2009;32:1–32. doi: 10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Everill B, Kocsis JD. Nerve growth factor maintains potassium conductance after nerve injury in adult cutaneous afferent dorsal root ganglion neurons. Neuroscience. 2000;100:417–422. doi: 10.1016/s0306-4522(00)00263-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ishikawa K, Tanaka M, Black JA, Waxman SG. Changes in expression of voltage-gated potassium channels in dorsal root ganglion neurons following axotomy. Muscle Nerve. 1999;22:502–507. doi: 10.1002/(sici)1097-4598(199904)22:4<502::aid-mus12>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 7.Kim DS, Choi JO, Rim HD, Cho HJ. Downregulation of voltage-gated potassium channel alpha gene expression in dorsal root ganglia following chronic constriction injury of the rat sciatic nerve. Brain Res Mol Brain Res. 2002;105:146–152. doi: 10.1016/s0169-328x(02)00388-1. [DOI] [PubMed] [Google Scholar]
- 8.Park SY, et al. Downregulation of voltage-gated potassium channel alpha gene expression by axotomy and neurotrophins in rat dorsal root ganglia. Mol Cells. 2003;16:256–259. [PubMed] [Google Scholar]
- 9.Rasband MN, et al. Distinct potassium channels on pain-sensing neurons. Proc Natl Acad Sci U S A. 2001;98:13373–13378. doi: 10.1073/pnas.231376298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang EK, Takimoto K, Hayashi Y, de Groat WC, Yoshimura N. Altered expression of potassium channel subunit mRNA and alpha-dendrotoxin sensitivity of potassium currents in rat dorsal root ganglion neurons after axotomy. Neuroscience. 2004;123:867–874. doi: 10.1016/j.neuroscience.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 11.Chabal C, Jacobson L, Russell LC, Burchiel KJ. Pain responses to perineuromal injection of normal saline, gallamine, and lidocaine in humans. Pain. 1989;36:321–325. doi: 10.1016/0304-3959(89)90091-2. [DOI] [PubMed] [Google Scholar]
- 12.Chien LY, Cheng JK, Chu D, Cheng CF, Tsaur ML. Reduced expression of Atype potassium channels in primary sensory neurons induces mechanical hypersensitivity. J Neurosci. 2007;27:9855–9865. doi: 10.1523/JNEUROSCI.0604-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Devor M. Potassium channels moderate ectopic excitability of nerve-end neuromas in rats. Neurosci Lett. 1983;40:181–186. doi: 10.1016/0304-3940(83)90299-9. [DOI] [PubMed] [Google Scholar]
- 14.Targ EF, Kocsis JD. Action potential characteristics of demyelinated rat sciatic nerve following application of 4-aminopyridine. Brain Res. 1986;363:1–9. doi: 10.1016/0006-8993(86)90652-9. [DOI] [PubMed] [Google Scholar]
- 15.Costa FF. Non-coding RNAs, epigenetics and complexity. Gene. 2008;410:9–17. doi: 10.1016/j.gene.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 16.Faghihi MA, Wahlestedt C. Regulatory roles of natural antisense transcripts. Nat Rev Mol Cell Biol. 2009;10:637–643. doi: 10.1038/nrm2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 18.Gibb EA, Brown CJ, Lam WL. The functional role of long non-coding RNA in human carcinomas. Mol Cancer. 2011;10:38. doi: 10.1186/1476-4598-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21:354–361. doi: 10.1016/j.tcb.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 20.Hsieh YH, et al. PKCalpha expression regulated by Elk-1 and MZF-1 in human HCC cells. Biochem Biophys Res Commun. 2006;339:217–225. doi: 10.1016/j.bbrc.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 21.Luo X, Zhang X, Shao W, Yin Y, Zhou J. Crucial roles of MZF-1 in the transcriptional regulation of apomorphine-induced modulation of FGF-2 expression in astrocytic cultures. J Neurochem. 2009;108:952–961. doi: 10.1111/j.1471-4159.2008.05845.x. [DOI] [PubMed] [Google Scholar]
- 22.Fulton S, et al. Contribution of Kv1.2 voltage-gated potassium channel to D2 autoreceptor regulation of axonal dopamine overflow. J Biol Chem. 2011;286:9360–9372. doi: 10.1074/jbc.M110.153262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castle NA, et al. Maurotoxin: a potent inhibitor of intermediate conductance Ca2+-activated potassium channels. Mol Pharmacol. 2003;63:409–418. doi: 10.1124/mol.63.2.409. [DOI] [PubMed] [Google Scholar]
- 24.Visan V, Fajloun Z, Sabatier JM, Grissmer S. Mapping of maurotoxin binding sites on hKv1.2, hKv1.3, and hIKCa1 channels. Mol Pharmacol. 2004;66:1103–1112. doi: 10.1124/mol.104.002774. [DOI] [PubMed] [Google Scholar]
- 25.Lavorgna G, et al. In search of antisense. Trends Biochem Sci. 2004;29:88–94. doi: 10.1016/j.tibs.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 26.Rehm H, Tempel BL. Voltage-gated K+ channels of the mammalian brain. FASEB J. 1991;5:164–170. doi: 10.1096/fasebj.5.2.2004663. [DOI] [PubMed] [Google Scholar]
- 27.Robbins CA, Tempel BL. Kv1.1 and Kv1.2: similar channels, different seizure models. Epilepsia. 2012;53 (Suppl 1):134–141. doi: 10.1111/j.1528-1167.2012.03484.x. [DOI] [PubMed] [Google Scholar]
- 28.Vacher H, Trimmer JS. Diverse roles for auxiliary subunits in phosphorylation-dependent regulation of mammalian brain voltage-gated potassium channels. Pflugers Arch. 2011;462:631–643. doi: 10.1007/s00424-011-1004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dodson PD, Barker MC, Forsythe ID. Two heteromeric Kv1 potassium channels differentially regulate action potential firing. J Neurosci. 2002;22:6953–6961. doi: 10.1523/JNEUROSCI.22-16-06953.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo JL, et al. Enhanced excitability and down-regulated voltage-gated potassium channels in colonic drg neurons from neonatal maternal separation rats. J Pain. 2011;12:600–609. doi: 10.1016/j.jpain.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 31.Cardenas CG, et al. Serotonergic modulation of hyperpolarization-activated current in acutely isolated rat dorsal root ganglion neurons. J Physiol. 1999;518 (Pt 2):507–523. doi: 10.1111/j.1469-7793.1999.0507p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng Q, et al. Enhanced excitability of small dorsal root ganglion neurons in rats with bone cancer pain. Mol Pain. 2012;8:24. doi: 10.1186/1744-8069-8-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rinke I, Artmann J, Stein V. ClC-2 voltage-gated channels constitute part of the background conductance and assist chloride extrusion. J Neurosci. 2010;30:4776–4786. doi: 10.1523/JNEUROSCI.6299-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicol GD, Vasko MR, Evans AR. Prostaglandins suppress an outward potassium current in embryonic rat sensory neurons. J Neurophysiol. 1997;77:167–176. doi: 10.1152/jn.1997.77.1.167. [DOI] [PubMed] [Google Scholar]
- 35.Kim YI, et al. Cell type-specific changes of the membrane properties of peripherally-axotomized dorsal root ganglion neurons in a rat model of neuropathic pain. Neuroscience. 1998;86:301–309. doi: 10.1016/s0306-4522(98)00022-0. [DOI] [PubMed] [Google Scholar]
- 36.Liu CN, et al. Tactile allodynia in the absence of C-fiber activation: altered firing properties of DRG neurons following spinal nerve injury. Pain. 2000;85:503–521. doi: 10.1016/S0304-3959(00)00251-7. [DOI] [PubMed] [Google Scholar]
- 37.Tal M, Wall PD, Devor M. Myelinated afferent fiber types that become spontaneously active and mechanosensitive following nerve transection in the rat. Brain Res. 1999;824:218–223. doi: 10.1016/s0006-8993(99)01190-7. [DOI] [PubMed] [Google Scholar]
- 38.Weissner W, Winterson BJ, Stuart-Tilley A, Devor M, Bove GM. Time course of substance P expression in dorsal root ganglia following complete spinal nerve transection. J Comp Neurol. 2006;497:78–87. doi: 10.1002/cne.20981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guan X, Zhu X, Tao YX. Peripheral nerve injury up-regulates expression of interactor protein for cytohesin exchange factor 1 (IPCEF1) mRNA in rat dorsal root ganglion. Naunyn Schmiedebergs Arch Pharmacol. 2009;380:459–463. doi: 10.1007/s00210-009-0451-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh OV, et al. Proteome of synaptosome-associated proteins in spinal cord dorsal horn after peripheral nerve injury. Proteomics. 2009;9:1241–1253. doi: 10.1002/pmic.200800636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang B, et al. Effect of knock down of spinal cord PSD-93/chapsin-110 on persistent pain induced by complete Freund’s adjuvant and peripheral nerve injury. Pain. 2003;106:187–196. doi: 10.1016/j.pain.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 42.Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 43.Park JS, et al. Persistent inflammation induces GluR2 internalization via NMDA receptor-triggered PKC activation in dorsal horn neurons. J Neurosci. 2009;29:3206–3219. doi: 10.1523/JNEUROSCI.4514-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Esumi S, Kaneko R, Kawamura Y, Yagi T. Split single-cell RT-PCR analysis of Purkinje cells. Nat Protoc. 2006;1:2143–2151. doi: 10.1038/nprot.2006.343. [DOI] [PubMed] [Google Scholar]
- 45.Gallagher PE, Diz DI. Analysis of RNA by northern-blot hybridization. Methods Mol Med. 2001;51:205–213. doi: 10.1385/1-59259-087-X:205. [DOI] [PubMed] [Google Scholar]
- 46.Dong X, Han S, Zylka MJ, Simon MI, Anderson DJ. A diverse family of GPCRs expressed in specific subsets of nociceptive sensory neurons. Cell. 2001;106:619–632. doi: 10.1016/s0092-8674(01)00483-4. [DOI] [PubMed] [Google Scholar]
- 47.Nehme B, Henry M, Mouginot D. Combined fluorescent in situ hybridization and immunofluorescence: limiting factors and a substitution strategy for slide-mounted tissue sections. J Neurosci Methods. 2011;196:281–288. doi: 10.1016/j.jneumeth.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 48.Xu JT, et al. Expression and distribution of mTOR, p70S6K, 4E-BP1, and their phosphorylated counterparts in rat dorsal root ganglion and spinal cord dorsal horn. Brain Res. 2010;1336:46–57. doi: 10.1016/j.brainres.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coggeshall RE, Lekan HA. Methods for determining numbers of cells and synapses: a case for more uniform standards of review. J Comp Neurol. 1996;364:6–15. doi: 10.1002/(SICI)1096-9861(19960101)364:1<6::AID-CNE2>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 50.Liang L, et al. Protein kinase B/Akt is required for complete Freund’s adjuvant-induced upregulation of Nav1.7 and Nav1.8 in primary sensory neurons. J Pain. 2013;14:638–647. doi: 10.1016/j.jpain.2013.01.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Han PJ, Shukla S, Subramanian PS, Hoffman PN. Cyclic AMP elevates tubulin expression without increasing intrinsic axon growth capacity. Exp Neurol. 2004;189:293–302. doi: 10.1016/j.expneurol.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 52.Xu Y, et al. Adeno-associated viral transfer of opioid receptor gene to primary sensory neurons: a strategy to increase opioid antinociception. Proc Natl Acad Sci U S A. 2003;100:6204–6209. doi: 10.1073/pnas.0930324100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Expression of Kcna2 antisense RNA in tissues and its whole sequence in rats.
Supplementary Figure 2. Expression of Kcna2 mRNA, Kcna2 protein, or Kcna2 antisense RNA in L4 DRG, L5 DRG, or L5 dorsal horn after peripheral nerve injury.
Supplementary Figure 3. MZF1 expression in Kcna2 antisense RNA-containing DRG neurons and MZF1 knockdown by its siRNA in HEK293T cells.
Supplementary Figure 4. EGFP expression is limited in L5 DRG and its fibers and terminals on the ipsilateral side 8 weeks after AAV5-EGFP injection into the left L5 DRG.
Supplementary Figure 5. Kcna2 antisense RNA overexpression in DRG reduces total Kv current density in medium, but not small, DRG neurons 8–12 weeks post-viral injection.
Supplementary Figure 6. Inhibition of Kcna2 function increases neuronal excitability in rat large and medium DRG neurons.
Supplementary Figure 7. SNL does not alter expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA in the injured DRG.
Supplementary Figure 8. Full-length pictures of the blots in the main figures
Supplementary Table 1. Effect of Kcna2 antisense RNA or MTX on membrane input resistance and other action potential parameters in the DRG
Supplementary Table 2. All primers and probes used