

Abstract
The transformation of colonic mucosal epithelium to adenocarcinoma requires progressive oncogene activation and tumor suppressor gene inactivation. Loss of chromosome 18q is common in colon cancer but not in precancerous adenomas. A few candidate tumor suppressor genes have been identified in this region, including CABLES1 at 18q11.2−12.1. This study investigates the role of CABLES1 in an in vivo mouse model of intestinal adenocarcinoma and in human colon cancer cell culture. ApcMin/+ mice were crossed with mice harboring targeted inactivation of the Cables1 gene (Cables1−/−). The intestinal tumor burden and tumor expression of β-catenin and PCNA was compared in Cables1+/+ApcMin/+ and Cables1−/−ApcMin/+ mice. β-catenin activity in human colon cancer cells with CABLES1 inactivation and intestinal progenitor cell function in Cables1−/− mice were assayed in vitro. The mean number of small intestinal tumors per mouse was 3.1 ± 0.6 in Cables1+/+ ApcMin/+ mice, compared with 32.4 ± 3.5 in the Cables1−/−ApcMin/+ mice (P < 0.0001). Fewer colonic tumors were observed in Cables1+/+ApcMin/+ mice (mean 0.6 ± 0.1) compared with the Cables1−/−ApcMin/+ mice (mean 1.3 ± 0.3, P = 0.01). Tumors from Cables1−/−ApcMin/+ mice demonstrated increased nuclear expression of β-catenin and an increased number of PCNA-positive cells. In vitro studies revealed that CABLES1 deficiency increased β-catenin dependent transcription and increased intestinal progenitor cell activity. Loss of Cables1 enhances tumor progression in the ApcMin/+ mouse model and activates the Wnt/β-catenin signaling pathway. Cables1 is a tumor suppressor gene on chromosome 18q in this in vivo mouse model and likely has a similar role in human colon cancer.
Keywords: ApcMinmice, Cables1, adenocarcinoma, chromosome 18q, colorectal cancer, tumor suppressor gene, β-catenin
Introduction
In the pathogenesis of colorectal cancer (CRC) via the classic chromosomal instability pathway, the colonic epithelium is thought to require an ordered activation of specific oncogenes and inactivation of tumor suppressor genes as the tumor progresses through an adenomatous intermediate prior to adenocarcinoma. Inactivation of the adenomatous polyposis coli (APC) tumor suppressor gene is one of the earliest events, followed by oncogenic KRAS mutations.1 Later events include activation of PIK3CA, inactivation of the tumor suppressor gene TP53 and loss of heterozygosity (LOH) on chromosome 18q.1
There is allelic deletion of chromosome 18q in approximately 70% of human CRCs, and several candidate tumor suppressor genes, including Deleted in Colorectal Carcinoma (DCC), SMAD4, and SMAD2, have been identified in this region.2,3 The expression of DCC is lost or markedly reduced in the majority of colorectal cancers.4 However, based on the observations that DCC encodes a cell surface receptor for the neuronal protein netrin-1, DCC mutations are rare in human colorectal cancers (6%), and mice with inactivating mutations of Dcc do not develop intestinal tumors, some doubts have been raised about the function of DCC as a tumor suppressor in colon cancer.4,5 The SMAD proteins are intracellular mediators of the transforming growth factor-β pathway that regulate cell growth, differentiation, and apoptosis. Although biallelic inactivation of SMAD4 occurs in greater than 60% of pancreatic tumors, mutations of SMAD4 and SMAD2 have been found in less than 20% and 10% of colon cancers, respectively.6,7 Thus, alterations in the DCC and SMAD genes are unlikely to fully account for all the 18q deletions in colorectal cancer, suggesting the existence of other tumor suppressor genes on chromosome 18q.
The CABLES1 gene maps to chromosome 18q11.2–12.1 and may be a proximal 18q gene involved in human colon cancer.8 Functionally, the Cables1 protein acts as a linker or “cable” enhancing cyclin-dependent kinase (CDKs 2, 3, and 5) tyrosine phosphorylation by non-receptor tyrosine kinases (Src, Abl, Wee1) to modulate CDK activity.8,9 Cables1 also interacts with p53 and has been reported to potentiate p53-induced cell death. In neurons, Cables1 links Robo-bound Abl kinase to N-cadherin bound β-catenin, resulting in a rapid loss of cadherin-mediated adhesion and activation of gene transcription.10 Loss of nuclear Cables1 expression has been observed in head and neck, lung, ovarian, endometrial and colon cancers.8,11,12 Cables1 expression, evaluated by immunohistochemistry, is decreased or absent in 65% of primary CRCs.13 Loss of Cables1 expression is likely due to hypermethylation of CpG islands in the promoter of CABLES1, coupled with 18q LOH.13 In Cables1-knockout mice (Cables1−/−), there is an increased incidence of both endometrial hyperplasia and endometrial cancer following prolonged exposure to unopposed estrogen.14 Furthermore, in response to the carcinogen 1,2-dimethylhydrazine, Cables1−/− mice have an increased incidence of intestinal tumors and reduced survival.15 Primary mouse embryonic fibroblasts from Cables1−/− mice exhibit an increased rate of cell proliferation and delayed onset of senescence.16
To determine whether the Cables1 gene modulates the progression of colonic tumors in a mouse model, we crossed Cables1−/− mice with mice carrying a truncating mutation in the Apc tumor suppressor gene (ApcMin/+ mice). Our analysis of tumor burden in Cables1+/+ ApcMin/+ mice compared with Cables1−/− ApcMin/+ mice indicates a role for Cables1 in intestinal tumor progression. In vitro assays suggest that loss of Cables1 expression results in activation of the Wnt/β-catenin signaling pathway.
Results
Identification of Cables1−/− ApcMin/+ and Cables1+/+ ApcMin/+ mice
To determine whether Cables1 modulates late events in colorectal cancer progression, Cables1−/− mice were crossed with ApcMin/+ mice, which are predisposed to the development of multiple adenomas in the small intestine and in the colon. Cables1 and Apc genotypes were determined by PCR analysis of tail-snip genomic DNA using specific primers (Fig. 1A). Nine Cables1−/− ApcMin/+ mice were identified following genotype analysis, forming the Cables1 gene knockout group. These were compared with 15 Cables1+/+ ApcMin/+ mice, which also had their genotypes confirmed by PCR. Immunohistochemistry confirmed loss of Cables1 nuclear staining in the Cables1−/− ApcMin/+ and intact Cables1 nuclear staining in Cables1+/+ ApcMin/+ mice (Fig. 1B and C). Seventeen mice heterozygous for Cables1 (Cables1+/− ApcMin/+ mice) were also identified as a control group for comparison with the two study groups.
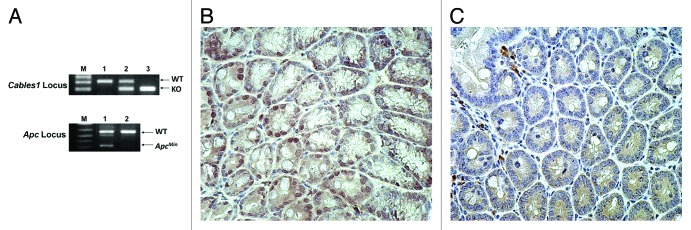
Figure 1. (A) PCR genotyping of Cables1 and Apc. In the upper panel, lanes 1, 2, and 3 represent the Cables1 wild-type, heterozygous, and homozygous genotypes, respectively. WT, wild-type fragment (~300 bp); KO, knockout fragment (~200 bp). In the lower panel, lanes 1 and 2 represent the ApcMin/+ and Apc wild type genotypes, respectively. WT fragment (~600 bp); ApcMin fragment (~340 bp). (B) Intact nuclear Cables1 expression by immunohistochemistry in Cables1+/+ ApcMin/+ mouse intestinal epithelial cells. (C) Loss of nuclear Cables1 expression by immunohistochemistry in Cables1−/−ApcMin/+ mouse intestinal epithelial cells. A few inflammatory cells retain Cables1 expression.
Quantification of tumor burden in Cables1−/− ApcMin/+ and Cables1+/+ ApcMin/+ mice
Nine Cables1−/− ApcMin/+ mice and 15 Cables1+/+ ApcMin/+ mice were euthanized at a mean age of 15.4 and 15.8 weeks respectively. Several mice in the Cables1−/− ApcMin/+ group died or became moribund before termination of the experiment as a consequence of intestinal obstruction and perforation. The number of intestinal and colonic tumors was counted histologically by assessment of a single H&E section of the intestine “Swiss” roll (Fig. 2A and B). A total of 304 tumors were found in the 9 Cables1−/− ApcMin/+ mice (292 in the small intestine and 12 in the colon), as compared with a total of 58 tumors in the 15 Cables1+/+ ApcMin/+ control mice (49 in the small intestine and 9 in the colon, respectively; P < 0.001). As shown in Table 1, the mean number of small intestinal tumors per mouse was significantly higher (P < 0.0001) in the Cables1−/− ApcMin/+ mice (mean 32.4 ± 3.5 tumors) compared with the Cables1+/+ ApcMin/+ mice (mean 3.1 ± 0.6 tumors). Similarly, the mean number of colonic tumors per mouse was increased (P = 0.01) in the Cables1−/− ApcMin/+ mice (mean 1.3 ± 0.3 tumors) compared with the Cables1+/+ ApcMin/+ mice (mean 0.6 ± 0.1 tumors). Seventeen mice heterozygous for Cables1 (Cables1+/− ApcMin/+) were sacrificed and there was no significant difference (P = 0.08) in mean tumor number between this group and mice with homozygous wild-type Cables1 genotype (Cables1+/+ ApcMin/+ mice). Furthermore, although histopathological analysis revealed that the majority of tumors in the Cables1−/− ApcMin/+ mice were adenomas, three adenocarcinomas, one in the small intestine and two in the colon, were observed in two Cables1 knockout mice (Fig. 2C). No adenocarcinomas were observed in the Cables1+/+ ApcMin/+ mice.
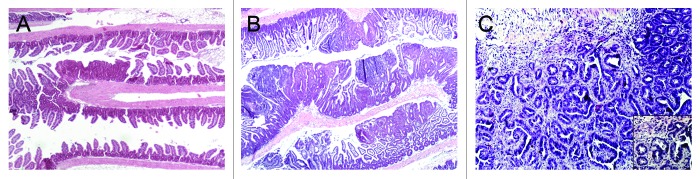
Figure 2. (A) Hematoxylin and eosin (H&E) stained small intestine from a Cables1+/+ApcMin/+ mouse with rare adenomas. (B) H&E stained small intestine from a Cables1−/− ApcMin/+ mouse with frequent adenomas. (C) An adenocarcinoma in the small intestine of a Cables1−/− ApcMin/+ mouse. Inset: higher magnification.
Table 1. Tumor burden in Cables1+/+ ApcMin/+ mice and Cables1−/− ApcMin/+mice.
| Group | Cables1+/+ ApcMin/+ | Cables1−/− ApcMin/+ | Significance (Student’s t-test) |
|---|---|---|---|
| Number of mice examined |
15 |
9 |
- |
| Mean number of small intestine tumors (± SE) |
3.1 ± 0.6 |
32.4 ± 3.5 |
P < 0.0001 |
| Mean number of colon tumors (± SE) | 0.6 ± 0.1 | 1.3 ± 0.3 | P = 0.01 |
β-catenin and PCNA IHC in tumor tissue from Cables1−/− ApcMin/+ and Cables1+/+ ApcMin/+ mice
Adenomas in the Cables1+/+ ApcMin/+ mice demonstrated membranous and weak cytoplasmic staining for β-catenin by IHC, with minimal nuclear immunopositivity (Fig. 3A). Tumor tissue from Cables1−/− ApcMin/+ mice revealed an increased percentage of cells with nuclear β-catenin staining and an increased intensity of nuclear β-catenin staining (Fig. 3B). When evaluated semi-quantitatively, there was a significantly higher mean nuclear β-catenin IHC score (P = 0.002) in the Cables1−/− ApcMin/+ mice (162 ± 25.2) compared with Cables1+/+ ApcMin/+ mice (20.6 ± 13.1), as shown in Figure 4A. The mean number of PCNA-positive cells per high power field of tumor tissue from the Cables1−/− ApcMin/+ mice was 310.2 ± 13.4 as compared with 119.2 ± 6.7 in the Cables1+/+ ApcMin/+ mice (P < 0.0001). Whereas proliferating cells were confined to the proliferative zone in the lower half of the crypt adjacent to the base (transient amplifying cell region) in the wild type mice, a more diffuse distribution of positive cells was observed in the Cables1-knockout mice. A significant correlation (r = 0.70, P = 0.0006) was observed between nuclear expression of β-catenin and PCNA.
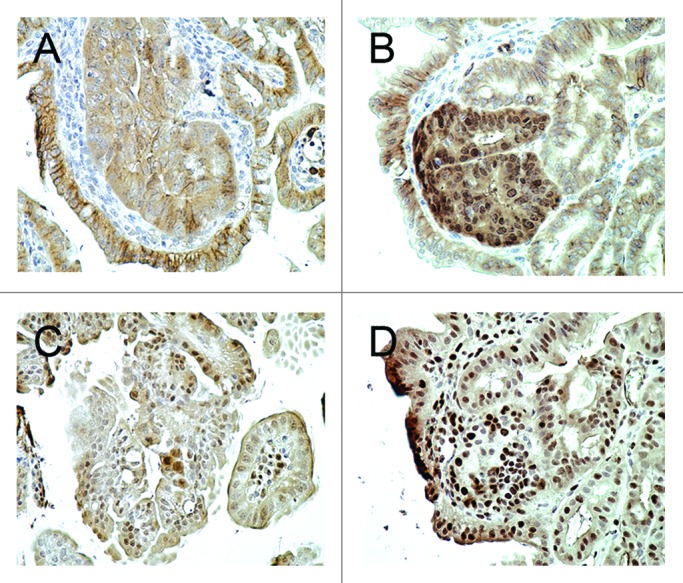
Figure 3. (A) Membranous and cytoplasmic β-catenin immunohistochemical staining was observed in the adenomas of Cables1+/+ ApcMin/+ mice, but there was minimal nuclear staining. (B) In the adenomas of Cables1−/− ApcMin/+ mice, there was nuclear immunopositivity for β-catenin. (C) A low proportion of epithelial cells were PCNA positive in adenomas from Cables1+/+ ApcMin/+ mice. (D) In the adenomas of Cables1−/− ApcMin/+ mice, a high proportion of epithelial cells were PCNA-positive by immunohistochemistry.
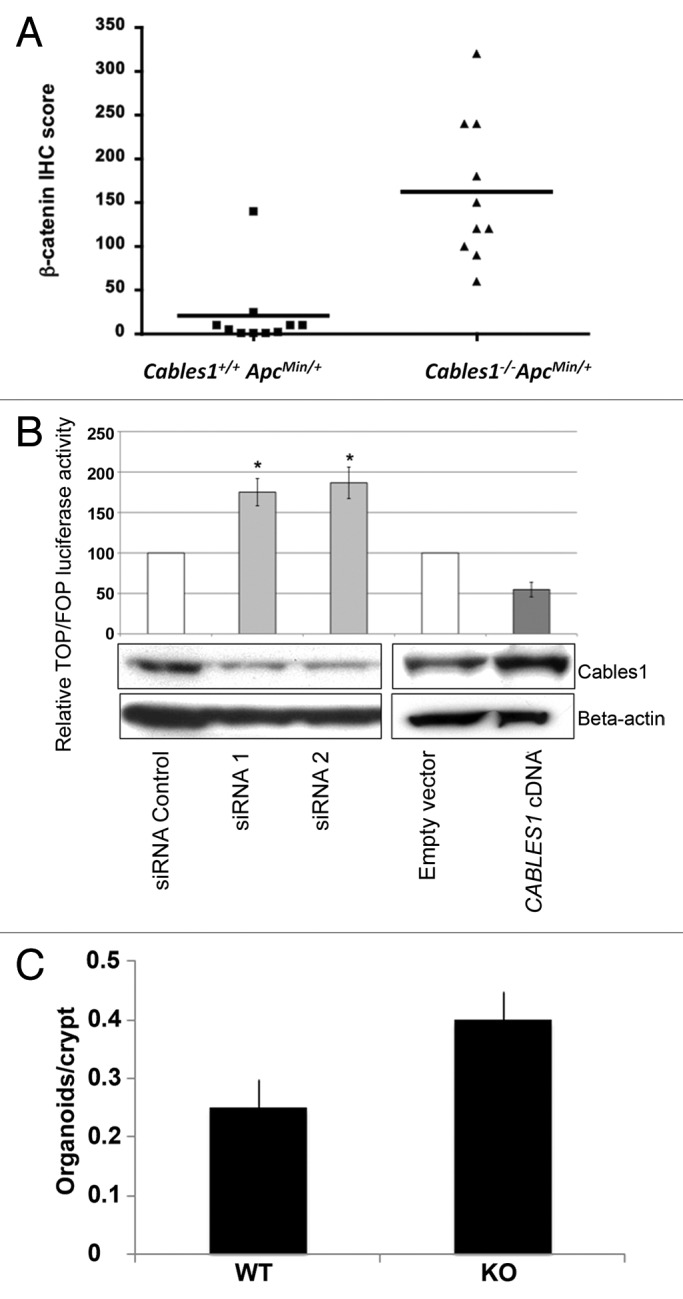
Figure 4. (A) β-catenin immunohistochemistry (IHC) scores of Cables1+/+ ApcMin/+ and Cables1−/− ApcMin/+ mice. A horizontal line indicates the median in each group. There was a significantly higher mean nuclear β-catenin IHC score (P = 0.002) in the Cables1−/− ApcMin/+ mice. (B) SW480 cells were transfected with either small interfering RNA (siRNA) targeting endogenous CABLES1 or with CABLES1 cDNA. Twelve hours later, cells were transfected with a β-catenin responsive (pGL3-OT) or a mutant (pGL3-OF) reporter construct and luciferase activity was measured. Results are expressed as the TOP/FOP ratio. The CABLES1 siRNA was associated with significantly increased TOP/FOP reporter activity compared with cells transfected with a control siRNA. Overexpression of Cables1 following CABLES1 cDNA transfection was associated with decreased TOP/FOP reporter activity compared with controls. Columns: average of at least two independent experiments; bars: standard error of mean (SEM). *P < 0.05 as compared with control cells. Protein levels of Cables1 were confirmed by western blot in lower panel. (C) Small intestinal crypts isolated from Cables1 knockout mice (KO) were capable of nearly 2-fold greater (P < 0.05) organoid formation than crypts from wild type mice (WT), indicating increased intestinal progenitor cell activity in the Cables1 knockout mice.
In vitro assessment of β-catenin expression with siRNA induced CABLES1 knockdown
We investigated whether Cables1 regulates β-catenin-dependent transcription in vitro in human colon cancer cell culture (SW480 cells) using a TOP/FOP reporter assay. Knockdown of CABLES1 gene expression by siRNA resulted in an increase in TOP/FOP reporter activity of almost 2-fold when compared with cells transfected with a control siRNA (Fig. 4B). Conversely, overexpression of CABLES1 led to a 46% decrease in TOP/FOP reporter activity compared with controls.
Intestinal progenitor cell function in Cables1−/− mice
To test how loss of Cables1 influenced intestinal progenitor function we isolated small intestinal crypts from one-year-old wild type and Cables1 knockout (Cables1−/−) mice and assayed the ability of these crypts to give rise to “mini-intestines” or organoid bodies in culture.17-19 These small intestinal crypts posses all of the intestinal stem and progenitor cells that give rise to all of the differentiated intestinal epithelial cell types. We found that Cables1-deficient crypts were nearly 2-fold more clonogenic in culture than crypts from wild-type mice, indicating that they contain more stem cell or progenitor cell activity (Fig. 4C).
Discussion
Loss of heterozygosity (LOH) for chromosome 18q has been implicated in the stepwise progression of human colonic mucosal epithelium to dysplasia and adenocarcinoma. Approximately 1/3 of colonic adenomas with low-grade dysplasia have LOH of 18q.20 The proportion of adenomas with high-grade dysplasia and adenocarcinomas with LOH of 18q has been reported to exceed 70%.20 However, the identity of the relevant tumor suppressor gene on 18q has remained elusive. DCC was one of the first candidate genes proposed, but multiple lines of evidence have not supported such a role. In particular, a Dcc null mutation does not affect intestinal polyp initiation, number, size, or morphology in ApcMin/+ mice.5 SMAD2 and SMAD4 also localize to chromosome 18q. One study demonstrated that tumor growth and invasion were accelerated by loss of Smad2 in Apc mutant mice, but another study failed to support a significant role in the malignant progression of polyps.21,22 Deletions of Smad4 appear to more consistently accelerate tumor progression in Apc mutant mice.23,24 However, SMAD4 mutations are present in less than 20% of human colorectal cancers.6 Cables1 expression, evaluated by immunohistochemistry, is decreased or absent in 65% of primary CRCs and there is evidence for frequent methylation induced epigenetic silencing of the CABLES1 gene in human colorectal cancer.13 LOH has been shown to be more frequent in the proximal portion of chromosome 18q containing the CABLES1 gene (18q11.1-q12.2) than in the more distal region, which is the location of DCC, SMAD2 and SMAD4 (18q21.1–q21.3).13
In this study, inactivation of Cables1 was associated with a marked (approximately 10-fold) increase in small intestinal tumor burden in vivo in the ApcMin/+ model compared with ApcMin/+ mice with wild type Cables1. The mean tumor burden of 32.4 ± 3.5 small intestinal tumors counted in the Cables1−/− ApcMin/+ mice may actually fall short of the true tumor burden in mice with this genotype, as several of these mice died due to intestinal obstruction and perforation prior to termination of the experiment and were not included in the final analysis. It was observed that mice heterozygous for Cables1 had no significant difference in mean number of intestinal tumors from the Cables1+/+ ApcMin/+ mice, suggesting that inactivation of both alleles of Cables1 is required to promote tumorigenesis. The finding of adenocarcinomas in the Cables1−/− ApcMin/+ mice, compared with none in the other groups, suggests that loss of Cables1 may accelerate intestinal tumorigenesis in the ApcMin/+ mouse model. Overall, these results suggest that Cables1 is a relevant 18q tumor suppressor gene in intestinal adenocarcinoma.
ApcMin/+ mice develop multiple intestinal adenomas by 16 weeks of age and indeed, all of the Cables1−/− ApcMin/+mice and Cables1+/+ ApcMin/+ mice in this study did develop intestinal adenomas. The mean number of colonic adenomas (0.6 tumors) was consistent with the literature.25 The mean number of small intestinal adenomas in the Cables1+/+ ApcMin/+ group (3.1) was lower than is typically reported in literature for the 16 week age group, which often number approximately 30.25 There are factors that may have contributed to the relatively low tumor burden observed in the Cables1+/+ ApcMin/+ group in this study, including a sterile housing facility and the method of counting polyps. Most studies report tumor counts using a dissecting microscope to analyze the entire mucosal surface. In contrast, our study did not analyze the entire mucosal surface, as polyps were only counted in the single plane sectioned from the “Swiss” roll for H&E staining. Since the tumor counting method and housing conditions were the same for both the Cables1−/− ApcMin/+mice and Cables1+/+ ApcMin/+ mice, these factors would have caused a similar degree of underestimation of the total tumor burden in both groups. The other possible explanation for lower than expected tumor burden is the presence of a modifier gene. Several genes, known as modifiers of Min (Mom1, Mom2, Mom3, and Mom7), have been identified that can modify the Min phenotype.26,27 Mutation of one of the Mom genes can result in a reduction of up to 90% in the polyp burden in ApcMin/+ mice.27 While it is possible that a modifier gene is responsible for the relatively low tumor burden observed in this study, this would have likely affected both the Cables1+/+ ApcMin/+ mice and the Cables1−/− ApcMin/+mice. Consequently, the difference in tumor burden between these two groups is of greater interest than the absolute tumor burden in either group.
In the Wnt signaling pathway, the APC gene product targets β-catenin for degradation by the proteosome, in association with GSK-3β, CK1 (casein kinase 1), and axin.28 Mutant APC disrupts the complex formation and creates a permissive condition whereby free, unphosphorylated β-catenin may accumulate in the cytoplasm, and following its translocation into the nucleus, drive the transcription of multiple genes implicated in tumor growth and invasion. In this study, increased β-catenin expression was detected in tumor tissue from Cables1−/− ApcMin/+ mice when compared with adenomas from Cables1+/+ ApcMin/+ mice. We also found that an siRNA induced CABLES1 knockdown in human colon cancer cell culture resulted in increased β-catenin-dependent transcription. Overall, this suggests that in colon cancer, inactivation of Cables1 may further deregulate Wnt pathway signaling, leading to increased accumulation of nuclear β-catenin and increased β-catenin-dependent transcription. Of note, this is in contrast with our current understanding of Cables1 function in neurons, in which Cables1 is thought to be an essential linker protein between Robo-associated Abl and N-cadherin-associated β-catenin, preventing nuclear targeting of β-catenin and activation of transcription.10
Because Cables1−/− ApcMin/+ mice exhibit increased tumor formation and Cables1 has been reported to play a role in regulating the activity of cyclin-dependent kinases, we measured proliferative activity by immunohistochemical staining for proliferating cell nuclear antigen.8,9 PCNA was significantly increased in tumor tissue from Cables1−/− ApcMin/+ mice. The strong correlation between nuclear expression of β-catenin and PCNA, suggests that nuclear accumulation of β-catenin increases during tumor progression and may have a direct effect on proliferation in vivo. Increased cell proliferation (shortened cell culture doubling time) has been previously identified in Cables1-deficient mouse embryonic fibroblasts.15 The Cables1-deficient cells in these experiments were shown to have decreased expression of cyclinD1 and the cyclin dependant kinase inhibitors p16 and p27. We have performed immunohistochemistry for cyclinD1, p16, and p27 on tissue from Cables1−/− ApcMin/+ mice and Cables1+/+ ApcMin/+ mice, but we have not identified differences in expression of these proteins in tumor tissue and background non-neoplastic tissue between the two groups (results not shown). The study of cyclins and cyclin dependent kinase inhibitors in a Cables1-deficient in vivo model, such as this, is an area of interest for future research, but likely requires more sensitive techniques than immunohistochemistry, such as western blotting, for assessment of cell cycle proteins.
Finally, to test how loss of Cables1 influences intestinal progenitor cell function, we isolated small intestinal crypts from one-year-old wild-type and Cables1 knockout mice and found that Cables1 deficient crypts were nearly twice as capable of forming organoids in culture than wild type crypts. This data suggests that there may be more stem cell or progenitor cell activity in Cables1 deficient crypts, since only long-lived stem cells can self-renew and differentiate into the various mature intestinal cell types required for organoid body maintenance in vitro.
In summary, loss of Cables1 enhances tumor progression in the ApcMin/+ mouse model. Inactivation of Cables1 results in activation of the Wnt/β-catenin signaling pathway, indicating that Cables1 is a tumor suppressor gene on chromosome 18q in this in vivo mouse model and likely has a similar role in human colon cancer.
Materials and Methods
Generation of Cables1+/+ ApcMin/+ and Cables1−/− ApcMin/+ mice and genotyping
C57BL/6J ApcMin/+ mice were obtained from the Jackson Laboratory. Mice harboring a targeted inactivation of the Cables1 gene (Cables1−/−) have been previously described.12,13 Cables1-knockout mice had been previously backcrossed with C57BL/6J mice before mating with C57BL/6J ApcMin/+ mice. Cables1−/− females were mated to ApcMin/+ males to obtain Cables1+/− ApcMin/+ breeding pairs, which were then intercrossed to generate both Cables1+/+ ApcMin/+ mice and Cables1−/− ApcMin/+ mice. All offspring were genotyped by PCR analysis of tail-snip genomic DNA using specific primers. The three primer pairs used for genotyping ApcMin/+ mice were: IMR033 5′-GCCATCCCTT CACGTTAG-3′, IMR034 5′-TTCCACTTTG GCATAAGGC-3′ and IMR758 5′-TTCTGAGAAA GACAGAAGTT A-3′. The PCR conditions used are those recommended by the Induced Mutant Resource of The Jackson Laboratory. The Cables1 PCR assay involved the use of three primers: 5′-TTGGTTATTG GGTCGGGTGC-3′, 5-GGAGGAAAAC AAAGCCGCAG G-3′ and 5′-GGATGTGGAA TGTGTGCGAG GC-3′. Reaction mixtures containing all three primers were subjected to 35 cycles of standard PCR condition with a 63 °C annealing temperature. Products were resolved using 1.5% agarose gel electrophoresis and stained with ethidium bromide. The wild type product is ~300 bp long and the disrupted product is ~200 bp long. By evaluating both of these PCR reaction products, the knockout status of the mice was determined. The proportion of mice with the desired Cables1−/− ApcMin/+ genotype was small, which was expected because Cables1 and Apc both map to mouse chromosome 18, whereas in humans they map to different chromosomes (5 and 18).
All of the animal protocols were reviewed and approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee and were performed in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Harvesting and preparation of intestinal tissue samples
Mice were euthanized at a mean of 15−16 weeks of age with isoflurane and dissected immediately. The colon and small intestines were removed and carefully opened longitudinally. The intestines were rolled with the mucosa outwards, fixed in 10% buffered formalin, processed and embedded in paraffin. A single section of each intestine roll was cut by microtome and stained with hematoxylin and eosin. Small intestinal and colonic tumors were counted by light microscopy by a pathologist (LZ), who was blinded to the genotypes of the animals.
Immunohistochemical analysis
Immunohistochemical stains (IHC) were applied to formalin-fixed, paraffin-embedded tissue sections. A mouse anti-Cables1 monoclonal antibody was produced using a GST fusion protein containing the C-terminal sequence of human Cables1 as the immunogen. Nuclear staining identified intact Cables1 expression, whereas loss of Cables1 expression was based on absence of nuclear staining. Immunohistochemistry for the following primary antibodies was also performed: anti-β-catenin (monoclonal; clone 14; BD Transduction Laboratories) and anti-proliferating cell nuclear antigen (PCNA polyclonal; DAKOTwo investigators (MSP and LZ) blinded to the genotypes of the animals evaluated β-catenin and PCNA staining. For β-catenin, two randomly chosen cases for each mouse genotype group were evaluated by light microscopy in five high power fields (400× magnification) of tumor tissue. β-catenin expression was scored semi-quantitatively and expressed as an IHC score by multiplying the percentage of immunopositive cells and the staining intensity, as described previously.29 Staining intensity was scored as follows: 0, negative; 1, weak; 2, moderate; 3, strong; and 4, very strong. The IHC score ranged from 0 to 400. To quantify PCNA expression, the number of positive cells in 10 randomly chosen high power fields (at 400 × magnification) of sections of tumor tissue were counted in one case for each genotype group.
Cell culture, transfections and luciferase assay
Human SW480 colon cancer cells, which carry an APC mutation, were obtained from the American Type Culture Collection and maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco-Invitrogen) supplemented with 10% fetal bovine serum (FBS; HyClone) and antibiotics (Penicillin-Streptomycin, Invitrogen). SW480 cells were transiently transfected with either CABLES1 cDNA, using FuGENE6 (Roche) or small interfering RNA (siRNA) targeting endogenous CABLES1, using Lipofectamine RNAiMAX (Invitrogen). Nucleotide sequences corresponding to siRNAs were as follows: siRNA1, 5′-GGAGACCCTG GAAGATATT-3′; siRNA2, 5′-GUAUCGCGAC AGUACCCAA-3′. To measure β-catenin reporter activity, cells were transfected with either the pGL3-OT or pGL3-OF luciferase reporter constructs containing three copies of wild-type or mutated consensus TCF-4 response elements (gifts of Bert Vogelstein, John Hopkins University), using FuGENE6 transfection reagent (Roche). Each well was cotransfected with a Renilla luciferase expression vector (pRL-CMV; Promega) to normalize for transfection efficiency. Twenty-four hours after transfection, cells were lysed and firefly and Renilla luciferase activities were measured using the Dual Luciferase Reporter kit (Promega). Firefly luciferase activity was normalized to Renilla luciferase activity.
Isolation and culture of intestinal stem cells from Cables1−/− mice
Intestinal stem cells were isolated from the intestinal tissue of one year old wild type and Cables1-deficient (Cables1−/−) mice according to a previously published protocol.17 Briefly, isolated crypts were counted and embeded in matrigel (BD Biosciences 356231 growth factor reduced) that contains 1 μM Jagged (ANASpec) at 5−10 crypts/μl and cultured in a modified form of medium as described by Sato et al.18,19 Briefly, DMEM/F12 (Gibco) was supplemented by EGF 40 ng/ml (R&D Systems), Noggin 200 ng/ml (Peprotech), R-spondin 500ng/ml (R&D or Sino Biological), N-acetyl-l-cysteine 1 μM (Sigma-Aldrich) and Y-27632 dihydrochloride monohydrate 20 ng/ml (Sigma-Aldrich). Thirty μl drops of matrigel with crypts were plated onto a flat bottom 48-well plate (Corning 3548) and allowed to solidify for 20 to 30 min in a 37 °C incubator. 300 μl of crypt culture medium was then overlaid onto the matrigel, changed every other day and maintained at 37 °C in fully humidified chambers containing 6% CO2. Clonogenicity (colony-forming efficiency) was calculated by plating 50 to 400 crypts and assessing organoid formation 3 to 10 d after initiation of cultures.
Statistical analysis
All data are reported as mean ± standard error of the mean (SEM), unless otherwise specified. Student’s t-test was used to compare the means of each group and differences were considered to be significant if p values were < 0.05. For correlations, Pearson’s correlation coefficients were calculated and considered statistically significant with an accompanying p value of < 0.05.
Acknowledgments
The study was funded through National Institute of Health Grant R01 CA103883 entitled “Cables and the colonocyte”. TA was funded through scholarships from the Royal College of Physicians and surgeons of Canada (Detweiler Traveling fellowship) and Dalhousie University (McLoughlin Scholarship).
Glossary
Abbreviations:
- APC
adenomatous polyposis coli
- CDK
cyclin-dependent kinase
- CRC
colorectal cancer
- H&E
hematoxylin and eosin
- IHC
immunohistochemistry
- LOH
loss of heterozygosity
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/25089
References
- 1.Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;135:1079–99. doi: 10.1053/j.gastro.2008.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–32. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 3.Tarafa G, Villanueva A, Farré L, Rodríguez J, Musulén E, Reyes G, et al. DCC and SMAD4 alterations in human colorectal and pancreatic tumor dissemination. Oncogene. 2000;19:546–55. doi: 10.1038/sj.onc.1203353. [DOI] [PubMed] [Google Scholar]
- 4.Keino-Masu K, Masu M, Hinck L, Leonardo ED, Chan SS, Culotti JG, et al. Deleted in Colorectal Cancer (DCC) encodes a netrin receptor. Cell. 1996;87:175–85. doi: 10.1016/S0092-8674(00)81336-7. [DOI] [PubMed] [Google Scholar]
- 5.Fazeli A, Dickinson SL, Hermiston ML, Tighe RV, Steen RG, Small CG, et al. Phenotype of mice lacking functional Deleted in colorectal cancer (Dcc) gene. Nature. 1997;386:796–804. doi: 10.1038/386796a0. [DOI] [PubMed] [Google Scholar]
- 6.Takagi Y, Kohmura H, Futamura M, Kida H, Tanemura H, Shimokawa K, et al. Somatic alterations of the DPC4 gene in human colorectal cancers in vivo. Gastroenterology. 1996;111:1369–72. doi: 10.1053/gast.1996.v111.pm8898652. [DOI] [PubMed] [Google Scholar]
- 7.Takagi Y, Koumura H, Futamura M, Aoki S, Ymaguchi K, Kida H, et al. Somatic alterations of the SMAD-2 gene in human colorectal cancers. Br J Cancer. 1998;78:1152–5. doi: 10.1038/bjc.1998.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu CL, Kirley SD, Xiao H, Chuang Y, Chung DC, Zukerberg LR. Cables enhances cdk2 tyrosine 15 phosphorylation by Wee1, inhibits cell growth, and is lost in many human colon and squamous cancers. Cancer Res. 2001;61:7325–32. [PubMed] [Google Scholar]
- 9.Zukerberg LR, Patrick GN, Nikolic M, Humbert S, Wu CL, Lanier LM, et al. Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron. 2000;26:633–46. doi: 10.1016/S0896-6273(00)81200-3. [DOI] [PubMed] [Google Scholar]
- 10.Rhee J, Buchan T, Zukerberg L, Lilien J, Balsamo J. Cables links Robo-bound Abl kinase to N-cadherin-bound beta-catenin to mediate Slit-induced modulation of adhesion and transcription. Nat Cell Biol. 2007;9:883–92. doi: 10.1038/ncb1614. [DOI] [PubMed] [Google Scholar]
- 11.Tan D, Kirley S, Li Q, Ramnath N, Slocum HK, Brooks JS, et al. Loss of cables protein expression in human non-small cell lung cancer: a tissue microarray study. Hum Pathol. 2003;34:143–9. doi: 10.1053/hupa.2003.26. [DOI] [PubMed] [Google Scholar]
- 12.DeBernardo RL, Littell RD, Luo H, Duska LR, Oliva E, Kirley SD, et al. Defining the extent of cables loss in endometrial cancer subtypes and its effectiveness as an inhibitor of cell proliferation in malignant endometrial cells in vitro and in vivo. Cancer Biol Ther. 2005;4:103–7. doi: 10.4161/cbt.4.1.1433. [DOI] [PubMed] [Google Scholar]
- 13.Park Y, Sakamoto H, Kirley SD, Ogino S, Kawasaki T, Kwon E, et al. The Cables gene on chromosome 18q is silenced by promoter hypermethylation and allelic loss in human colorectal cancer. Am J Pathol. 2007;171:1509–19. doi: 10.2353/ajpath.2007.070331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zukerberg LR, DeBernardo RL, Kirley SD, D’Apuzzo M, Lynch MP, Littell RD, et al. Loss of cables, a cyclin-dependent kinase regulatory protein, is associated with the development of endometrial hyperplasia and endometrial cancer. Cancer Res. 2004;64:202–8. doi: 10.1158/0008-5472.CAN-03-2833. [DOI] [PubMed] [Google Scholar]
- 15.Kirley SD, D’Apuzzo M, Lauwers GY, Graeme-Cook F, Chung DC, Zukerberg LR. The Cables gene on chromosome 18Q regulates colon cancer progression in vivo. Cancer Biol Ther. 2005;4:861–3. doi: 10.4161/cbt.4.8.1894. [DOI] [PubMed] [Google Scholar]
- 16.Kirley SD, Rueda BR, Chung DC, Zukerberg LR. Increased growth rate, delayed senescense and decreased serum dependence characterize cables-deficient cells. Cancer Biol Ther. 2005;4:654–8. doi: 10.4161/cbt.4.6.1732. [DOI] [PubMed] [Google Scholar]
- 17.Yilmaz OH, Katajisto P, Lamming DW, Gültekin Y, Bauer-Rowe KE, Sengupta S, et al. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature. 2012;486:490–5. doi: 10.1038/nature11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469:415–8. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–5. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 20.Boland CR, Sato J, Appelman HD, Bresalier RS, Feinberg AP. Microallelotyping defines the sequence and tempo of allelic losses at tumour suppressor gene loci during colorectal cancer progression. Nat Med. 1995;1:902–9. doi: 10.1038/nm0995-902. [DOI] [PubMed] [Google Scholar]
- 21.Takaku K, Wrana JL, Robertson EJ, Taketo MM. No effects of Smad2 (madh2) null mutation on malignant progression of intestinal polyps in Apc(delta716) knockout mice. Cancer Res. 2002;62:4558–61. [PubMed] [Google Scholar]
- 22.Hamamoto T, Beppu H, Okada H, Kawabata M, Kitamura T, Miyazono K, et al. Compound disruption of smad2 accelerates malignant progression of intestinal tumors in apc knockout mice. Cancer Res. 2002;62:5955–61. [PubMed] [Google Scholar]
- 23.Takaku K, Oshima M, Miyoshi H, Matsui M, Seldin MF, Taketo MM. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell. 1998;92:645–56. doi: 10.1016/S0092-8674(00)81132-0. [DOI] [PubMed] [Google Scholar]
- 24.Alberici P, Jagmohan-Changur S, De Pater E, Van Der Valk M, Smits R, Hohenstein P, et al. Smad4 haploinsufficiency in mouse models for intestinal cancer. Oncogene. 2006;25:1841–51. doi: 10.1038/sj.onc.1209226. [DOI] [PubMed] [Google Scholar]
- 25.Thompson MB. The Min mouse: a genetic model for intestinal carcinogenesis. Toxicol Pathol. 1997;25:329–32. doi: 10.1177/019262339702500312. [DOI] [PubMed] [Google Scholar]
- 26.McCart AE, Vickaryous NK, Silver A. Apc mice: models, modifiers and mutants. Pathol Res Pract. 2008;204:479–90. doi: 10.1016/j.prp.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 27.Silverman KA, Koratkar R, Siracusa LD, Buchberg AM. Identification of the modifier of Min 2 (Mom2) locus, a new mutation that influences Apc-induced intestinal neoplasia. Genome Res. 2002;12:88–97. doi: 10.1101/gr.206002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.White BD, Chien AJ, Dawson DW. Dysregulation of Wnt/β-catenin signaling in gastrointestinal cancers. Gastroenterology. 2012;142:219–32. doi: 10.1053/j.gastro.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong SC, Chan AT, Chan JK, Lo YM. Nuclear beta-catenin and Ki-67 expression in choriocarcinoma and its pre-malignant form. J Clin Pathol. 2006;59:387–92. doi: 10.1136/jcp.2005.026666. [DOI] [PMC free article] [PubMed] [Google Scholar]