

Abstract
To assess the statistical relationship between tumor growth and copper metabolism, we performed a metaanalysis of studies in which patients with neoplasms were characterized according to any of the copper status indexes (atomic copper serum concentration, serum oxidase activity, ceruloplasmin protein content). Our metaanalysis shows that in the majority of cases (more than 3100 patients), tumor growth positively correlates with the copper status indexes. Nude athymic CD-1 nu/nu mice with subcutaneous tumors of human origin, C57Bl/6J mice with murine melanoma and ApcMin mice with spontaneously developing adenomas throughout the intestinal tract were studied to experimentally determine the relationship between tumor progression, liver copper metabolism, and copper status indexes. We showed that the copper status indexes increased significantly during tumor growth. In the liver tissue of tumor-bearing mice, ceruloplasmin gene expression, as well as the expression of genes related to ceruloplasmin metallation (CTR1 and ATP7B), increased significantly. Moreover, the presence of an mRNA splice variant encoding a form of ceruloplasmin anchored to the plasma membrane by glycosylphosphatidyl inositol, which is atypical for hepatocytes, was also detected. The ATP7A copper transporter gene, which is normally expressed in the liver only during embryonic copper metabolism, was also activated. Depletion of holo-ceruloplasmin resulted in retardation of human HCT116 colon carcinoma cell growth in nude mice and induced DNA fragmentation in tumor cells. In addition, the concentration of cytochrome c increased significantly in the cytosol, while decreasing in the mitochondria. We discuss a possible trans-effect of developing tumors on copper metabolism in the liver.
Keywords: copper status indexes, tumor progression, liver copper metabolism, copper deficiency, animal models, gene expression, metaanalysis
Introduction
Copper is an essential nutrient that serves as a structural and catalytic cofactor for cuproenzymes. These enzymes utilize the unique property of copper to coordinate and activate dioxygen. Copper ions can also act as toxic agents and can induce the formation of reactive oxygen species (ROS), which are capable of provoking carcinogenesis.1 Normally copper is tightly coordinated by the active sites of cuproenzymes in both redox states (Cu(I)↔Cu(II)), and it cannot easily leave its coordination sphere.2 Copper from the environment is delivered to cuproenzymes by the dedicated transport system.3,4 The members of this system contain high affinity Cu(I)-binding sites. They transfer Cu(I) to one another down the free energy gradient until it reaches the apo-cuproenzymes. The system guarantees that no “free” copper ions are present in cells.5 Disturbances in the copper transportation system resulting in the accumulation of uncoordinated copper might cause malignant transformation (Wilson disease,6-9 genetic copies of Wilson disease in rats,10,11 and mice12). Increasing evidence indicates that abnormalities in copper metabolism are also linked to tumor progression, invasion and metastasis.1,13-16
The physiological role of copper is not limited to its function as a redox cofactor, and other copper-dependent processes have been described. First, neovascularization is dependent on the copper-mediated activation of vascular endothelial growth factor (VEGF), which acts through a specific VEGF receptor (VEGFR).17-19 The activity of this system was also conclusively shown to be associated with invasion and migration of tumor cells.20-22 Second, the levels of the copper-dependent intracellular lysyl oxidase-like proteins (LOXLs, LOXL1–4) have been reported to be upregulated in various tumors compared with their normal or less aggressive neoplastic counterparts, where LOXLs acted as promoters of motility/migration.23,24 The suppression of LOXLs was shown to impede tumor growth.25,26 Third, copper also participates in signaling pathways. So, the interaction between XIAP (X-linked inhibitor of apoptosis protein) and the caspases weakens, when intracellular copper level increases, leading to induction of apoptosis.27,28 In addition, copper is required for signaling in pathways regulated by receptor tyrosine kinases that play major roles in development and cancer.16 Finally, copper stabilizes the HIF1-α (hypoxia inducible factor 1) transcription factor,29,30 which controls the activity of genes responsible for reprogramming energy metabolism during neoplastic transformation known as the Warburg effect.31 Most of the listed processes are advantageous to tumor cells, so that copper deficiency might be viewed as an anti-oncofactor.
Tumors have elevated copper requirements, as newly generated cells need cuproenzymes and copper-dependent proteins, mentioned above. For most cells of mammals bloodstream is the main source of copper. In human, 95% of this copper is bound to ceruloplasmin (Cp, the blue multicopper (ferr)oxidase),32 which is synthesized in the liver. In Cp, 6 tightly bound copper atoms have catalytic functions, but there are also 1–3 loosely bound atoms;33 therefore, Cp is both an enzyme and a copper transporter. Copper pool of the bloodstream is usually characterized by a set of indicator variables known as copper status indexes (CSIs). CSIs include atomic copper concentration, Cp oxidase activity and immunoreactive Cp content, which are usually correlated with one another.34 Intensive consumption of copper by a tumor should cause the depletion of the copper pool in the bloodstream. However, an increase in bloodstream copper levels has been observed in patients with tumors.35,36 This effect is favorable to the tumor and might be a target of antitumor strategies. The intuitive attempts have been made to use approved copper-lowering agents (D-penicillamine, tetrathiomolybdate, trientine) to retard tumor growth;37-40 these attempts achieved some success.
Thus, the link between copper metabolism and carcinogenesis is well documented. But the exact biochemical nature of this link and the origin of increase of CSIs in case of neoplastic disorders remain unclear, which prevents their rational use in practice. The aims of the present work were to perform a metaanalysis of the published data to estimate the representativeness and significance of the observation that human CSIs increase with tumor growth, to study copper metabolism in the livers of model mice with growing non-hepatic tumors of various origins and to trace the influence of depletion of holo-Cp (the major marker of copper status) on tumor growth.
Results
Metaanalysis of literature references on the relationship between copper status indexes and cancer
Our initial search of the NCBI PubMed database retrieved 249 literature references dating from 1962. Applying our study selection criteria narrowed the list to 51 references dating from 1976. At least 3111 cases of tumors were reported in these studies. Four studies focused on hepatocellular carcinoma (in which the tumor is located directly in the organ controlling copper homeostasis for the entire body). The other studies covered a wide range of non-hepatic neoplastic disorders with various origins and localizations. Atomic copper concentration, Cp protein concentration, and Cp activity in the blood serum were assessed in 42, 21, and 24 studies, respectively. The vast majority of the studies reported a significant increase in CSIs in tumor patients (Fig. 1). In 1 paper, differential effects were observed for two types of cancer (increased CSIs in stomach cancer and no change in colon cancer). In one paper, the CSIs did not change. It should be noted that the latter paper dealt with mucous fibrosis caused by chronic exposure of the mucous membranes to elevated copper concentrations in areca nut chewers.41 Therefore this work differs from the general set. The remaining reference did not make any statistical inferences regarding the change of CSIs. In 6 studies, elevated CSIs were reported to decrease after successful therapy. In one study, CSIs were elevated in cancer patients but did not decrease with therapy. Twenty-six studies stated that at least one of the CSI parameters had diagnostic and prognostic value for tumor detection and/or treatment. Although they observed a significant increase in CSIs associated with tumor growth, 6 papers considered these indexes to be of poor diagnostic value because of either low sensitivity or selectivity. Of the 37 papers in which both copper and Cp concentration or activity were measured, four found that the [Cu]/[Cp] serum ratio increased in the tumor cases, six stated that the [Cu]/[Cp] ratio did not change significantly, and the others did not focus on this question. No relationship was observed between these inferences and tumor type. It was noted in several studies that changes in CSIs induced by tumor growth were more distinct and had better diagnostic value in males. This observation may be because CSIs in females are affected by the estrogen cycle or contraceptive drugs, which also increase Cp levels.42,43
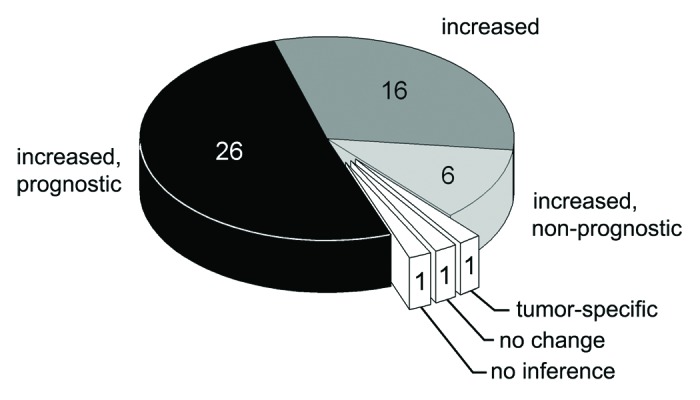
Figure 1. Metaanalysis of changes in CSI levels in adult humans with neoplastic diseases and the prognostic value of CSI values. The number of articles is indicated.
The results of the metaanalysis clearly show that there is a link between tumor growth and elevated CSI values. However, a causal link between them cannot be unequivocally established, because the data we analyzed were obtained from patients who had already developed tumors and in whom progressive tumor growth was already underway. We devised two hypotheses that can explain these results. First, Cp levels could be increased because Cp is an acute phase protein. In the case of late-stage tumors, Cp may be induced by the acute response. This possibility was mentioned in the analyzed literature, though no data to support it was presented. The other possibility is that the increase of CSIs occurs in response to the increased Cp requirements of the growing tumor. In this case, the increase of CSIs should precede active tumor progression. This hypothesis was tested in the experimental portion of this work.
CSIs changes in mice with growing tumors
Three models of laboratory mice with growing non-hepatic tumors were utilized: Min mice with spontaneously developing adenomas of the intestine (autologous tumor), nude mice bearing xenotransplanted human colon tumors (heterologous tumor), and mice with transplanted murine melanoma (homologous tumor). CSI values were measured in the sera of these animals and compared with those in the 3 respective control groups of animals without tumors. The data are presented in Figure 2. These experiments show that in nude mice and in C57Bl/6J mice with growing tumors, holo-Cp enzyme activity and Cp protein content increased approximately 3-fold compared with baseline (Fig. 2A and B). Concurrently, copper concentrations increased 2-fold (Fig. 2C). Holo-Cp accounts for ~60% of the serum copper in mice.44,45 Therefore, increased serum copper concentrations are likely to be caused by an increase in the Cp pool alone. Cp activity and copper concentrations also increased in Min mice (Fig. 2A, B and C). In mice with HCT116 tumors, CSI values increased significantly at 7 d after transplantation (Fig. 2A and B) when tumor growth was still in the latent phase (Fig. 2D).

Figure 2. Growing tumors induce increased copper status indexes in mice. HCT(7d), HCT(24d), nude mice on the 7th and 24th day after the transplantation of HCT116 cells; B16, C57Bl/6J mice on the 21st day after the implantation of B16 melanoma cells; Min, 70-d-old Min mice. Sera from animals of the respective lineages without tumors were used as the controls. (A) Oxidase Cp content, % of respective control. (B) Relative Cp protein content as measured by immunoblotting, % of respective control. Cp protein content was not measured in Min mice. (C) Atomic copper concentration in sera, μg/g tissue. (D) Quartile plots (n = 5–7) of HCT116 tumor growth curve in nude mice, g.
However, it is known that some carcinoma lines express the Cp gene,46 so there was a possibility that the increased CSI values were partly caused by Cp synthesis by the tumor cells. As shown by western blot (WB) analysis, HCT116 cells produced a secretory form of Cp, which could be detected in the culture medium as 65- and 48-kDa fragments of full-length 130 kDa Cp (Fig. 3A). Direct quantification of hCp in murine serum is difficult because human and murine Cp have almost identical electrophoretic mobility (Fig. 3B). In addition, rabbit antibodies to human and rat Cp cross-react with murine Cp, but antibodies to rat Cp do not react with human Cp (Fig. 3C and D). Therefore, we applied the dot-spot technique with radiolabeled monospecific polyclonal murine antibodies to hCp to determine the concentration of hCp in the bloodstream of mice with HCT116 tumors (Fig. 3E). The hCp concentration comprised less than 2–3 mg/100 ml of serum, which is approximately 10% of the basal serum Cp level in control mice.

Figure 3. HCT116 cells produce human Cp in vitro and in tumor xenografts. (A) WB with antibodies to human Cp. Lane M, molecular weight marker (prestained proteins, Fermentas, code SM0671). Lane 1, culture medium in which HCT116 cells were grown; lane 2, human serum (positive control); lane 3, bovine serum (negative control). The two WB panes are sections of a single blot. (B) Oxidase activity in human (H), rat (R), and murine (M) sera, o-dianisidine stain of a non-denaturing 8% PAGE, 1 μl of serum per lane; WB of the same samples with antibodies to human (C) and rat (D) Cp, 0.5 μl of serum per lane. Immune complexes were detected using a goat anti-rabbit IgG-conjugated peroxidase/α-naphthol assay. (E) Human Cp content as measured by dot-spot hybridization with [125I]-mAB-hCp. One, human serum; 2, serum of HCT116-bearing nude mice (n = 6).
Changes in copper transporter gene expression in the livers of mice bearing non-hepatic tumors
To characterize copper metabolism in the liver, the activities of genes that are intimately linked to CSIs were studied. CTR1 gene encodes a high-affinity copper importer that is critical for copper import into all cell types.3 ATP7A and ATP7B genes encode copper transporting ATPases through which copper is pumped into the lumen of the secretory machinery in preparation for loading onto apo-cuproenzymes including Cp.3 ATP7B gene is expressed primarily in the liver of adult mammals. On the contrary, ATP7A gene is expressed in adult animals in the cells of all organs, but not in hepatocytes. Two splice variants, which are tissue-specific products of the primary transcript of the Cp gene, were analyzed: the mRNA encoding the secretory isoform of Cp, which is typically found in the liver, and the non-hepatic mRNA encoding the membrane-anchored GPI-Cp generated by alternative RNA splicing.47 This set of genes allowed us to trace a transport route for Cp metallation: CTR1 → ATP7B → Cp. The activities of these genes in the liver were measured in the 3 mouse tumor models and the respective control mouse lineages. The changes in the experimental groups with respect to the controls were similar for the three models. The Min mice proved to be the most adequate model of natural carcinogenesis, and the data for this group are shown in Figure 4. The activity of Cp, CTR1 and ATP7B genes was analogously increased in the liver of mice with growing HCT116 or B16 transplants, as compared with nude and C57Bl/6J mice without tumors respectively. (Fig. S1A and B). These data demonstrate that the levels of secretory Cp mRNA, CTR1 mRNA and ATP7B mRNA in liver tissue were increased more than 2-fold (P < 0.0005). In these experiments ATP7A and GPI-Cp mRNAs were also assayed to evaluate the specificity and selectivity of the method. Unexpectedly, Cp mRNA for the GPI-anchored form of Cp and ATP7A mRNA were found in the total RNA extracts of liver cells of tumor-bearing mice, but not of the respective control group (Fig. 4). GPI-Cp and ATP7A mRNAs also appeared in the liver of HCT116- and B16-bearing mice (Fig. S1A and B).
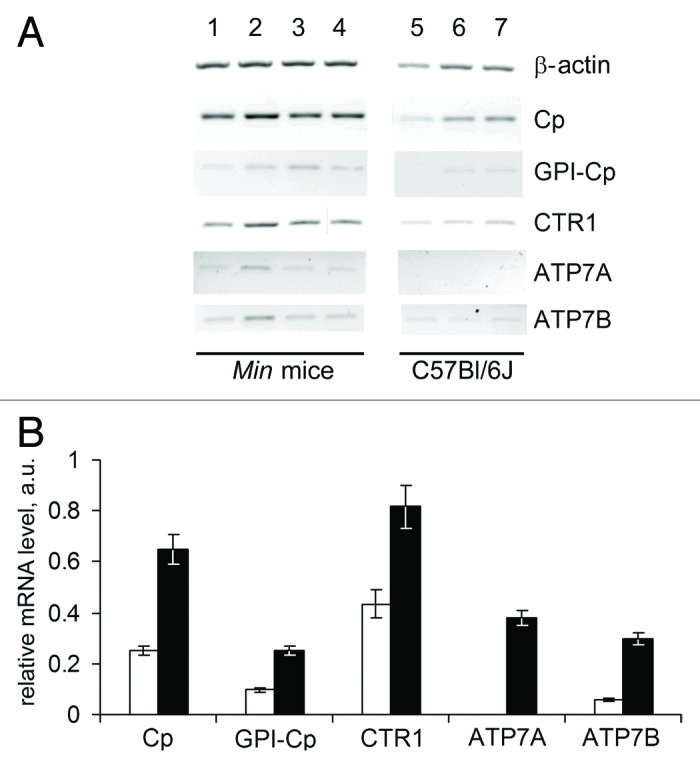
Figure 4. Altered copper transporter gene expression profile in the liver of 70-day-old Min mice. (A) RT-PCR results: negative image of 1% agarose gels stained with EtBr. Lanes 1–4, Min mice; lanes 5–7, C57Bl/6J mice. (B) Semi-quantitative analysis of the gels. White bars, C57Bl/6J mice; black bars, Min mice. Changes are significant at P < 0.0005.
Expression of copper transporter genes in tumors
The relative gene expressions in the adenomas of 70-d-old Min mice are presented in Figure 5. These data show that the CTR1 gene expression level in adenomas was significantly higher than in the homologous tissue of the control mice. Interestingly, CTR1 mRNA levels in tissues surrounding the adenomas were higher than those in the intestinal tissue of age-matched mice of the parent line. Of the two copper transporter ATPases, only the ATP7A gene was expressed. However, its mRNA levels were close to the detection limit of the method. Neither of the two mature mRNA products of the Cp gene was detected in the adenomas of Min mice. The CTR1 gene was actively expressed by both B16 melanomas and tumors developing from HCT116 cells (Fig. S1C). The Cp and ATP7B mRNAs were not detected in the melanomas; and the level of mRNA-ATP7A was low. Only secretory Cp mRNA and ATP7B mRNA (low level) were found in the HCT116 tumors (Fig. S1C). These data are in good agreement with the previously observed levels of hCp in the bloodstreams of mice with HCT116 tumors. In cultured HCT116 and B16 cells, the expression levels of the CTR1, ATP7A, ATP7B, and Cp genes (Cp is expressed only in HCT116) were much lower than in the respective tumors generated by these cells in mice (Fig. S1C).
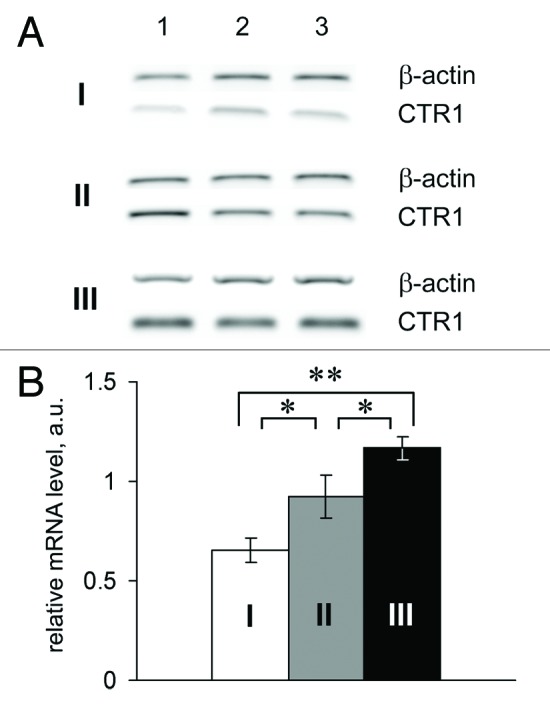
Figure 5. Relative CTR1 mRNA level in the small intestines of Min mice. I and white bar, control; II and gray bar, tissues surrounding the adenoma; III and black bar, adenomas of Min mice. *P < 0.05. **P < 0.0001.
Holo-Cp depletion induced by Ag diet is correlated with tumor growth retardation
In previous studies, we have shown that feeding mice with Ag(I) ions, which have similar coordination properties to Cu(I) ions, caused the appearance of inactive silver-loaded Cp in the circulation, resulting in a Cp-associated blood serum copper deficiency. The deficiency of holo-Cp developed completely by the seventh day on the Ag diet, the levels and activity of intracellular cuproenzymes did not change.44 In Ag mice, the level of Cp protein did not decrease as determined by rocket immunoelectrophoresis. In WB assay in non-denaturing PAGE Cp of Ag mice is revealed as two bands with different mobility, both of which correspond to the 130 kDa protein.44,48 Ag mice were utilized to assess the influence of circulatory copper levels on tumor growth. The effect of the Cp copper deficit on tumor growth was tested in two models: the HCT116 xenograft model and the B16 melanoma model. The HCT116 xenograft experiments were conducted in two subgroups. In the first subgroup, mice began to receive Ag diet one week prior to the implantation of HCT116 cells (Ag(+) mice). In the other subgroup, the Ag diet began at the time of cell implantation (Ag(P) mice). The tumor progression in these subgroups is shown in Figure 6A (left). In the Ag(P) mice, tumor development was almost completely repressed in five out of six mice, and large tumor developed in only one animal (Fig. 6A, right). For this reason, the range of tumor sizes within the group was very high (0.39 ± 0.56 g compared with 0.76 ± 0.45 g for controls). At the same time, HCT116 cells transplanted in Ag(+) mice grew progressively, with no lag period, and the tumors grew faster than in mice fed a normal diet (1.05 ± 0.64 g, Fig. 6A). Despite the large variance, the difference between tumor growth in Ag(+) and Ag(P) mice is significant (P < 0.05). A characteristic deficiency of Cp oxidase developed and persisted in both Ag groups until the end of the experiment (Fig. 6B), as firmly evidenced by the drop of oxidase activity and the two-banded WB.

Figure 6. Effects of Ag diet on the development of tumor xenografts. (А) Left, dynamics of human colon tumor growth in control (◊), Ag(+) (■), and Ag(P) (●) mice. Ordinate, weight of tumors, g; abscissa, days. Average weights for five mice in the control group, 6 Ag(+) mice and 6 Ag(P) mice are shown; right, weights of the 24-d-old tumors in the individual mice. (B) Serum oxidase activity (upper) and Cp protein (lower) detected in the same mice. (C) Serum oxidase activity (upper) and Cp protein (lower) detected in B16 melanoma-bearing mice. For oxidase activity measurements, serum aliquots (1.0 µl) were resolved by 8% native PAGE, and the gel was stained with o-dianisidine; the bands are dark orange. For Cp protein measurements, serum aliquots (0.5 µl) were fractioned by 8% native PAGE followed by immunoblotting with antibodies to human Cp. (D) Copper and silver content in the livers and tumors of the mice bearing implanted tumors. *P < 0.05, **P < 0.005, ***P < 0.0001.
In C57Bl mice, the Ag diet was initiated at the time of injection of the B16 murine melanoma cells. In three weeks, tumor progression and tumor size were approximately the same in the control and Ag groups, although the Ag mice also demonstrated a characteristic serum Cp oxidase deficit (Fig. 6C).
Atomic copper and silver concentrations were measured in the livers and tumors of all groups (Fig. 6D). The data showed that silver accumulated to considerable amounts in the liver tissue of all Ag mice, whereas the silver concentration was 25 to 50 times lower in all of the studied tumors obtained from Ag mice than in their livers. In HCT116 tumors, the copper concentration was significantly reduced by 30% in the Ag(P) and Ag(+) subgroups. The Ag diet also resulted in a significant change in the copper concentration of melanomas. It is worth noting that Ag-fed mice displayed an increased copper concentration in the liver.
Tumor tissues from the Ag(P) subgroup were then examined in more detail. A low molecular weight DNA ladder with the step of ~150 bp was clearly detected in the cytosolic fraction of tumor homogenates (Fig. 7A). The expression levels of the copper transporter genes listed above were measured in the tumors of three control mice and four Ag(P) mice. Sample 4 in Figure 7B corresponds to the Ag(P) mouse with the largest tumor. There were no significant changes in the expression profile of copper metabolism genes that may have been associated with tumor growth retardation. The enzymatic activity of SOD1, which is a major cytosolic cuproenzyme, did not change either in the cytosol or the mitochondria (Fig. 7C). In mitochondria, the relative level of the Cox4i1 subunit of COX did not change (Fig. 7C), showing that the formation of mature COX was not disturbed. Cytosolic p53 levels in tumors from Ag mice and control mice were similar (Fig. 7C); however, cytochrome C was significantly elevated in the cytosol (Fig. 7C). There was a general increase in cytosolic levels of HTRA2, but it was not significant at P < 0.05 level (P = 0.087, t test) (Fig. 7C). The possibility that the cytosol was contaminated with mitochondria was excluded (Fig. 7B, lane 8, horizontal section 5′). The redistribution of cytochrome C was not pronounced in sample 4, which was the only well-developed tumor observed in an Ag(P) mouse.

Figure 7. Apoptotic markers and cuproenzyme expression in tumors from Ag(P) mice. (A) DNA in the cytosol fraction of 24-d-old tumors, 2% agarose gel stained with EtBr. Lanes 1–3, control mice; lanes 4–6, Ag(P) mice; lane 7, 100–1500 bp DNA ladder. (B) Expression and intracellular distribution of cuproenzymes and selected apoptotic markers in HCT116 tumors. Vertical lanes: control (lanes 1–3) and Ag(P) mice (lanes 4–7). Horizontal sections: 1′, WB of cytosolic proteins blotted with antibodies to SOD1; 2′, SOD1 activity in the same samples; 3′, WB of mitochondrial proteins with antibodies to SOD1 (Lane 8, cytosolic protein from lane 2 diluted 10-fold); 4′, WB of mitochondrial proteins with antibodies to COX ; 5′, WB of the same samples with antibodies to VDAC (Lane 8, cytosol protein from lane 2); 6′, WB of cytosolic proteins with antibodies to p53; 7′, WB of cytosolic proteins with antibodies to cytochrome c; 8′, WB of mitochondrial proteins with antibodies to cytochrome c; 9′, WB of the same samples with antibodies to VDAC; 10′, WB of cytosolic proteins with antibodies to HTRA2; 11′, WB of cytosolic proteins with antibodies to β-actin. Gels were loaded with 20 μg of total cytosolic or mitochondrial protein per lane. For western blots, 12% denaturing PAGE was used, and 8% non-denaturing PAGE was used to assess SOD activity (2′). (C) Quantification of data, relative units: protein content as measured by WB, white, control mice; black, Ag(P) mice; gray and spotted bars, cytosolic SOD1 enzymatic activity in control and Ag(P) mice, respectively. Cytosolic protein levels are expressed relative to β-actin levels, and mitochondrial protein levels are expressed relative to VDAC levels. The relative cytochrome c content was determined from two independent experiments (the protocol is given for a single experiment). *P < 0.01.
Discussion
In the present work, we examined the connection between CSIs and cancer growth. A metaanalysis of the literature over the past 40 years revealed a distinct positive correlation between CSIs and cancer growth (Fig. 1). This relationship was experimentally tested in 3 mouse models bearing tumors of different origins. Significant increases in CSIs was detected in all of the models studied, and CSIs increased during the phase of tumor growth in which the tumor was proceeding from the lag period to the exponential phase (Fig. 2). This profound increase in CSIs cannot be explained by the potential production of Cp by tumors. B16 melanomas and adenomas from Min mice did not produce Cp, and HCT116-tumors produced relatively small amounts of Cp, which could be detected as hCp (Fig. 3). Moreover, the levels of mature mRNAs of CTR1, ATP7B, and secretory Cp in the livers of mice bearing tumors were found to be increased (Fig. 4, Fig. S1A and B). Therefore, tumor growth is able to induce changes in the expression of copper metabolism genes, which meet the increased copper requirements of the tumor. Previously, Kim et al.49 showed that, in cardiomyopathic mice with Ctr1 gene knockout in the heart, heart copper deficiency provokes the activity of genes of copper transporters in the liver. They suggested that a copper-deficient organ might secrete a factor stimulating liver copper metabolism. So the changes observed by us might be part of a general response of the liver to the increased copper requirements. Unexpectedly, mRNAs that encode ATP7A and GPI-Cp were detected in the livers of all mice with growing tumors (Fig. 4, Fig. S1A and B). Therefore, expression profile in the liver is changed both at the transcription and the splicing level. Additional studies are required to understand the physiological role of the coordinated induction of ATP7A and GPI-Cp expression in the adult liver under the trans-influence of growing tumors.
We measured the relative expression levels of copper metabolism genes in adenomas that develop “naturally” in Min mice (Fig. 4), as well as in tumors originating from the HCT116 and B16 cell lines. In each of the tumors, the CTR1 gene and at least one of the copper transporting ATPases were expressed. In this group of experiments, we considered valuable only the data showing an increase in CTR1 expression in adenomas, since the internal reference was available only for this group (i.e., the native tissue from which the adenomas originated). The increase in CTR1 expression in adenomas may be explained by the enhanced requirement for copper of these tumors (Fig. 5). This finding agrees with data from other studies reporting increased expression of CTR1 in tumors as compared with homologous native tissue.50 In tumors, we observed the low expression of ATP7A/B; possibly tumor cells do not excrete copper and synthesize very small amounts of extracellular cuproenzymes.
Because growing tumors stimulate holo-Cp synthesis, the question naturally arises as to whether tumor growth can be suppressed by lowering serum copper availability. To test this idea, we fed an Ag-rich diet to C57Bl/6J mice with B16 melanoma and nude mice with HCT116 carcinoma. Ag diet was chosen as an experimental copper-lowering approach that causes Cp-associated blood serum copper deficiency, which is more synchronized, profound and reproducible than those induced by copper-chelating agents. Advantageously, Ag diet does not disturb intracellular copper metabolism and produces no toxic side effects, which limit the use of copper chelators. In planning this experiment, we assumed that a Cp-associated copper deficiency would likely cause little or no effect in Ag-fed mice with melanoma, as B16 cells have a murine origin and may therefore also exploit a non-ceruloplasmin copper pool that is specific to murine copper metabolism.45 This assumption was found to be true. We also predicted that HCT116 tumor growth would directly depend on the duration of the Ag diet. However, human tumor growth was only inhibited in Ag(Р) mice (Fig. 6A) although copper concentrations were reduced in all tumors (Fig. 6D). In tumors from Ag(Р) mice, mitochondrial caspase-mediated apoptosis was triggered, as demonstrated by redistribution of cytochrome C into the cytosol and the presence of nucleosome-sized fragments of nuclear DNA (Fig. 7).
The unexpected effect of the Ag diet on the growth of HCT116 tumors in Ag(+) mice may be explained by the fact that in the Ag(+) and Ag(Р) groups, tumors began their development under different copper status conditions. In Ag(+) mice, the implanted HCT116 cells started to grow in Cp copper-deficient conditions, whereas in Ag(P) mice, the tumors started to develop under normal copper conditions. Although we have insufficient data to conclusively provide a reason for the higher tumor growth rate in Ag(+) mice, several possibilities can be proposed. It is possible that a mammalian copper-dependent oncosuppressor linked to copper metabolism exists and that its activity is lost during copper deficiency. It is also possible that a week of silver treatment induces a reorganization of copper metabolism and that this new copper traffic is then used by the human tumor cells, resulting in the potential for CTR1-independent copper transport.51 Alternatively, the copper deficiency might completely suppress the residual immunity of nude mice,52 thereby permitting faster tumor growth (e.g., Cu/VEGFR/VEGF system has been shown to participate in the early immune response in the macrophages of adult mammals53). It is possible that all of these factors operate together, but further experiments are required to help clarify this possibility.
In general, our results show that tumor growth is suppressed in response to Ag-induced depletion of holo-Cp. This finding is in agreement with studies investigating possible antitumor applications of copper-chelating agents.37-40
Based on the data presented here, we propose that the link between copper metabolism and carcinogenesis is multifactorial and may depend on different copper pools at different stages of tumor development. Locating a tumor-related intracellular copper pool that participates in signaling and finding a way to control this pool are important goals.
The use of copper-lowering strategies for anticancer therapy is very promising. However, detailed studies of liver copper metabolism and tumor copper metabolism as it relates to tumor origin and stage of development are needed to optimize this type of treatment approach.
Materials and Methods
Animals and cell lines
Female nude athymic CD-1 nu/nu mice (7 weeks old) and C57Bl/6J mice (7 weeks old) were obtained from Harlan (Italy). Male and female C57Bl/6J ApcMin/+ mice were bred at the Laboratory Animal Center in Viikki, Helsinki, from inbred mice originally obtained from the Jackson Laboratory. The mice were screened for the Min genotype using a PCR assay described by Dietrich et al.54 Seventy-day-old Min mice with fully formed adenomas of the intestine were used. In experiments with Min mice, animals from the parental C57Bl/6J line of matched age were used as the control group. The animals were maintained under specific pathogen-free conditions in polycarbonate cages with wood shavings in a temperature-controlled facility (23–25 °C). They were housed with a 12:12 h light-dark cycle in 80% humidity and were given suitable food and water ad libitum. Procedures involving animals and their care were conducted in conformity with institutional guidelines that are in compliance with national and international laws.
The HCT-116 human colon cancer cell line was maintained in IMDM supplemented with 1% glutamine (Cambrex) and 10% fetal bovine serum (Sigma). Exponentially growing cells were expanded in vitro and injected subcutaneously (7 × 106 cells/mouse) into athymic CD-1 nu/nu mice. The length (L) and width (W) of the resulting tumor masses were measured twice weekly using calipers, and the tumor volumes (TV) were calculated as: TV = (L × W2) / 2, with W being < L. Murine B16 melanoma cells (106 cells/mouse) were injected intramuscularly into C57Bl mice. Specific groups of nu/nu and C57Bl mice received 50 mg AgCl per kg of body weight daily (Ag diet), which was added into ground and moistened standard fodder.48 Biological samples were collected, snap frozen, and stored at -80 °C until being used in biochemical tests.
Experimental assays
The sequences of specific primers, the procedures for total RNA isolation, RT-PCR, determination of relative mRNA levels, isolation of subcellular fractions, WB analysis, measurement of specific enzymatic activities of Cp and SOD1, and measurement of the copper and silver concentrations have been described previously.44,48 Murine Cp was visualized using polyclonal monospecific rabbit antibodies raised against electrophoretically pure recrystallized hCp preparation (A610/280 = 0.047). Rabbit polyclonal anti-HTRA2 (mitochondrial high-temperature requirement serine protease) antibody (R&D Systems), goat polyclonal anti-actin antibody, mouse monoclonal anti-cytochrome C antibody, goat polyclonal anti-VDAC (voltage-dependent anion channel) antibody, and mouse monoclonal anti-p53 antibody (Santa Cruz Biotechnology) were used in these analyses. Immune complexes were revealed using corresponding peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) and visualized by enhanced chemiluminescence assay (ECL; Amersham). For quantification, cytosolic and mitochondrial protein levels were related to β-actin and VDAC in the same nitrocellulose membranes respectively.
DNA were isolated from the cytosolic fraction obtained by differential centrifugation of tumor homogenate using a modified version of the method described by Oridate et al.55 Briefly, ~20 mg of total cytosolic protein per 1 ml was treated with RNase A (50 μg/ml) at 37 °C for 1 h, followed by treatment with proteinase K (100 μg/ml) in 0.5% SDS at 50 °C for 2 h. The residual material was purged with phenol/chloroform and precipitated using 70% ethanol. The pellets were dried, dissolved, fractionated in 2% agarose gel, and stained with EtBr.
Data are expressed as means ± SD. Statistical inferences were made using the 2-tailed Student t test. Differences were considered significant at P values < 0.05.
Identification of Cp protein in cell culture media and blood sera from xenografted mice
Proteins from cell-conditioned medium were precipitated by adding 1.6 ml of cold acetone (-20 °C) to 0.4 ml of medium. The medium was then shaken and stored at -20 °C for 1 h. Pellets were collected by centrifugation (13000 × g, 10 min), dried, resuspended in 40 µl of electrophoresis buffer and fractionated by electrophoresis in an 8% PAG with 0.1% SDS, followed by WB analysis.
Monospecific polyclonal murine antibodies to hCp (mAB-hCp) were obtained by immunizing mice with hCp using a standard procedure. The IgG fraction was radiolabelled with [125I]Na with chloramine T to a specific activity of 105 µCi/µg. Serum samples (1 µl) from the xenografted mice were placed on a nitrocellulose membrane. The membrane was then treated with a 5% solution of skim milk proteins in PBS containing 0.05% Tween 20. The membrane was then incubated with 100 µl of [125I]-mAB-hCp in 1 ml of PBS overnight at +4 °C. The membrane was then washed and radioautographed. Human serum containing Cp at a concentration of 35 mg/100 ml was serially diluted and used as a quantitative standard. The X-ray film was scanned and the Cp concentrations estimated by assessing the optical density of the resulting spots.
Metaanalysis
The metaanalysis was performed using typical methodology.56 The initial search was performed using the NCBI PubMed open database (http://www.ncbi.nlm.nih.gov/pubmed/) on November 2012. The search criterion was the simultaneous presence of the terms “copper”, “ceruloplasmin”, and one of the terms “cancer”, “tumor”, or “neoplasms” in any of the database fields. References were then manually reviewed by 2 reviewers to meet the following criteria:
1) Studies in groups of human adult or adolescent individuals with tumors as compared with a representative group of apparently healthy individuals and/or individuals with non-tumor diseases;
2) At least 1 of the 3 major CSIs was measured: atomic copper concentration (by atomic absorption spectroscopy, inductively coupled plasma mass spectrometry or colorimetric techniques), Cp protein content (by the Mancini method, rocket immunoelectrophoresis, nephelometric assay with monoclonal antibodies or by electron spin resonance of Cp-specific Cu(II)), Cp oxidase activity (determined by colorimetric, kinetic or in-gel assays with p-phenylenediamine or o-dianisidine);
3) Patients were not receiving metal-chelating therapy;
4) Patients did not have Wilson disease or liver cirrhosis, as these patients may have genetic or acquired defects in holo-Cp formation.
Selected references were analyzed for the following meta-parameters:
A) Number of cases in the study;
B) Tumor type/localization, if available;
C) Set of measured CSIs (atomic copper concentration, Cp protein concentration, and Cp activity);
D) Change of CSI values in tumor cases with respect to control (increase, decrease or not significant—this ranked parameter was used to eliminate the bias of different methods of copper status estimation);
E) Change in CSI levels during successful therapy (increase, decrease, not significant, or not studied);
F) Diagnostic and/or prognostic value of CSI in tumor cases, as stated by the authors (“valuable”, “not valuable”, or “no inference”).
All retrieved literature data were considered equally trustworthy. Gender or ethnic factors were not specially accounted for.
Supplementary Material
Acknowledgments
This work was supported by SIMO (to PB, a CARIPLO Foundation fellowship grant (to LP) and the Russian Foundation for Basic Research (09-04-01165a, 12-04-01530a to LP). We also gratefully acknowledge the generous contribution of the Italian Association for Cancer Research (AIRC to MB).
Glossary
Abbreviations:
- CSIs
copper status indexes
- Cp
ceruloplasmin
- WB
western blot
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/24594
References
- 1.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 2.Rubino JT, Franz KJ. Coordination chemistry of copper proteins: how nature handles a toxic cargo for essential function. J Inorg Biochem. 2012;107:129–43. doi: 10.1016/j.jinorgbio.2011.11.024. [DOI] [PubMed] [Google Scholar]
- 3.Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. 2008;4:176–85. doi: 10.1038/nchembio.72. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Hodgkinson V, Zhu S, Weisman GA, Petris MJ. Advances in the understanding of mammalian copper transporters. Adv Nutr. 2011;2:129–37. doi: 10.3945/an.110.000273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O’Halloran TV. Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science. 1999;284:805–8. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- 6.Polio J, Enriquez RE, Chow A, Wood WM, Atterbury CE. Hepatocellular carcinoma in Wilson’s disease. Case report and review of the literature. J Clin Gastroenterol. 1989;11:220–4. doi: 10.1097/00004836-198904000-00022. [DOI] [PubMed] [Google Scholar]
- 7.Walshe JM, Waldenström E, Sams V, Nordlinder H, Westermark K. Abdominal malignancies in patients with Wilson’s disease. QJM. 2003;96:657–62. doi: 10.1093/qjmed/hcg114. [DOI] [PubMed] [Google Scholar]
- 8.Iwadate H, Ohira H, Suzuki T, Abe K, Yokokawa J, Takiguchi J, et al. Hepatocellular carcinoma associated with Wilson’s disease. Intern Med. 2004;43:1042–5. doi: 10.2169/internalmedicine.43.1042. [DOI] [PubMed] [Google Scholar]
- 9.Kumagi T, Horiike N, Abe M, Kurose K, Iuchi H, Masumoto T, et al. Small hepatocellular carcinoma associated with Wilson’s disease. Intern Med. 2005;44:439–43. doi: 10.2169/internalmedicine.44.439. [DOI] [PubMed] [Google Scholar]
- 10.Masuda R, Yoshida MC, Sasaki M, Dempo K, Mori M. High susceptibility to hepatocellular carcinoma development in LEC rats with hereditary hepatitis. Jpn J Cancer Res. 1988;79:828–35. doi: 10.1111/j.1349-7006.1988.tb00044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Powell CJ. Copper-overload causes cancer? The LEC rat: a model for human hepatitis, liver cancer, and much more. Hum Exp Toxicol. 1994;13:910–2. [PubMed] [Google Scholar]
- 12.Huster D, Finegold MJ, Morgan CT, Burkhead JL, Nixon R, Vanderwerf SM, et al. Consequences of copper accumulation in the livers of the Atp7b-/- (Wilson disease gene) knockout mice. Am J Pathol. 2006;168:423–34. doi: 10.2353/ajpath.2006.050312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupte A, Mumper RJ. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat Rev. 2009;35:32–46. doi: 10.1016/j.ctrv.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 14.Tsubota A, Matsumoto K, Mogushi K, Nariai K, Namiki Y, Hoshina S, et al. IQGAP1 and vimentin are key regulator genes in naturally occurring hepatotumorigenesis induced by oxidative stress. Carcinogenesis. 2010;31:504–11. doi: 10.1093/carcin/bgp313. [DOI] [PubMed] [Google Scholar]
- 15.Jomova K, Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 2011;283:65–87. doi: 10.1016/j.tox.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 16.Tsai CY, Finley JC, Ali SS, Patel HH, Howell SB. Copper influx transporter 1 is required for FGF, PDGF and EGF-induced MAPK signaling. Biochem Pharmacol. 2012;84:1007–13. doi: 10.1016/j.bcp.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finney L, Mandava S, Ursos L, Zhang W, Rodi D, Vogt S, et al. X-ray fluorescence microscopy reveals large-scale relocalization and extracellular translocation of cellular copper during angiogenesis. Proc Natl Acad Sci U S A. 2007;104:2247–52. doi: 10.1073/pnas.0607238104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gérard C, Bordeleau LJ, Barralet J, Doillon CJ. The stimulation of angiogenesis and collagen deposition by copper. Biomaterials. 2010;31:824–31. doi: 10.1016/j.biomaterials.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 19.Easter RN, Qilin Chan, Lai B, Ritman EL, Caruso JA, Zhenyu Qin Vascular metallomics: copper in the vasculature. Vasc Med. 2010;15:61–9. doi: 10.1177/1358863X09346656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fan F, Wey JS, McCarty MF, Belcheva A, Liu W, Bauer TW, et al. Expression and function of vascular endothelial growth factor receptor-1 on human colorectal cancer cells. Oncogene. 2005;24:2647–53. doi: 10.1038/sj.onc.1208246. [DOI] [PubMed] [Google Scholar]
- 21.Wey JS, Fan F, Gray MJ, Bauer TW, McCarty MF, Somcio R, et al. Vascular endothelial growth factor receptor-1 promotes migration and invasion in pancreatic carcinoma cell lines. Cancer. 2005;104:427–38. doi: 10.1002/cncr.21145. [DOI] [PubMed] [Google Scholar]
- 22.Yang AD, Camp ER, Fan F, Shen L, Gray MJ, Liu W, et al. Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in human pancreatic carcinoma cells. Cancer Res. 2006;66:46–51. doi: 10.1158/0008-5472.CAN-05-3086. [DOI] [PubMed] [Google Scholar]
- 23.Payne SL, Hendrix MJ, Kirschmann DA. Paradoxical roles for lysyl oxidases in cancer--a prospect. J Cell Biochem. 2007;101:1338–54. doi: 10.1002/jcb.21371. [DOI] [PubMed] [Google Scholar]
- 24.Schietke R, Warnecke C, Wacker I, Schödel J, Mole DR, Campean V, et al. The lysyl oxidases LOX and LOXL2 are necessary and sufficient to repress E-cadherin in hypoxia: insights into cellular transformation processes mediated by HIF-1. J Biol Chem. 2010;285:6658–69. doi: 10.1074/jbc.M109.042424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li W, Nellaiappan K, Strassmaier T, Graham L, Thomas KM, Kagan HM. Localization and activity of lysyl oxidase within nuclei of fibrogenic cells. Proc Natl Acad Sci U S A. 1997;94:12817–22. doi: 10.1073/pnas.94.24.12817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giampuzzi M, Oleggini R, Albanese C, Pestell R, Di Donato A. beta-catenin signaling and regulation of cyclin D1 promoter in NRK-49F cells transformed by down-regulation of the tumor suppressor lysyl oxidase. Biochim Biophys Acta. 2005;1745:370–81. doi: 10.1016/j.bbamcr.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 27.Mufti AR, Burstein E, Duckett CS. XIAP: cell death regulation meets copper homeostasis. Arch Biochem Biophys. 2007;463:168–74. doi: 10.1016/j.abb.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burstein E, Ganesh L, Dick RD, van De Sluis B, Wilkinson JC, Klomp LW, et al. A novel role for XIAP in copper homeostasis through regulation of MURR1. EMBO J. 2004;23:244–54. doi: 10.1038/sj.emboj.7600031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wenger RH. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002;16:1151–62. doi: 10.1096/fj.01-0944rev. [DOI] [PubMed] [Google Scholar]
- 30.Martin F, Linden T, Katschinski DM, Oehme F, Flamme I, Mukhopadhyay CK, et al. Copper-dependent activation of hypoxia-inducible factor (HIF)-1: implications for ceruloplasmin regulation. Blood. 2005;105:4613–9. doi: 10.1182/blood-2004-10-3980. [DOI] [PubMed] [Google Scholar]
- 31.Gogvadze V, Zhivotovsky B, Orrenius S. The Warburg effect and mitochondrial stability in cancer cells. Mol Aspects Med. 2010;31:60–74. doi: 10.1016/j.mam.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 32.Prohaska JR. Impact of copper limitation on expression and function of multicopper oxidases (ferroxidases) Adv Nutr. 2011;2:89–95. doi: 10.3945/an.110.000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zaitsev VN, Zaitseva I, Papiz M, Lindley PF. An X-ray crystallographic study of the binding sites of the azide inhibitor and organic substrates to ceruloplasmin, a multi-copper oxidase in the plasma. J Biol Inorg Chem. 1999;4:579–87. doi: 10.1007/s007750050380. [DOI] [PubMed] [Google Scholar]
- 34.Harvey LJ, Ashton K, Hooper L, Casgrain A, Fairweather-Tait SJ. Methods of assessment of copper status in humans: a systematic review. Am J Clin Nutr. 2009;89:2009S–24S. doi: 10.3945/ajcn.2009.27230E. [DOI] [PubMed] [Google Scholar]
- 35.Cooper EH. [Plasma protein profile in neoplastic diseases] Ric Clin Lab. 1983;13(Suppl 1):57–69. [PubMed] [Google Scholar]
- 36.Vavilova TP, Gusarova IuN, Koroleva OV, Medvedev AE. [The role of ceruloplasmin in neoplastic processes] Biomed Khim. 2005;51:263–75. [PubMed] [Google Scholar]
- 37.Brewer GJ, Merajver SD. Cancer therapy with tetrathiomolybdate: antiangiogenesis by lowering body copper--a review. Integr Cancer Ther. 2002;1:327–37. doi: 10.1177/1534735402238185. [DOI] [PubMed] [Google Scholar]
- 38.Goodman VL, Brewer GJ, Merajver SD. Copper deficiency as an anti-cancer strategy. Endocr Relat Cancer. 2004;11:255–63. doi: 10.1677/erc.0.0110255. [DOI] [PubMed] [Google Scholar]
- 39.Lowndes SA, Harris AL. Copper chelation as an antiangiogenic therapy. Oncol Res. 2004;14:529–39. doi: 10.3727/0965040042707952. [DOI] [PubMed] [Google Scholar]
- 40.Fu S, Naing A, Fu C, Kuo MT, Kurzrock R. Overcoming platinum resistance through the use of a copper-lowering agent. Mol Cancer Ther. 2012;11:1221–5. doi: 10.1158/1535-7163.MCT-11-0864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trivedy CR, Warnakulasuriya KA, Peters TJ, Senkus R, Hazarey VK, Johnson NW. Raised tissue copper levels in oral submucous fibrosis. J Oral Pathol Med. 2000;29:241–8. doi: 10.1034/j.1600-0714.2000.290601.x. [DOI] [PubMed] [Google Scholar]
- 42.Mondorf AW, Mackenrodt G, Halberstadt E. [Ceruloplasmin. I. Biochemistry of ceruloplasmin. II. Influence of estrogens on the ceruloplasmin content of the serum] Klin Wochenschr. 1971;49:61–70. doi: 10.1007/BF01497302. [DOI] [PubMed] [Google Scholar]
- 43.Kluft C, Leuven JA, Helmerhorst FM, Krans HM. Pro-inflammatory effects of oestrogens during use of oral contraceptives and hormone replacement treatment. Vascul Pharmacol. 2002;39:149–54. doi: 10.1016/S1537-1891(02)00304-X. [DOI] [PubMed] [Google Scholar]
- 44.Zatulovskiy EA, Skvortsov AN, Rusconi P, Ilyechova EY, Babich PS, Tsymbalenko NV, et al. Serum depletion of holo-ceruloplasmin induced by silver ions in vivo reduces uptake of cisplatin. J Inorg Biochem. 2012;116:88–96. doi: 10.1016/j.jinorgbio.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 45.Cabrera A, Alonzo E, Sauble E, Chu YL, Nguyen D, Linder MC, et al. Copper binding components of blood plasma and organs, and their responses to influx of large doses of (65)Cu, in the mouse. Biometals. 2008;21:525–43. doi: 10.1007/s10534-008-9139-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoshimura S, Tamaoki N, Ueyama Y, Hata J. Plasma protein production by human tumors xenotransplanted in nude mice. Cancer Res. 1978;38:3474–8. [PubMed] [Google Scholar]
- 47.Patel BN, Dunn RJ, David S. Alternative RNA splicing generates a glycosylphosphatidylinositol-anchored form of ceruloplasmin in mammalian brain. J Biol Chem. 2000;275:4305–10. doi: 10.1074/jbc.275.6.4305. [DOI] [PubMed] [Google Scholar]
- 48.Ilyechova E, Skvortsov A, Zatulovsky E, Tsymbalenko N, Shavlovsky M, Broggini M, et al. Experimental switching of copper status in laboratory rodents. J Trace Elem Med Biol. 2011;25:27–35. doi: 10.1016/j.jtemb.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 49.Kim BE, Turski ML, Nose Y, Casad M, Rockman HA, Thiele DJ. Cardiac copper deficiency activates a systemic signaling mechanism that communicates with the copper acquisition and storage organs. Cell Metab. 2010;11:353–63. doi: 10.1016/j.cmet.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holzer AK, Varki NM, Le QT, Gibson MA, Naredi P, Howell SB. Expression of the human copper influx transporter 1 in normal and malignant human tissues. J Histochem Cytochem. 2006;54:1041–9. doi: 10.1369/jhc.6A6970.2006. [DOI] [PubMed] [Google Scholar]
- 51.Lee J, Petris MJ, Thiele DJ. Characterization of mouse embryonic cells deficient in the ctr1 high affinity copper transporter. Identification of a Ctr1-independent copper transport system. J Biol Chem. 2002;277:40253–9. doi: 10.1074/jbc.M208002200. [DOI] [PubMed] [Google Scholar]
- 52.Chandra RK. Nutrition and immunoregulation. Significance for host resistance to tumors and infectious diseases in humans and rodents. J Nutr. 1992;122(Suppl):754–7. doi: 10.1093/jn/122.suppl_3.754. [DOI] [PubMed] [Google Scholar]
- 53.Cursiefen C, Chen L, Borges LP, Jackson D, Cao J, Radziejewski C, et al. VEGF-A stimulates lymphangiogenesis and hemangiogenesis in inflammatory neovascularization via macrophage recruitment. J Clin Invest. 2004;113:1040–50. doi: 10.1172/JCI20465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dietrich WF, Lander ES, Smith JS, Moser AR, Gould KA, Luongo C, et al. Genetic identification of Mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell. 1993;75:631–9. doi: 10.1016/0092-8674(93)90484-8. [DOI] [PubMed] [Google Scholar]
- 55.Oridate N, Lotan D, Xu X-C, Hong WK, Lotan R. Differential induction of apoptosis by all-trans-retinoic acid and N-(4-hydroxyphenyl)retinamide in human head and neck squamous cell carcinoma cell lines. Clin Cancer Res. 1996;2:855–63. [PubMed] [Google Scholar]
- 56.Hooper L, Ashton K, Harvey LJ, Decsi T, Fairweather-Tait SJ. Assessing potential biomarkers of micronutrient status by using a systematic review methodology: methods. Am J Clin Nutr. 2009;89:1953S–9S. doi: 10.3945/ajcn.2009.27230A. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.