

Abstract
Chordoma is a rare, slow growing malignant tumor arising from remnants of the fetal notochord. Surgery is the first choice for chordoma treatment, followed by radiotherapy, although postoperative complications remain significant. Recurrence of the disease occurs frequently due to the anatomy of the tumor location and violation of the tumor margins at the initial surgery. Currently, there are no effective drugs available for patients with chordoma. Due to the rarity of the disease, there is limited opportunity to test agents in clinical trials and no concerted effort to develop agents for chordoma in the pharmaceutical industry. To rapidly and efficiently identify small molecules that inhibit chordoma cell growth, we screened the NCGC Pharmaceutical Collection (NPC) containing approximately 2800 clinically approved and investigational drugs at 15 different concentrations in chordoma cell lines, U-CH1 and U-CH2. We identified a group of drugs including bortezomib, 17-AAG, digitoxin, staurosporine, digoxin, rubitecan, and trimetrexate that inhibited chordoma cell growth, with potencies from 10 to 370 nM in U-CH1 cells, but less potently in U-CH2 cells. Most of these drugs also induced caspase 3/7 activity with a similar rank order as the cytotoxic effect on U-CH1 cells. Cantharidin, digoxin, digitoxin, staurosporine, and bortezomib showed similar inhibitory effect on cell lines and 3 primary chordoma cell cultures. The combination treatment of bortezomib with topoisomerase I and II inhibitors increased the therapeutic potency in U-CH2 and patient-derived primary cultures. Our results provide information useful for repurposing currently approved drugs for chordoma and potential approach of combination therapy.
Keywords: chordoma, NCGC Pharmaceutical Collection, cell viability, caspase 3/7, U-CH1, U-CH2, qHTS
Introduction
Chordoma is a rare, slow growing malignant tumor arising from remnants of the fetal notochord, an embyological structure that guides the growth of the bony skull and spine.1 Chordomas occur predominantly at base of the skull, in the vertebral bodies and the sacrum.2 The annual incidence rate of chordoma is approximately 1 in 1 000 000 per year in the United States3 and the median survival is 7 years.3
Initial treatment of chordoma is most commonly surgical removal of the tumors, which may result in significant perioperative morbidity and long-term sequelae.4 Radiation therapy is often used postoperatively, but proximity of the tumor to vital structures frequently prevents delivery of sufficiently high radiation doses and causes heterogeneous coverage of the target volume.5 Currently, no effective drugs are available to treat chordoma, although increasing knowledge of the molecular biology of chordoma may lead to future targeted therapeutics.6
To identify and repurpose currently approved drugs that inhibit the growth of chordoma, we screened approximately 2800 clinically approved or investigational drugs from the NIH Chemical Genomics Center Pharmaceutical Collection7 in a quantitative high-throughput screening (qHTS) format.8 U-CH19 and U-CH210 cell lines were used in the screening because they are immortal tumor cell lines derived from human chordoma with typical characteristics of chordoma tumors at molecular (Brachyury expression) and morphological (physaliferous cells) levels.10 From this study, we have identified several small molecule drugs that have a cytotoxic effect on chordoma cell lines and primary human chordoma cell cultures. The combination treatments of these drugs in U-CH2 and primary chordoma cell cultures showed increase of the drug potency. These findings suggest that these drugs may be repurposed for chordoma treatment.
Results
Drug screen to identify potential novel chemotherapeutics for chordoma
To search for small molecule drugs that inhibit chordoma cell growth, we screened the NPC library of 2816 drugs for inhibition of cell viability of U-CH1 and U-CH2 cell lines using a qHTS format. The non-neoplastic chordoma-derived cell line CCL4 was tested in parallel as a counter-screen to identify compounds that are selectively cytotoxic against U-CH1 and U-CH2. The primary screens using cell viability assay (CellTiter-Glo) in these 3 cell lines performed well in all 34 plates, with average Z’ factors11 of 0.75 ± 0.09, 0.67 ± 0.05, and 0.82 ± 0.04 for U-CH1, U-CH2, and CCL4 lines, respectively. The signal to background ratio was 21.2 ± 3.6, 32.7 ± 1.6, and 28.5 ± 5.9 for U-CH1, U-CH2, and CCL4 lines, respectively. The concentration response curves of potassium dichromate and staurosporine, used as positive controls, were performed in each plate to monitor plate-to-plate variations during the primary screening. Both control compounds replicated well across all the plates with average IC50 values of 2.54 ± 0.95 µM, 2.48 ± 0.39 µM, and 3.31 ± 1.01 µM for potassium dichromate and IC50 values of 2.61 ± 1.21 µM, 9.89 ± 2.00 µM, and 6.19 ± 3.44 µM for staurosporine in U-CH1, U-CH2 and CCL4 lines, respectively. These results suggest the robustness of the screen.
Based on potency and selectivity, mode of compound action and compound availability, 35 compounds, including compounds with novel mechanisms of action and some compounds in clinical use or trials for cancer, were selected for further study. Powder samples of these compounds were purchased from commercial vendors and tested in U-CH1 and U-CH2 viability assays (Table 1). Among these compounds, bortezomib (Fig. 1A) was the most potent with IC50 of 10 nM, followed by 17-AAG (0.11 µM, Fig. 1B), digitoxin (0.21 µM, Table 1), bleomycin (0.22 µM, Fig. 1C), staurosporine (0.23 µM, Figure 1D), digoxin (0.27 µM, Fig. 1E), rubitecan (0.32 µM, Fig. 1F), and trimetrexate (0.37 µM, Fig. 1G) in U-CH1 cells. However, most compounds tested were less potent or efficacious in U-CH2 cells than in U-CH1 cells (Fig. 1), except for oxyphenisatin (Fig. 1H). Two cardiac glycoside drugs, digoxin and digitoxin, had similar inhibitory effect on all the cell types (Table 1).
Table 1. Compound potency (µM) in cytotoxicity assay in U-CH1, U-CH2, and CCL4 cells.
| Name | U-CH1, IC50, µM (% inhibition) | U-CH2, IC50, µM (% inhibition) | CCL4, IC50, µM (% inhibition) | Mode of action, inhibitor |
|---|---|---|---|---|
| 17-Allylamino-geldanamycin |
0.11 ± 0.03 |
1.17 ± 0.77 (18%) |
0.89 (29%) |
Heat shock protein 90 |
| Acidum bromebricum |
32.77 ± 2.22 |
Inactive |
Inactive |
|
| BEZ-235 |
2.29 ± 2.9 |
0.18 ± 0.22 (21%) |
Inconclusive |
PI3 kinase and mTOR |
| BIBX 1382 dihydrochloride |
12.08 ± 0.79 |
62.64 ± 27.49 |
Inactive |
EGFR tyrosine kinase |
| Bleomycin |
0.23 ± 0.05 (42%) |
Inactive |
Inactive |
Glycopeptide antibiotic |
| Bortezomib |
0.010 ± 0 |
4.16 ± 3.74 |
Inactive |
Proteasome |
| Cantharidin |
2.81 ± 0 |
4.71 ± 1.33 |
0.44 (40%) |
Protein phosphatase 2A |
| Daunorubicin |
4.14 ± 0.53 |
21.83 ± 19.55 |
10.0 (42%) |
Topoisomerase |
| Digitoxin |
0.21 ± 0.04 |
0.06 ± 0.02 (24%) |
0.18 |
Na+/K+ ATPase |
| Digoxin |
0.27 ± 0.07 |
0.11 ± 0.02 (24%) |
0.58 |
Na+/K+ ATPase |
| Ecteinascidin 743 |
42.06 ± 3.42 |
32.65 ± 20.53 |
0.02 (20%) |
DNA binding and repair pathways |
| Erlotinib |
5.26 ± 3.29 |
37.84 ± 23.65 (28%) |
4.95 (27%) |
EGFR tyrosine kinase |
| GDC-0941 |
2.58 ± 0.71 |
31.93 ± 31.66 |
Inconclusive |
PI3 kinase |
| Gefitinib |
17.77 ± 9.03 |
48.04 ± 38.88 |
Inactive |
EGFR tyrosine kinase |
| Homoharringtonine |
0.53 ± 0.53 (21%) |
Inactive |
0.79 |
Protein synthesis |
| Idarubicin hydrochloride |
3.05 ± 0.42 |
56.46 ± 28.42 |
15.84 |
Topoisomerase II |
| Imatinib mesylate |
26.03 ± 1.76 |
55.19 ± 13.57 |
Inactive |
Receptor tyrosine kinase |
| Irinotecan hydrochloride |
31.14 ± 8.68 |
38.98 ± 18.40 (29%) |
Inactive |
Topoisomerase I |
| MG-132 |
1.52 ± 0.10 |
24.14 ± 6.97 |
Inconclusive |
Proteasome |
| Narasin |
2.58 ± 0.83 |
4.47 ± 2.65 |
35.5 |
Antibacterial |
| Oxyphenisatin |
6.71 ± 3.89 |
1.21 ± 0.48 (35%) |
Inactive |
Laxative |
| Plicamycin |
0.02 ± 0.002 (20%) |
Inactive |
0.35 (32%) |
Protein synthesis |
| Rapamycin |
35.48 ± 4.08 |
64.09 ± 20.64 |
Inactive |
mTOR pathway |
| Rubitecan |
0.32 ± 0.04 |
0.83 ± 0.21 (17%) |
7.7 (27%) |
Topoisomerase I |
| SB 202190 |
27.73 ± 13.16 |
Inactive |
Inactive |
P38 MAPK |
| SB 203580 |
51.38 ± 14.31 |
Inactive |
Inactive |
P38 MAPK |
| SB431542 |
Inactive |
Inactive |
Inactive |
Activin receptor-like kinase receptors |
| SF1126 (LY294002 hydrochloride) |
13.48 ± 3.99 |
14.54 ± 10.95 |
Inconclusive |
PI3 kinase/mTOR |
| Staurosporine |
0.24 ± 0.05 |
3.96 ± 4.08 |
7.0 |
ATP-competitive kinase |
| Suberoylanilide hydroxamic acid (SAHA) |
11.22 ± 4.08 |
Inactive |
Inactive |
HDAC |
| Sunitinib malate |
30.34 ± 1.98 |
48.92 ± 16.15 |
Inactive |
Receptor tyrosine kinase |
| Trichostatin A |
1.04 ± 0.07 |
6.46 ± 7.51 (23%) |
Inconclusive |
HDAC |
| Trimetrexate glucuronate |
0.38 ± 0.11 |
Inactive |
Inactive |
Dihydrofolate reductase |
| Vandetanib |
11.31 ± 2.11 |
41.01 ± 24.09 |
24.84 |
EGFR and VEGFR tyrosine kinase |
| Vincristine | 14.70 ± 1.87 | 62.82 ± 0 | 0.45 (32%) | Microtubule |
Each IC50 value (concentration of half the maximal inhibition) is the mean ± SD of the results from 3 to 5 experiments. Efficacy (inhibition as % positive control) of drugs was listed in parenthesis if it is less 50%.

Figure 1. Concentration response curves of chordoma inhibitors in U-CH1 (■) and U-CH2 cell lines (▲). Cell viability was measured after the cells were incubated with various concentrations of bortezomib (A), 17-AAG (B), bleomycin (C), staurosporine (D), digoxin (E), rubitecan (F), trimetrexate (G), oxyphenisatin (H), MG-132 (I), and trichostatin A (J) for 48 h. Data are expressed as mean ± SD from 3 to 5 experiments.
Effect of chordoma inhibitors on caspase 3/7 activity
Apoptosis of cancer cells is known to play a critical role in modulating their sensitivity to chemotherapy. Anticancer agents including tumor necrosis factor-related apoptosis-inducing ligands eventually kill cancer cells by inducing apoptosis.12 To study the mechanism of drug action on the inhibition of chordoma cell growth, the apoptosis inducing potential of the drugs was investigated. Caspase 3/7 activity was measured in U-CH1 cells after compound treatment at 5, 16, 24, and 48 h. Among the compounds tested (Table 2), bortezomib was the most potent stimulator of caspase 3/7 activity, with an EC50 value of 60 nM and 5 nM after 24 and 48 h treatment, respectively. Cantharidin (EC50 of 0.63 µM at 16 h, 0.79 µM at 24 h, and 0.34 µM at 48 h), digoxin (EC50 of 1.37 µM at 16 h, 1.14 µM at 24 h, and 0.48 µM at 48 h), narasin (EC50 of 9.20 µM at 16 h, 11.72 µM at 24 h, and 0.19 µM at 48 h), plicamycin (EC50 of 0.12 µM at 16 h, 90 nM at 24 h, and 50 nM at 48 h), staurosporine (used as a positive control for apoptosis; EC50 of 0.96 µM at 16 h, 0.94 µM at 24 h, and 70 nM at 48 h) and vincristine (EC50 of 7.08 µM at 16 h, 9.73 µM at 24 h, and 20 nM at 48 h) also showed significantly increased caspase 3/7 activity after the treatment. The potency rank order of the compounds tested in this study is very similar between the compound cytotoxicity and caspase 3/7 induction in U-CH1 cells (Table 2).
Table 2. Compound potency (µM) in cytotoxicity and caspase 3/7 assays in U-CH1 cells.
| Name | Cytotoxicity | Caspase, 5 h | Caspase, 16 h | Caspase, 24 h | Caspase, 48 h |
|---|---|---|---|---|---|
| 17-Allylamino-geldanamycin |
0.11 ± 0.03 |
N/D |
N/D |
N/D |
N/D |
| Acidum bromebricum |
32.77 ± 2.22 |
Inactive |
Inactive |
Inactive |
Inactive |
| BEZ-235 |
2.29 ± 2.9 |
N/D |
N/D |
N/D |
N/D |
| BIBX 1382 dihydrochloride |
12.08 ± 0.79 |
0.77 ± 0.05 (20%) |
3.47 ± 1.85 (44%) |
5.44 ± 0.75 |
11.66 ± 5.82 |
| Bleomycin |
0.23 ± 0.05 (42%) |
Inactive |
Inactive |
Inactive |
17.59 ± 4.90 (35%) |
| Bortezomib |
0.10 ± 0 |
Inactive |
0.93 ± 0.4 |
0.06 ± 0.011 |
0.005 ± 0.0005 |
| Cantharidin |
2.81 ± 0 |
Inactive |
0.63 ± 0 |
0.79 ± 0.09 |
0.34 ± 0.04 |
| Daunorubicin |
4.14 ± 0.53 |
N/D |
N/D |
N/D |
N/D |
| Digitoxin |
0.21 ± 0.04 |
N/D |
N/D |
N/D |
N/D |
| Digoxin |
0.27 ± 0.07 |
Inactive |
1.37 ± 0.18 (32%) |
1.14 ± 0.24 |
0.48 ± 0.03 |
| Ecteinascidin 743 |
42.06 ± 3.42 |
N/D |
N/D |
N/D |
N/D |
| Erlotinib (393.4) |
5.26 ± 3.29 |
0.26 ± 0.12 (20%) |
Inactive |
Inactive |
Inactive |
| GDC-0941 |
2.58 ± 0.71 |
N/D |
N/D |
N/D |
N/D |
| Gefitinib |
17.77 ± 9.03 |
1.00 ± 0.12 (27%) |
8.43 ± 3.67 (49%) |
15.27 ± 0.99 |
10.75 ± 3.18 |
| Homoharringtonine |
0.53 ± 0.53 (21%) |
N/D |
N/D |
N/D |
N/D |
| Idarubicin hydrochloride |
3.05 ± 0.42 |
N/D |
N/D |
N/D |
N/D |
| Imatinib mesylate |
26.03 ± 1.76 |
Inactive |
Inactive |
Inactive |
Inactive |
| Irinotecan hydrochloride |
31.14 ± 8.68 |
Inactive |
Inactive |
Inactive |
Inactive |
| MG-132 |
1.52 ± 0.10 |
N/D |
N/D |
N/D |
N/D |
| Narasin |
2.58 ± 0.83 |
14.76 ± 1.88 (26%) |
9.20 ± 3.85 |
11.72 ± 1.49 |
0.19 ± 0.03 |
| Oxyphenisatin |
6.71 ± 3.89 |
Inactive |
Inactive |
Inactive |
4.22 ± 1.84 |
| Plicamycin |
0.02 ± 0.002 (20%) |
Inactive |
0.12 ± 0.03 (35%) |
0.09 ± 0 |
0.05 ± 0.003 |
| Rapamycin |
35.48 ± 4.08 |
N/D |
N/D |
N/D |
N/D |
| Rubitecan |
0.32 ± 0.04 |
Inactive |
1.12 ± 0.13 (36%) |
0.71 ± 0.08 |
1.25 ± 0 |
| SB 202190 |
27.73 ± 13.16 |
Inactive |
Inactive |
Inactive |
31.83 ± 5.16 |
| SB-203580 |
51.38 ± 14.31 |
Inactive |
Inactive |
Inactive |
Inactive |
| SB431542 |
Inactive |
Inactive |
Inactive |
Inactive |
Inactive |
| SF1126 (LY294002 hydrochloride) |
13.48 ± 3.99 |
N/D |
N/D |
N/D |
N/D |
| Staurosporine |
0.24 ± 0.05 |
4.34 ± 0.74 |
0.96 ± 0.06 |
0.94 ± 0.17 |
0.07 ± 0.04 |
| Suberoylanilide hydroxamic acid |
11.22 ± 4.08 |
Inactive |
5.10 ± 1.16 (23%) |
11.00 ± 4.14 |
8.95 ± 1.02 |
| Sunitinib malate |
30.34 ± 1.98 |
Inactive |
Inactive |
Inactive |
Inactive |
| Trichostatin A |
1.04 ± 0.07 |
N/D |
N/D |
N/D |
N/D |
| Trimetrexate glucuronate |
0.38 ± 0.11 |
N/D |
N/D |
N/D |
N/D |
| Vandetanib |
11.31 ± 2.11 |
7.35 ± 4.79 |
14.19 ± 1.63 |
21.81 ± 11.86 |
18.51 ± 1.25 |
| Vincristine | 14.70 ± 1.87 | Inactive | 7.08 ± 0 (37%) | 9.73 ± 7.67 (49%) | 0.02 ± 0.004 (41%) |
N/D, not determined. Each IC50 or EC50 value (concentration of half the maximal inhibition or activation) is the mean ± SD of the results from 3 to 5 experiments. Efficacy (inhibition or activation as % positive control) of drugs was listed in parenthesis if it is less 50%.
Effect of chordoma inhibitors on primary chordoma cell cultures
To further investigate the effect of the drugs on primary chordoma cell cultures, 3 independent primary chordoma cell cultures, C24, C25, and C32, obtained from 3 chordoma patient donors, were used for the present study. To ensure that these primary cultures were composed of neoplastic chordoma cells and not contaminating stromal cells, we first measured the expression of Brachyury, a highly specific biomarker for chordoma cells.13 Immunoblot analysis of whole-cell extracts from primary chordoma cells with anti-Brachyury antibody shows that Brachyury is expressed in C24, C25, and C32 cells, but not in U87MG, a negative control (Fig. 2). U87MG is a line established from malignant glioma, a tumor that does not express Brachyury.
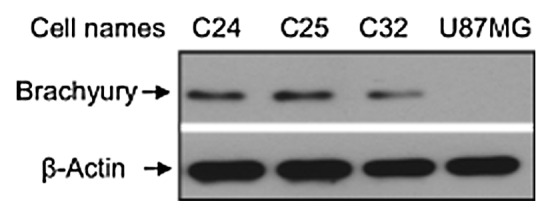
Figure 2. western blot analysis of Brachyury expression in chordoma primary culture cells. Protein samples (40 µg/lane) were prepared from the lysed cells, and western blot was performed with a specific Brachyury antibody. The Brachyury protein band was identified at 49 kDa.
To confirm the effect of compounds found to be active in chordoma cell lines on primary chordoma cells, we determined cell viability by measuring intracellular ATP content in primary chordoma cells, C24, C25, and C32, after 48 h of compound treatment. Compound activities in the chordoma lines U-CH1 and U-CH2 and individual primary chordoma cell cultures are compared in Figure 3. Some drugs, including cantharidin, digoxin, digitoxin, staurosporine, and bortezomib, showed similar inhibitory effect on all the cell types, while others had inhibitory effect on cell growth only in certain primary cultures. For example, homoharringtonine showed significant cytotoxicity in primary chordoma cells, C24 (IC50 = 0.69 µM), C25 (IC50 = 0.34 µM) and C32 (IC50 = 0.91 µM), with efficacy more than 80%, but marginal inhibition on U-CH1 (IC50 = 0.53 µM, efficacy 21%) and no inhibition on U-CH2 cells. Daunorubicin and idarubicin had a 10-fold more potent effect in U-CH1 (IC50 = 4.14 µM for daunorubicin and 3.05 µM for idarubicin) and C25 (IC50 = 2.32 µM for daunorubicin and 1.50 µM for idarubicin) cells than in U-CH2 (IC50 = 21.83 µM for daunorubicin and 56.45 µM for idarubicin), C24 (IC50 = 20.18 µM for daunorubicin and 65.65 µM for idarubicin) and C32 (IC50 = 55.27 µM for daunorubicin and 21.26 µM for idarubicin) cells. Receptor tyrosine kinases (RTKs) and their downstream signaling cascades have been implicated as possible targets involved in chordoma.6 In this study, we found that a group of tyrosine kinase inhibitors including erlotinib, gefitinib, imatinib, sunitinib, and vandetanib had modest growth inhibitor activity in most of these cells, except in C24 cells where erlotinib and vandetanib had significant inhibitory effect with IC50 of 5.26 µM and 5.05 µM, respectively.

Figure 3. Inhibitory effect of drugs on chordoma cell lines and patient cells. The drug activity in each cell line is colored according to potency (IC50) and efficacy (% inhibition) ranges.
Synergistic effect of chordoma inhibitors in combination treatment
Given the finding that some drugs, including bortezomib (Fig. 1C) and rubitecan (Fig. 1G), were less effective in U-CH2 than U-CH1, we evaluated the cytotoxic effect of the compounds in combination with selected anticancer compounds and found that the potency of bortezomib against U-CH2 improved significantly in the presence of topoisomerase inhibitors including rubitecan, camptothecin, topotecan, and doxorubicin; combination drug exposure had little differential effect on growth of U-CH1 cell line. The U-CH2 cell line was exposed to the 35 compounds in combination with 500 nM rubitecan, 500 nM camptothecin, 500 nM topotecan, or 500 nM doxorubicin for 48 h. Figure 4A−D shows that the potency of bortezomib improved significantly in the presence of rubitecan (IC50 = 68.6 nM, Fig. 4A, P = 0.036), camptothecin (IC50 = 84.7 nM, Fig. 4B, P = 0.036), topotecan (IC50 = 131.1 nM, Fig. 4C, P = 0.037), or doxorubicin (IC50 = 377.8 nM, Fig. 4D, P = 0.044). There was a 10- to 50-fold increase in bortezomib’s potency in the combination treatments relative to bortezomib treatment alone (IC50 = 3.93 µM) in the U-CH2 cell line. Similar combination studies were also performed in the primary chordoma cells, C24, C25, and C32. As shown in Figure 5A−L, bortezomib in combination with rubitecan, camptothecin, daunorubicin, or doxorubicin showed improved potencies in all 3 primary cell cultures, with an average 2- to 9-fold increase in bortezomib’s potency compared with bortezomib treatment alone. However, these effects in the primary chordoma cells, C24, C25, and C32 were not as pronounced as that found in U-CH2 cells. This may be due to the fact that bortezomib treatment alone was less potent in U-CH2 cells (IC50 = 3.93 µM) than in these primary chordoma cells (average IC50 = 0.4 µM).

Figure 4. Synergistic inhibitory effect of bortezomib and topoisomerase inhibitors on U-CH2 chordoma cell line. Cell viability was measured in the U-CH2 cells after the treatment of bortezomib in the presence of medium (■, DMSO control), (A) rubitecan (▲), (B) camptothecin (●), (C) topotecan (♦), or (D) doxorubicin (▼) for 48 h. Data are expressed as mean ± SD from 3 to 5 experiments.

Figure 5. Synergistic inhibitory effect of bortezomib and topoisomerase inhibitors on chordoma patient cells. Cell viability was measured in the C24 (A–D), C25 (E–H), or C32 (I–L) cells after the treatment of bortezomib in the presence of medium (■, DMSO control), rubitecan (▲), camptothecin (●), daunorubicin (♦), or doxorubicin (▼) for 48 h. Data are expressed as mean ± SD from 3 to 5 experiments.
Discussion
In the present study, we have identified more than 20 drugs (IC50 < 10 µM) that inhibited chordoma cell growth by screening a collection of approximately 2800 small molecule drugs that have been approved by regulatory agencies for human use or tested in clinical trials. Most of these drugs identified as chordoma inhibitors also activated caspase 3/7, in a similar rank order of potency to their cytotoxicity in a chordoma cell line. The correlation between the growth inhibition and caspase 3/7 activation potencies of these drugs suggests that the drugs may exert their cytotoxic effect on chordoma cell lines via apoptosis. Among these drugs, bortezomib, cantharidin, digoxin, digitoxin, and staurosporine showed an inhibitory effect on both chordoma cell lines and all 3 primary chordoma cell cultures, while others had selective inhibitory effect on cell growth in certain primary cell cultures. The inhibitory potencies of bortezomib against the U-CH2 human chordoma cell line and 3 primary chordoma cell cultures improved significantly when combined with some topoisomerase I (rubitecan, camptothecin, and topotecan) or topoisomerase II (doxorubicin) inhibitors. The differential drug effects on these primary cells may provide important information for developing personalized medicine for chordoma treatment. The results from this study indicate that the drugs identified from this screening could be further validated with in vivo animal model testing and may be potentially repurposed for chordoma treatment.
Bortezomib, a proteasome inhibitor, has been approved for the treatment of relapsed or refractory multiple myeloma and mantle-cell lymphoma.14 One possible mechanism of bortezomib, associated with its anti-myeloma cell growth and survival, is its ability to inhibit NFκB signaling. Bortezomib inhibits the 26S subunit of the proteasome, preventing IκB degradation and NFκB activation.15 Bortezomib was found to induce apoptosis in melanoma and myeloma cells via a p53-independent induction of NOXA,16 a pro-apoptotic member of the Bcl-2 family.17 Our previous study also demonstrated that bortezomib inhibited the NFκB signaling pathway and induced caspase 3/7 activity in two human cervical cancer cell lines, ME180 and HeLa cells.8
The current study showed that the combination treatment of bortezomib with DNA topoisomerase inhibitors, rubitecan, camptothecin, topotecan, and doxorubicin showed significant inhibition on cell growth of the U-CH2 cell line as well as 3 primary chordoma cell cultures. Camptothecin was first discovered to have anticancer activity in the 1960s.18 It was discovered later that the mechanism of its anticancer action is due to the inhibition of topoisomerase I.19 The other topoisomerase I inhibitors, topotecan and rubitecan, which are camptothecin analogs, are currently in clinical trials for ovarian cancer.19,20 There are two types of topoisomerases, the essential enzymes in DNA replication, topoisomerase I and II. Topoisomerase I binds to super-coiled DNA and cleaves a phosphodiester bond, causing single-stranded breaks and religation of DNA. Topoisomerase I also forms a cleavable complex between the strands to allow for relaxation of the DNA. Camptothecin and its analogs bind to the cleavable complex at topoisomerase I sites and inhibit the re-ligation reaction of topoisomerase I.21 Topoisomerase II functions similar to topoisomerase I except that it causes double-stranded breaks of the DNA in an ATP-dependent manner.21 Doxorubicin, an anthracycline antibiotic, targets topoisomerase II and is used clinically to treat multiple cancers including leukemia, soft tissue, and bone sarcoma, as well as breast cancer.22 Combination therapies using multiple anticancer drugs have become popular in the cancer field because these drugs have different mechanisms of action. In an age-stratified phase I trial, a combination of bortezomib, gemcitabine, and liposomal doxorubicin in patients with advanced malignancies is well tolerated and demonstrated effective antitumor activity in T-cell lymphoma and small cell carcinoma.23 Recently, a phase II trial demonstrated that the botezomib-containing combination treatment in elderly mantle cell lymphoma patients results in high complete responses and prolonged progression-free survival with predictable and manageable toxic effect.24 We also found a synergistic inhibitory activity when using a combination of botezomib and topoisomerase inhibitors in human chordoma cell lines and primary cell cultures, suggesting that this combination drug treatment might be a future direction for chordoma therapy.
Recent research efforts have begun to focus on the molecular targets and signaling pathways of chordoma in order to efficiently find anti-chordoma therapies.6 The primary possible targets involved in chordoma are receptor tyrosine kinases (RTKs) and their downstream signaling cascades including the PI3K/AKT/mTOR pathway.6 In this study we tested several RTK inhibitors including imatinib, sunitnib, vandetanib, erlotinib, and gefitinib, PI3K/mTOR inhibitors, BEZ-235 and rapamycin, and p38 MAP kinase inhibitors including SB 202190 and SB 203580 on chordoma cell lines and primary chordoma cell cultures. The targets of these broad spectrum kinase inhibitors include the vascular endothelial growth factor receptor, epidermal growth factor receptor and platelet derived growth factor receptor (α and β), all of which are expressed in chordomas.25 Most of these drugs have been approved for clinical cancer treatments or clinical trials. Imatinib has been approved for the treatment of chronic myeloid leukemia (CML) and gastrointestinal stromal tumors (GISTs) by inhibiting BCR-Abl protein and mutant c-kit, respectively. Sunitinib has been used in phase I–II clinical trials for treatment of acute myeloid leukemia by targeting VEFGR and FLT325 and is approved for treatment of renal cell cancer. BEZ-235 showed potent antitumor activity in vivo by dual inhibition of the PI3K and mTOR pathways26 and is currently in phase I-II clinical trials for treatment of advanced solid tumors. Rapamycin and its analogs, which target mTOR, are already in clinical use for treating cancers including renal cell carcinoma.27 In this study we found that BEZ-235 had a more potent inhibitory effect on chordoma cell growth than rapamycin, suggesting that dual inhibition of PI3K and mTOR may have a synergistic inhibitory effect on chordomas. However, RTK inhibitors, imatinib, sunitnib, vandetanib, erlotinib, and gefitinib, had only moderate inhibitory effects on chordoma cells. SB 202190 and SB 203580, which are p38 MAPK inhibitors and SB431542, a TGF-β receptor inhibitor, had minimal or no effect on the growth of chordoma cells. These results suggest that chordoma cell growth is dependent on signaling through an as yet unidentified kinase upstream of PI3K and mTOR.
In summary, we have identified several clinically approved drugs and combinations of drugs that may have potential clinical application for the treatment of chordomas. In the next step, the drugs identified in this study that inhibited chordoma cell growth will be further investigated in chordoma xenograft models currently under development and validation. The significance of this study is to identify clinically approved drugs and drug combinations that can be repurposed as chordoma therapies. These results support designing the clinical trials for treatment of chordoma patients utilizing these drugs singly or in combination with the therapies guided by publicly available human safety data.
Materials and Methods
Cell line and culture conditions
Human chordoma cell lines U-CH19 and U-CH210 were kindly provided by Dr Silke Bruderlein (Institute of Pathology, Ulm University). Cell line CCL4, which is human cell line not derived from chordoma,10 was kindly provided by Dr Chris Hunter (University of Calgary). Human primary chordoma cell cultures C24, C25, and C32 were obtained from chordoma patient donors through an Institutional Review Board approved protocol at the University of Pittsburgh Medical Center. Both U-CH1 and U-CH2 cells were seeded in gelatin-coated flasks and cultured in IMDM/RPMI (4:1) medium supplemented with 10% heat inactivated fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin. Primary chordoma cells were cultured in DMEM medium supplemented with 10% heat inactivated fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin and 20 ng/mL human PDGF AB (R&D Systems). All cell culture media and reagents were obtained from Invitrogen. The cells were maintained in a 37 °C incubator with 5% CO2 and under a humidified atmosphere, except for the human primary chordoma cells that were cultured in a 37 °C incubator with 5% CO2/1% O2.
NCGC Pharmaceutical Collection and additional compounds used in this study
The NCGC Pharmaceutical Collection (NPC) was constructed and prepared as previously described.7 Briefly, the NPC at the time of this study consisted of 2816 small molecule compounds, of which 52% are drugs approved for human or animal use by the United States Food and Drug Administration (FDA), 22% are drugs approved in Europe, Canada, or Japan, and the remaining 25% are drugs approved in other countries or compounds that have been tested in clinical trials.
The compounds were prepared in dimethylsulfoxide (DMSO) to a stock concentration of either 4.5 or 10 mM stock solutions. The compounds were prepared first in 96 or 384 well plates and then consolidated into 1536 well plates using an Evolution P3 system (PerkinElmer, Inc.). For use in quantitative high throughput screening, each compound in the NPC compound library was prepared at 15 2.23-fold serial dilutions. After dilutions, the NPC plates were stored desiccated at room temperature for as long as 6 mo when in use, or heat sealed and stored at -80 °C for long-term storage.8
Acidum bromebericum, cantharidin, daunorubicin, digitoxin, digoxin, idarubicin hydrochloride, narasin, plicamycin, rubitecan, SF1126, and staurosporine were purchased from Sigma-Aldrich. BIBX 1382 dihydrochloride, homoharringtonine, rapamycin, SB202190, SB431542, trichostatin A, and trimeterxate glucuronate were purchased from Tocris Bioscience. Erlotinib, imatinib, and vandetanib were purchased from Sequoia Research Products. Allylamino-17-demethoxy-geldanamycin (17-AAG), bortezomib, and sunitinib malate were purchased from LC Laboratories. BEZ-235 and suberoylanilide hydroxamic acid were purchased from Toronto Research Chemicals, Inc. SB203580 and vincristine were purchased from Enzo Life Sciences, Inc. Bleomycin was purchased from MicroSource Discovery Systems, Inc. GDC-0941 was purchased from Axon Medchem. Gefitinib was purchased from AKos Consulting & Solutions GmBH. Irinotecan was purchased from Chem Pacific Corp. Oxyphenisatin was purchased from Labotest. MG-132 was purchased from AG Scientific Inc. Ectinascidin 743 was provided by the National Cancer Institute.
Cell viability assay and qHTS
Cell viability of U-CH1, U-CH2 and CCL4 cells, was measured using a luciferase-coupled ATP quantitation assay (CellTiter-Glo viability assay, Promega) after compound treatment. The change in intracellular ATP content indicates the number of metabolically competent cells. The cells were suspended in culture medium and dispensed at 500 to 1000 cells in 5 μL media per well in 1,536-well white/solid bottom plates (Greiner Bio-One North America) using a Flying Reagent Dispenser (FRD) (Aurora Discovery). After the cells were incubated at 37 °C overnight, 23 nL of compounds at 15 concentrations from the NPC was transferred to the assay plate by a pin tool (Kalypsys). The final concentration of the compounds in the 5 μl assay volume ranged from 0.6 nM to 46 μM. The positive control plate format was as follows: Column 1, potassium dichromate, a cytotoxic agent,28 ranged from 2.8 nM to 92 µM; column 2, staurosporine, a caspase 3/7 inducer,29 ranged from 2.8 nM to 92 µM; column 3, DMSO only; and columns 4, 92 µM potassium dichromate. The plates were incubated for 48 h at 37 °C, followed by the addition of 5 μL per well of CellTiter-Glo reagent. After 30 min incubation at room temperature, the luminescence intensity of the plates was measured using a ViewLux plate reader (PerkinElmer).
In the follow-up study, 35 selected active drugs were re-tested in U-CH1 and U-CH2 cell lines and evaluated in primary human chordorma cells C24, C25, and C32 using 24 drug concentrations ranging from 5.5 pM to 46 µM in the cell viability assay protocol described above except that the 24 point titrations were within one 1536-well plate.
In the drug combination study, these 35 compounds were tested in U-CH1, U-CH2, C24, C25, and C32 cells using the same protocol as above in the presence of a single concentration around the IC20 value of one of the following compounds: BIBX (300 nM), bortezomib (5.2 nM), rapamycin (20 nM), SAHA (950 nM), erlotinib (120 nM), topotecan (500 nM), camptothecin (500 nM), rubitecan (500 nM), daunorubicin (500 nM), and doxorubicin (500 nM). The combination treatment with two drugs was performed simultaneously. The statistical significance of the IC50 differences between single and combination treatment was evaluated by a Student t test. Values of P < 0.05 were considered significant.
Caspase 3/7 assay
Caspase 3/7 activity was measured as previously described.30 U-CH1 and U-CH2 cells were dispensed in culture medium at 500 to 1000 cells in 5 μL per well in 1536-well white/solid-bottom assay plates. The cells were incubated a minimum of 5 h at 37 °C. The compounds (23 nL per well) were added via the pin tool. Treated cells were incubated for 16 h at 37 °C, followed by the addition of the Caspase-Glo 3/7 (Promega) reagent at 5 μL per well. After 30 min incubation at room temperature, the luminescence intensity of the assay plates was measured using a ViewLux plate reader.
Immunoblot and expression analyses of Brachyury in human primary chordoma cells
Protein immunoblot analyses were performed as previously described.31 Briefly, proteins were separated by SDS-PAGE and transferred to nitrocellulose membrane. Membrane was blocked with 5% nonfat dry milk in PBS-Tween-20 (0.1%, v/v) at 4 °C overnight and incubated for 2 h with Brachyury antibody (R&D Systems, AF2085), at a dilution recommended in the manufacturer’s instructions. Horseradish peroxidase conjugated anti-mouse IgG was used as the secondary antibody. Immunoreactive proteins were visualized by chemiluminescence (ECL).
qHTS data analysis
Concentration response data were analyzed as previously described.28 Briefly, data from raw plate reads for each titration point were normalized to the neutral (DMSO, 0%) and positive (92 µM potassium dichromate, −100%) controls and corrected by applying a NCGC in-house pattern correction algorithm. Concentration response curves were fitted to the Hill equation yielding concentrations of half-maximal inhibition (IC50) and maximal cytotoxicity (efficacy) values. The concentration response curves were classified into four major classes (1−4) using previously published criteria.28 Curve classes were further subdivided to provide more detailed classification. Briefly, compounds with curve class 1.1, 1.2, 2.1, or 2.2 (efficacy >50%) were considered active. Class 4 compounds with no concentration response were defined as inactive; and compounds with other curve classes were defined as inconclusive. The compounds active in any chordoma lines were further prioritized based on their activity, selectivity relative to the CCL4 control cell line (i.e., more potent in the chordoma line than the CCL4 line), and the mechanism of action, resulting in a total of 60 compounds selected for confirmation in U-CH1 and U-CH2 cells. Among these 60 compounds, staurosporine, SB-202190, SB-203580, and SB-431542, which were not tested in the primary screening, were also selected based on their mode of action. In addition, 35 compounds were purchased from commercial vendors and re-tested in all the chordoma cells.
Acknowledgments
We gratefully acknowledge Dr Gurusingham Sittampalam for critical reading of the manuscript. These studies were supported by the Intramural Research Program of the National Center for Advancing Translational Sciences, National Institutes of Health.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/24596
References
- 1.Soo MY. Chordoma: review of clinicoradiological features and factors affecting survival. Australas Radiol. 2001;45:427–34. doi: 10.1046/j.1440-1673.2001.00950.x. [DOI] [PubMed] [Google Scholar]
- 2.Chugh R, Tawbi H, Lucas DR, Biermann JS, Schuetze SM, Baker LH. Chordoma: the nonsarcoma primary bone tumor. Oncologist. 2007;12:1344–50. doi: 10.1634/theoncologist.12-11-1344. [DOI] [PubMed] [Google Scholar]
- 3.McMaster ML, Goldstein AM, Bromley CM, Ishibe N, Parry DM. Chordoma: incidence and survival patterns in the United States, 1973-1995. Cancer Causes Control. 2001;12:1–11. doi: 10.1023/A:1008947301735. [DOI] [PubMed] [Google Scholar]
- 4.Casali PG, Stacchiotti S, Sangalli C, Olmi P, Gronchi A. Chordoma. Curr Opin Oncol. 2007;19:367–70. doi: 10.1097/CCO.0b013e3281214448. [DOI] [PubMed] [Google Scholar]
- 5.Amichetti M, Cianchetti M, Amelio D, Enrici RM, Minniti G. Proton therapy in chordoma of the base of the skull: a systematic review. Neurosurg Rev. 2009;32:403–16. doi: 10.1007/s10143-009-0194-4. [DOI] [PubMed] [Google Scholar]
- 6.Barry JJ, Jian BJ, Sughrue ME, Kane AJ, Mills SA, Tihan T, et al. The next step: innovative molecular targeted therapies for treatment of intracranial chordoma patients. Neurosurgery. 2011;68:231–40, discussion 240-1. doi: 10.1227/NEU.0b013e3181fd2ac5. [DOI] [PubMed] [Google Scholar]
- 7.Huang R, Southall N, Wang Y, Yasgar A, Shinn P, Jadhav A, et al. The NCGC pharmaceutical collection: a comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics. Sci Transl Med. 2011;3:80ps16. doi: 10.1126/scitranslmed.3001862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller SC, Huang R, Sakamuru S, Shukla SJ, Attene-Ramos MS, Shinn P, et al. Identification of known drugs that act as inhibitors of NF-kappaB signaling and their mechanism of action. Biochem Pharmacol. 2010;79:1272–80. doi: 10.1016/j.bcp.2009.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheil S, Brüderlein S, Liehr T, Starke H, Herms J, Schulte M, et al. Genome-wide analysis of sixteen chordomas by comparative genomic hybridization and cytogenetics of the first human chordoma cell line, U-CH1. Genes Chromosomes Cancer. 2001;32:203–11. doi: 10.1002/gcc.1184. [DOI] [PubMed] [Google Scholar]
- 10.Brüderlein S, Sommer JB, Meltzer PS, Li S, Osada T, Ng D, et al. Molecular characterization of putative chordoma cell lines. Sarcoma. 2010;2010:630129. doi: 10.1155/2010/630129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 12.Wang S. The promise of cancer therapeutics targeting the TNF-related apoptosis-inducing ligand and TRAIL receptor pathway. Oncogene. 2008;27:6207–15. doi: 10.1038/onc.2008.298. [DOI] [PubMed] [Google Scholar]
- 13.Vujovic S, Henderson S, Presneau N, Odell E, Jacques TS, Tirabosco R, et al. Brachyury, a crucial regulator of notochordal development, is a novel biomarker for chordomas. J Pathol. 2006;209:157–65. doi: 10.1002/path.1969. [DOI] [PubMed] [Google Scholar]
- 14.Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets. 2011;11:239–53. doi: 10.2174/156800911794519752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pascal L, Gay J, Willekens C, Wemeau M, Balkaran S, Robu D, et al. Bortezomib and Waldenstrom’s macroglobulinemia. Expert Opin Pharmacother. 2009;10:909–16. doi: 10.1517/14656560902800160. [DOI] [PubMed] [Google Scholar]
- 16.Qin JZ, Ziffra J, Stennett L, Bodner B, Bonish BK, Chaturvedi V, et al. Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res. 2005;65:6282–93. doi: 10.1158/0008-5472.CAN-05-0676. [DOI] [PubMed] [Google Scholar]
- 17.Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–8. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 18.Oberlies NH, Kroll DJ. Camptothecin and taxol: historic achievements in natural products research. J Nat Prod. 2004;67:129–35. doi: 10.1021/np030498t. [DOI] [PubMed] [Google Scholar]
- 19.Pizzolato JF, Saltz LB. The camptothecins. Lancet. 2003;361:2235–42. doi: 10.1016/S0140-6736(03)13780-4. [DOI] [PubMed] [Google Scholar]
- 20.Wethington SL, Wright JD, Herzog TJ. Key role of topoisomerase I inhibitors in the treatment of recurrent and refractory epithelial ovarian carcinoma. Expert Rev Anticancer Ther. 2008;8:819–31. doi: 10.1586/14737140.8.5.819. [DOI] [PubMed] [Google Scholar]
- 21.Mathijssen RH, Loos WJ, Verweij J, Sparreboom A. Pharmacology of topoisomerase I inhibitors irinotecan (CPT-11) and topotecan. Curr Cancer Drug Targets. 2002;2:103–23. doi: 10.2174/1568009023333890. [DOI] [PubMed] [Google Scholar]
- 22.Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol. 1999;57:727–41. doi: 10.1016/S0006-2952(98)00307-4. [DOI] [PubMed] [Google Scholar]
- 23.Falchook GS, Duvic M, Hong DS, Wheler J, Naing A, Lim J, et al. Age-stratified phase I trial of a combination of bortezomib, gemcitabine, and liposomal doxorubicin in patients with advanced malignancies. Cancer Chemother Pharmacol. 2012;69:1117–26. doi: 10.1007/s00280-011-1808-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Houot R, Le Gouill S, Ojeda Uribe M, Mounier C, Courby S, Dartigeas C, et al. French GOELAMS group Combination of rituximab, bortezomib, doxorubicin, dexamethasone and chlorambucil (RiPAD+C) as first-line therapy for elderly mantle cell lymphoma patients: results of a phase II trial from the GOELAMS. Ann Oncol. 2012;23:1555–61. doi: 10.1093/annonc/mdr450. [DOI] [PubMed] [Google Scholar]
- 25.Petrelli A, Giordano S. From single- to multi-target drugs in cancer therapy: when aspecificity becomes an advantage. Curr Med Chem. 2008;15:422–32. doi: 10.2174/092986708783503212. [DOI] [PubMed] [Google Scholar]
- 26.Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–63. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 27.Ballou LM, Lin RZ. Rapamycin and mTOR kinase inhibitors. J Chem Biol. 2008;1:27–36. doi: 10.1007/s12154-008-0003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia M, Huang R, Witt KL, Southall N, Fostel J, Cho MH, et al. Compound cytotoxicity profiling using quantitative high-throughput screening. Environ Health Perspect. 2008;116:284–91. doi: 10.1289/ehp.10727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yue TL, Wang C, Romanic AM, Kikly K, Keller P, DeWolf WE, Jr., et al. Staurosporine-induced apoptosis in cardiomyocytes: A potential role of caspase-3. J Mol Cell Cardiol. 1998;30:495–507. doi: 10.1006/jmcc.1997.0614. [DOI] [PubMed] [Google Scholar]
- 30.Huang R, Southall N, Cho MH, Xia M, Inglese J, Austin CP. Characterization of diversity in toxicity mechanism using in vitro cytotoxicity assays in quantitative high throughput screening. Chem Res Toxicol. 2008;21:659–67. doi: 10.1021/tx700365e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD, et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene. 2009;28:3949–59. doi: 10.1038/onc.2009.252. [DOI] [PubMed] [Google Scholar]