

Abstract
Cytoskeletal and focal adhesion abnormalities are observed in several types of cancer, including lung cancer. We have previously reported that paxillin (PXN) was mutated, amplified, and overexpressed in a significant number of lung cancer patient samples, that PXN protein was upregulated in more advanced stages of lung cancer compared with lower stages, and that the PXN gene was also amplified in some pre-neoplastic lung lesions. Among the mutations investigated, we previously found that PXN variant A127T in lung cancer cells enhanced cell proliferation and focal adhesion formation and colocalized with the anti-apoptotic protein B Cell Lymphoma 2 (BCL-2), which is known to localize to the mitochondria, among other sites. To further explore the effects of activating mutations of PXN on mitochondrial function, we cloned and expressed wild-type PXN and variants containing the most commonly occurring PXN mutations (P46S, P52L, G105D, A127T, P233L, T255I, D399N, E423K, P487L, and K506R) in a GFP-tagged vector using HEK-293 human embryonic kidney cells. Utilizing live-cell imaging to systematically study the effects of wild-type PXN vs. mutants, we created a model that recapitulates the salient features of the measured dynamics and conclude that compared with wild-type, some mutant clones confer enhanced focal adhesion and lamellipodia formation (A127T, P233L, and P487L) and some confer increased association with BCL-2, Dynamin-related Protein-1 (DRP-1), and Mitofusion-2 (MFN-2) proteins (P233L and D399N). Further, PXN mutants, through their interactions with BCL-2 and DRP-1, could regulate cisplatin drug resistance in human lung cancer cells. The data reported herein suggest that mutant PXN variants play a prominent role in mitochondrial dynamics with direct implications on lung cancer progression and hence, deserve further exploration as therapeutic targets.
Keywords: mitochondria, paxillin, gene mutation, cell motility, fission, fusion, mitochondrial dynamics
Introduction
Lung cancer is a devastating illness with over 220 000 patients diagnosed and more than 160 000 patients dying in the United States this year alone.1 Lung cancer is typically divided into small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC), with histologies including adenocarcinoma, bronchoalveolar carcinoma, and squamous cell carcinoma. The overall 5-year survival of NSCLC, which accounts for 85% of all lung cancers, is ~16%. The traditional therapy for early stage NSCLC is surgery and potentially chemotherapy, whereas in advanced disease chemotherapy remains the mainstay of treatment. Novel targeted therapies have been utilized, such as inhibitors of the epidermal growth factor receptor (EGFR), echinoderm microtubule-associated protein-like 4 (EML-4) anaplastic lymphoma kinase (ALK) inhibitors, and anti-angiogenic agents.2 However, even with the most novel agents, the response is relatively short-lived in most patients. In order to make an impact on this disease, we need to understand the basic biology of lung cancer and subsequently target the particular pathway(s) involved.
It has been demonstrated that cell motility, migration, metastasis, and cytoskeletal structures (especially actin and actin binding proteins) play an important role in lung cancer.3 The focal adhesion protein PXN is heterogeneously expressed in lung cancer cell lines and NSCLC primary tumors,4 and there appears to be a key role for PXN in lung cancer and its metastasis.5 PXN is an adaptor protein and a target of many oncogenes such as BCR/ABL, v-Src, and human papillomavirus E6 protein.6-8 In transformation mechanisms through these oncogenes, PXN is routinely recruited and phosphorylated.8 One of the outcomes of PXN phosphorylation is the generation of specific SH2 and SH3 interaction sites that bridge integrin receptors to downstream kinases such as FAK and cytoskeletal elements actin and vinculin.7,9 PXN interacts with FAK through its LIM and LD domains and thereby facilitates phosphorylation of FAK at Y397 that is critical for binding to downstream signaling molecules such as Src, PI3K, and SHC. Additionally, PXN recruits actin to its C-terminus LIM3 domain at sites of cell adhesion to the extracellular matrix and undergoes extensive phosphorylation during integrin-mediated cell adhesion.10-12 There is considerable energy needed for PXN to affect cell migration, which may be elicited from the mitochondria.
Mitochondria are classically believed to play a role in formation of reactive oxygen species (ROS) and ATP production. The dynamics of the mitochondria involve fission or fusion. Fission is primarily controlled by the GTPase DRP-1 and fusion by the mitofusins MFN-1 and 2. Previously, we have shown higher DRP-1 and lower MFN-2 expression in lung cancers.13 Biologically, there is more fission (“fragmentation”) of the mitochondria than fusion in lung cancer cell lines as well as in lung cancer tumor tissues.13 The regulation of these mitochondria dynamics could possibly be through the actin cytoskeleton.
In this study, we have systematically investigated the effects of PXN mutations on in vitro cellular functions utilizing various mutations of PXN that we previously described to be present in NSCLC patient samples. Based on the differential biological functions, six mutations (P52L, A127T, P233L, T255I, D399N, and P487L) were selected for investigating their role in the potential regulation of focal adhesions and mitochondrial dynamics. It seems clear that PXN’s reach extends beyond focal adhesions and affects mitochondrial physiology. A better understanding of the intricate nature of regulation of mitochondrial morphology and dynamics by PXN variants is essential for understanding the role of mitochondrial dysregulation in cancer cell metabolism, apoptosis and tumor progression.
Results
Identification of effects of GFP-tagged PXN mutations on cell morphology
Previously, we identified several mutations of PXN in lung cancer specimens (Fig. 1A).5 The most frequently occurring mutation was at the A127T position of PXN coding sequence, with other most prevalent mutations identified in the LD and LIM domains. For this study, we cloned human paxillin coding sequence into pAcGFP-N1 vector and by site-directed mutagenesis introduced some of the previously identified mutations. The mutant constructs thus established were PXN S32L, P52L, G105D, A127T, P233L, T255I, D399N, E423K, P487L, and K506R (Fig. 1A). Initially, to understand the biology of PXN and mutant PXN, transient transfections were performed utilizing the well-established HEK-293 cell line with transfection efficiency at approximately 30%, and cellular properties were visualized under a confocal microscope (Fig. 1B). The transfection strength, focal adhesion formation, filopodia formation, lamellipodia formation, mobility, and displacement were analyzed by three different observers and reported as low (+), moderate (++), and high (+++) (Table 1; Supplemental Movies). Even at low power magnification, the morphology of control cells (empty vector), PXN overexpressing wild-type (WT), and mutant cells was dramatically different. Noticeable differences were observed in various properties, and dramatic differences were seen for P52L (increased lamellipodia and mobility), A127T (scoring highest in all of the parameters evaluated), P233L (not as strong in filopodia formation as compared with A127T; nevertheless, strong overall), D399N (weak focal adhesions), and P487L (increased focal adhesions and lamellipodia) as compared with WT. Based on these characteristics, we identified five PXN mutants P52L, A127T, P233L, D399N, and P487L, to focus on for further analysis.

Figure 1. (A) Diagram showing the structure of PXN and the position of mutations found in NSCLC patient samples. (B) Confocal images of HEK-293 cells transiently transfected with GFP tagged plasmid DNA containing various mutants of PXN. The panel represents photographs taken at a single time point of empty vector, wild-type, and each mutant.
Table 1. The chart represents visual estimations of cell size, transfection strength, appearance of focal adhesions, filopodia, lamellipodia, cell mobility, and displacement over the period imaged.
| Mutant | Transfection strength | Focal Adhesion | Filopodia | Lamellipodia | Mobility | Displacement |
|---|---|---|---|---|---|---|
| WT |
+ + |
+ |
+ + |
+ + |
+ + |
+ |
| P46S |
+ + + |
+ |
+ + |
+ + |
+ + |
+ |
| P52L |
+ + |
+ + |
+ + |
+ + + |
+ + + |
+ + |
| G105D |
+ + |
+ |
+ |
+ + |
+ + + |
+ |
| A127T |
+ + |
+ + + |
+ + + |
+ + + |
+ + + |
+ + + |
| P233L |
+ + |
+ + + |
+ + |
+ + + |
+ + + |
+ + + |
| T255I |
+ + |
+ |
+ + |
+ + + |
+ + |
- |
| D399N |
+ + + |
- |
+ |
+ + |
+ + |
+ |
| E423K |
+ |
+ |
+ |
+ |
+ + |
+ |
| P487L |
+ + |
+ + + |
+ + |
+ + + |
+ + |
- |
| K506R | + + + | - | + | + + | + + + | + |
At least 6–13 cells were tracked for each mutant.
Effect of PXN mutations on mitochondria and mitochondrial dynamics
During investigation of these various morphological differences, we observed that the mitochondria were localized differently in wild-type PXN expressing cells as compared with that of various mutant-expressing cells. To further dissect the differences in mitochondrial localization, we stained WT and six PXN mutant cells (P52L, A127T, P233L, T255I, D399N, and P487L) with MitoTracker Red and imaged the cells using a spinning disc confocal microscope at different z-planes. At first, cells were compared visually, and a semi-quantitative evaluation was performed to determine mitochondrial appearance and distribution pattern in the cellular compartments. Four to eight individual cells were evaluated for each transfected construct. Although all mitochondria were localized in the cytoplasm, interestingly, there was a differential cytoplasmic pattern visible in the different mutants (Fig. 2A). In WT PXN and A127T, the mitochondria were arranged in the perinuclear region in a ring-like pattern, whereas in the other mutants they were predominantly distributed in the entire cytoplasm. In addition, the mitochondria were starkly visible in the moving front of the cells in the lamellipodia in all types, except in P52L (the P52L mutant cells were particularly affected by the transfection and their morphology difficult to assess visually). The complexity of the mitochondria was distinct among the various mutants. In the WT and A127T and P487L mutants, the mitochondria appeared dense and were distributed in network-like structures in the cytoplasm. In P52L and D399N, there was less network formation, and the mitochondria appeared more punctate, forming discrete circles (Fig. 2A and Table 2). These images were further analyzed with a macro developed for measurement of different cellular and mitochondrial parameters. Three parameters, number of mitochondria (approximate total number of individual mitochondrion in each cell), mean mitochondrial intensity (the mean brightness or intensity of red mitochondrial staining in the entire cell indicating mitochondrial energy levels), and mean volume (mitochondrial volume in an image divided by the number of nuclei, indicating how much mitochondria are present in the cell) were measured (Fig. 2B). P52L and P487L mutants had the highest number of mitochondria compared with all others. Parental HEK-293 as well as T255I, D399N, and P487L expressing cells showed low mitochondrial energy levels, while A127T and WT exhibited the highest levels of energy. The Mean Volume showed that WT had the highest volume of cellular mitochondria and P52L and P487L had the lowest (Fig. 2B). These observations clearly, and unexpectedly, show PXN expression affects mitochondrial localization and dynamics.

Figure 2. (A) Live-cell confocal images of HEK-293 cells expressing various mutants of PXN labelled with MitoTracker Red. The panel represents single time point photographs of untransfected parental HEK-293 cells, wild-type and mutant PXN. (B) Multiple images analysed with Imaris software show the number of mitochondria, mean intensity of MitoTracker Red indicating mitochondrial energy levels, and mean volume (indicating the level of average mitochondrial volume).
Table 2. The table represents visual estimations of mitochondrial appearance and localization within the cellular compartment of wild-type and mutant PXN.
| Mutant | Cellular Distribution | Appearance | Comments | |
|---|---|---|---|---|
| WT |
Cytoplasmic |
Perinuclear |
Dense; Network; Punctate |
punctate at moving front of cell; some in lamellipodia |
| P52L |
Cytoplasmic |
Extranuclear |
Discrete; Punctate |
cells are not easy to define |
| A127T |
Cytoplasmic |
Perinuclear |
Dense; Network; Punctate |
some adjacent normal cells also similar in appearance |
| P233L |
Cytoplasmic |
Extranuclear |
Dense; Network; Punctate |
in lamellipodia at moving front of cell; dense in moving direction |
| D399N |
Cytoplasmic |
Extranuclear |
Discrete; Punctate |
more pronounced at moving front (not in lamellipodia/filopodia) |
| P487L | Cytoplasmic | Extranuclear | Dense; Network; Punctate | in lamellipodia |
Four to eight individual cells were evaluated for each construct.
HGF/MET axis activation of PXN and effects on the mitochondria
Since PXN serves as a downstream adaptor target for various receptor tyrosine kinases, we investigated the role of PXN as a mediator between stimulation of the MET receptor tyrosine kinase and localization of mitochondria. Hepatocyte growth factor (HGF), the ligand for MET, has been shown to rapidly induce phosphorylation of PXN.14,15 Stable transfectants of wild-type and PXN mutants in HEK-293 cells were created for further cellular functional analysis. GFP fusion constructs of PXN WT, P52L, A127T, P233L, D399N, and P487L HEK-293 cells were stimulated with HGF (100 ng/ml, 15 min) and stained with MitoTracker Red (3-D reconstructed images, Fig. 3A). Upon quantitation of confocal images, it was determined that HGF induced mitochondrial swelling and increased or decreased mitochondrial staining intensity, suggesting changes in mitochondrial respiratory activity. Mitochondrial PXN content (calculated as GFP-PXN intensity at/in the fit mitochondrial surface; not total cell GFP-PXN content), mean MitoTracker intensity, mitochondrial surfaces (structures)/cell (count of fitted mitochondria in the volume of the cell), and mean mitochondrial volume/cell were calculated using the Imaris software. The baseline volume/cell for unstimulated mitochondria was higher in WT and D399N compared with most of the PXN mutants. The number of separate mitochondria was higher in P52L, P233L, and D399N; about the same in P487L; and lower in A127T compared with WT under basal conditions. Overall, there was sizable and differential response to HGF stimulation in the WT and mutants (Fig. 3B). Whereas no significant difference in PXN-GFP intensity and MitoTracker Red intensity between stimulated and unstimulated cells was observed, there was a substantial mitochondrial volume change in response to HGF in WT, P52L, P233L, and D399N. Mitochondrial volume increases indicate swelling and/or increased fusion events; a decrease in volume indicates shrinkage and/or more fission events. Fission/fusion changes are detected in the numbers of separate mitochondrial structures per cell (surfaces fit per cell). For example, an increase in volume with no change in the number of structures per cell indicates swelling only. A decrease in volume with an increase in the number of structures per cell indicates fragmentation and shrinkage. No change in volume with a decrease in structures per cell indicates increased fusion. Cells expressing PXN WT, P233L, and D399N showed decrease in volume with decrease in the numbers of separate mitochondria following HGF stimulation, indicating mitochondria fusion and shrinkage in response to MET activation. P52L volume and surfaces increased with stimulation indicating swelling and more fissions following HGF stimulation. A127T and P487L mutants generally had the lowest mitochondrial volume per cell and did not change in volume after HGF treatment, but the number of mitochondria per cell increased in A127T, but not in P487L, indicating increased fission in A127T.

Figure 3. Effect of HGF stimulation on HEK-293 wild-type PXN and mutants shown in (A) confocal images after treatment with HGF and stained with MitoTracker Red and Hoechst and (B) measurements of mitochondrial functions by Imaris.
Expression of mitochondrial functional proteins
Based on the above findings on mitochondrial dynamics, we screened untransfected HEK-293 cells, WT, A127T, P233L, and D399N PXN stable mutants for expression of mitochondrial function related proteins with and without HGF stimulation. Expression levels were plotted according to densitometry analyses analyzed and the results are shown in Figures S1 and S2. Our results indicate that parental HEK-293 cells express endogenous PXN, the level of which did not change with HGF stimulation. These results are indicated by the lower and upper bands in the PXN blot in Figure 4A respectively and in Figure S1. WT transfectants expressed both endogenous and exogenous forms of PXN at the same level as untransfected cells, but PXN expression was doubled in all three mutants. The PXN level was not altered significantly with HGF stimulation. The expression levels of total MFN-2 did not change with PXN overexpression or HGF stimulation (Fig. 4A, MFN-2 blot) whereas total DRP-1 expression was reduced by 50% after PXN overexpression in WT and all three mutants. There was no change seen with HGF stimulation (Fig. 4A, DRP-1 blot). Phosphorylation of DRP-1 occurs at two sites, serine 616 and serine 637, which affect DRP-1 in opposing ways. Phosphorylation at serine 616 leads to increased fission during mitosis, whereas phosphorylation at serine 637 leads to inhibition of fission.16 We found that compared with untransfected controls, cells overexpressing the WT, P233L and D399N mutants increased activated DRP-1(616) expression by 50%. Treating the cells with HGF reduced the levels of phospho-DRP-1(616) in untransfected and WT cells, but not the mutants. On the other hand, although parental HEK-293 did not express detectable levels of phospho-DRP-1(637), we found baseline expression in all stably transfected cells as well as increased levels with HGF treatment in parental, WT, A127T, P233L, and D399N cells indicating that PXN and its mutations caused inhibition of fission in these cells. We also assessed the expression of total and phosphorylated BCL-2 in these cells. Compared with the baseline expression in parental cells, all transfectants expressed more or less the same levels of BCL-2, except P233L, which expressed slightly higher levels. We found no induction of BCL-2 by acute HGF treatment in any of the cells; in A127T, HGF reduced BCL-2 expression by 50%. Expression of phosphorylated BCL-2 (p-BCL2) was significantly higher in transfected cells compared with parental control except in A127T, which had mild increase (Fig. 4A, p-BCL-2 blot, Fig. S1). In all cells except A127T, HGF stimulated higher expression of p-BCL2.

Figure 4. (A) HEK-293 cells were treated with HGF, and total protein lysates were immunoassayed to detect expression levels of PXN, DRP-1, MFN-2, phospho-DRP 616, phospho-DRP 637, total BCL-2, and phospho-BCL-2. (B) BCL-2 was immunoprecipitated and then blotted for PXN to assess association between BCL-2 and PXN. These blots show wild-type and mutant PXN binding to BCL-2 and their association with DRP-1 and MFN-2 in response to HGF treatment. The BCL-2 blot shows levels of BCL-2 immunoprecipitated from each sample.
Differential binding of BCL-2 with PXN mutants, DRP-1, and MFN-2
In order to explore the effect of PXN mutation on their ability to bind to BCL-2 as well as their subsequent effect on the expression of mitochondrial proteins DRP-1 and MFN-2 in response to HGF stimulation, we looked at BCL-2 binding in HEK-293 parental, WT, A127T, P233L, and D399N mutants by immunoprecipitation assay. All three PXN mutants A127T, P233L, and D399N were associated with BCL-2, although interaction of D399N with BCL-2 was minimal. There was no binding of parental HEK-293 or WT (Fig. 4B). HGF treatment (100 ng/ml, 15 min) decreased association of A127T with BCL-2 (Fig. 4B), with much less significant effect on the other mutants. The DRP-1 and MFN-2 blots show that each mutant was differentially bound to BCL-2. Parental HEK-293 cells showed no BCL-2 association with DRP-1, but they did show significant association with MFN-2 and no change after HGF treatment. In WT PXN overexpressing cells, there was minimal association of BCL-2 with DRP-1, but there was significant association with MFN-2, which was inhibited with HGF treatment. A127T showed interesting association patterns; although the strength of association was lower, A127T was associated with both DRP-1 and MFN-2; of further interest, MFN-2 binding increased with HGF treatment, whereas DRP-1 binding decreased. Since the number of mitochondrial structures per cell increased, perhaps this association with the fusion protein MFN-2 explains the increased fission observed (the binding may have inhibited function). P233L showed the highest association of BCL-2 with DRP-1 and MFN-2 compared with the other mutants. The association was inhibited by HGF treatment. D399N showed significant association with DRP-1 but minimal association with MFN-2. HGF treatment inhibited binding to both. The BCL-2 blot on Figure 4B serves to show that equal levels of BCL-2 were precipitated from each sample.
Sensitivity to cisplatin
Lung cancer is a heterogeneous disease, and cisplatin is a commonly used drug in therapy. Since BCL-2 and the mitochondria play an important role in sensitivity to cisplatin,17,18 we determined if the various PXN mutants exhibited a differential response to cisplatin. Compared with control, HEK-293 cells overexpressing PXN mutations A127T and P233L were found to be more sensitive. D399N cells exhibited highest resistance to cisplatin (Fig. 5). These data support a major role of PXN in cisplatin sensitivity.
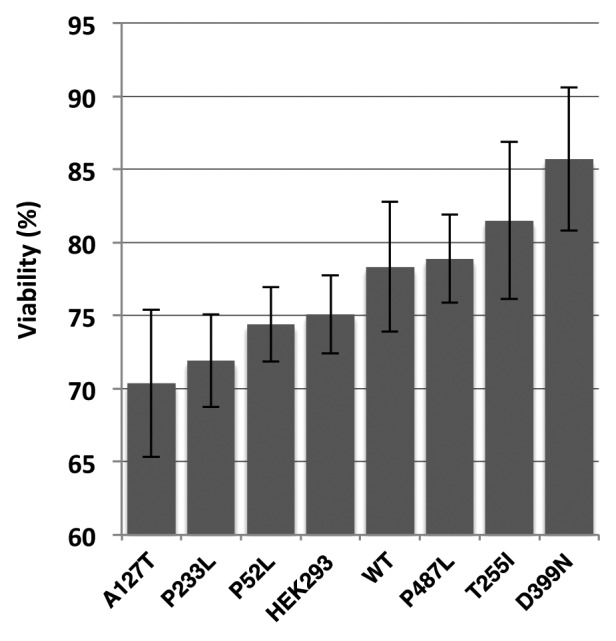
Figure 5. Viability of HEK-293 stably expressing wild-type PXN or mutant cells treated with cisplatin for 72 h. The graph represents percentage of cells viable after cisplatin treatment as compared to cells treated with vehicle (DMSO) only.
PXN mutation status, protein expression, and survival outcomes for patients in NSCLC
As mentioned previously, we had identified a number of somatic mutations in different domains of PXN in NSCLC tumor tissues at the rate of 9.4% samples bearing mutation and 12.1% having PXN gene amplification.5 Here, we analyzed the relevance between clinical outcomes of patients with mutations vs. those with wild-type PXN (WT). Since each of the PXN mutations was non-repeating, i.e., found only in a single patient sample, we treated all of the samples with mutations as a group (MT, n = 7) and compared them to a set of 60 wild-type PXN samples (WT, n = 60). There were no obvious differences in the patient demographics (race, ethnicity, sex, and age). However, the patients with PXN mutations had poorer survival rate relative to patients with wild-type PXN (P 0.0001), with the median survival of 2.8 mo for the mutant patients compared with 33.3 mo for the wild-type patients (Fig. 6A).

Figure 6. (A) Survival outcome analysis in NSCLC patients with wild-type versus mutated PXN. Left and right graphs show overall and individual mutant survival curves, respectively. The relative expression of PXN, p-PXN, BCL-2, FAK, phospho-FAK, Cox IV, DRP-1, and MFN-2 was evaluated by IHC on two sets of samples; seven NSCLC patient samples bearing wild-type PXN and six NSCLC patient samples that were identified to have various PXN mutations as shown in (B). (C and D) represent the averaged staining intensity scores among all WT and all mutants. (E) Shows graph of staining intensity scores of the proteins assayed as expressed in the cytoplasm of the cells. (F)The panel shows photographs of IHC stains represented in one wild-type and one mutant sample.
Expression of PXN, phospho-PXN, FAK, phospho-FAK, and mitochondrial proteins BCL-2, DRP-1, and MFN-2 in patient samples with wild-type and mutated PXN
A subset of the previously identified NSCLC patient tissue samples with wild-type and somatic PXN mutations were used to determine expression of mitochondrial function related proteins. The samples selected for this were from six patient samples bearing the mutations P46S, P47L, A127T, E355K, K506R, and P487L and seven samples bearing no mutations in PXN (Fig. 6B). Unfortunately, patient tissue samples bearing P52L, P233L and D399N were completely depleted and not available. We determined the tissue protein expression levels of PXN, phospho-PXN, FAK, phospho-FAK, BCL-2, DRP-1, and MFN-2 by IHC. Since each of the PXN mutations was non-repeating, i.e., found only in a single patient sample, the data shown represents single patient samples harbouring mutations (Fig. 6B). The staining score represents only the intensity of stain in each subcellular compartment (cytoplasm or membrane) without regard to extent of staining. In tumors with wild-type PXN, most samples expressed high intensity of PXN in the cytoplasm, as well as high expression of FAK, COX-IV, DRP-1, and MFN-2. Moreover, the wild-type samples showed no or low expression of phospho-PXN, BCL-2 and phospho-FAK (Fig. 6C and F). In contrast, there was intense phospho-PXN expression in the cytoplasm of mutated samples as well as high expression of BCL-2 and FAK (in a subset of the samples) with very low or no expression of the other factors such as DRP-1 or MFN-2 (Fig. 6D and F). When all wild-type staining scores were averaged and compared with the average scores of all mutants, a distinct pattern of differential expression of these proteins in the cytoplasm of the cells was evident (Fig. 6E).
In the nuclei of wild-type PXN tumors, phospho-PXN and FAK were the only proteins found to be expressed, whereas in most of the mutant samples, all proteins except phospho-PXN and FAK were expressed in the nuclei. The membranous expression pattern did not differ between the two groups (data not shown).
Discussion
PXN is a unique adaptor protein with association with the actin cytoskeleton. It is classically thought to be involved in cancer cell motility, migration, and invasion, and eventually with metastasis. We previously shown there were somatic mutations of PXN in lung cancer.5 Utilizing constructs of wild-type and mutant PXN, we show dramatic effects of the mutations of PXN on actin cytoskeletal function, as well as alteration of mitochondrial localization and dynamics. In particular, the A127T mutation of PXN exhibited considerable biological changes in vitro.
Cancer progression and its resistance to treatment depend, at least in part, on suppression of apoptosis. Mitochondria are recognized as regulators of apoptosis, and their involvement in regulation of cancer progression and metastasis is currently gaining much focus. In 1930, Warburg suggested that mitochondrial dysfunction in cancer results in a characteristic metabolic phenotype, anaerobic glycolysis, which is characterized by a decrease in oxidative phosphorylation and a high glycolytic activity.19 Positron emission tomography (PET) imaging has now confirmed that most malignant tumors have increased glucose uptake and metabolism.20 Gatenby and Gillies have proposed that because early carcinogenesis occurs in a hypoxic microenvironment, the transformed cells initially have to rely on glycolysis for energy production.12 These metabolic adaptations allow for a proliferative advantage, suppression of apoptosis and the breakdown of extracellular matrix, facilitate cell mobility, and increase the metastatic potential.21 Mitochondria are motile, dynamic organelles that continuously change in morphology through the process of fusion and fission. Mitochondria play a central role in complex physiological processes including cell proliferation, differentiation, and apoptosis and in cellular processes of glucose sensing/insulin regulation,22 cellular Ca2+ and ROS homeostasis.23-26 Moreover, mitochondria are at the core of the intrinsic apoptotic pathway. The loss of mitochondrial fusion has been shown to result in embryonic lethality in mice.27 Mitochondrial filaments can form a hyperfused structure in response to stress by UV exposure that in turn lead to improved cell survival.28 The anti-apoptotic BCL-2 protein plays an important role in regulating mitochondrial fusion in healthy cells, with apoptosis and mitochondrial outer membrane permeabilization (MOMP) negatively regulating mitochondrial fusion.30 Upon apoptotic stimuli, mitochondria display dramatic shape change before releasing apoptotic factors cytochrome c mediated by DRP-1.31 DRP-1 is activated at two phosphorylation sites, serine 616 and serine 637, which act in opposing ways. Phosphorylation at Ser616 leads to increased fission during mitosis, whereas phosphorylation at Ser637 leads to inhibition of fission.16
Mitochondrial dysfunctions have been found to be associated with aging, several degenerative diseases, and cancer.32,33 Members of the BCL-2 apoptotic pathway have also been shown to modulate mitochondrial morphology. Findings by Karbowski et al.29 suggest that BAX and BAK are involved in the regulation of mitochondrial fusion through interactions with MFN-2 in healthy cells. Bax/Bak double knockout cells exhibited fragmented mitochondria, as well as abnormal localization of MFN-2. Delivani et al.34 reported that the BCL-2 homolog in C. elegans, CED-9, interacts with MFN-2 and induces mitochondrial fusion when expressed in HeLa cells. On the other hand, the anti-apoptotic protein BCL-xL was found to stimulate DRP-1 GTPase activity and to promote mitochondrial fission.
PXN was originally identified as a focal adhesion protein that complexes with the actin cytoskeleton.9 The cloning of PXN in 1995 lead to considerable advances in understanding the biology of this protein.35 In particular, genetic aberrations were identified that lead to speculations that there could be a strong role for PXN in certain tumors.5 We were the first to show that PXN is mutated in large cell lung cancer (18%), adenocarcinoma (9%), and squamous cell carcinoma (6%), and that these mutations are somatic.5 In addition, we have demonstrated that one of the most frequently occurring PXN mutations is a gain-in-function mutation (A127T) that confers cell survival advantage and co-localizes with the anti-apoptotic protein BCL-25. Herein, we systematically studied 10 mutations of PXN as described, and showed important differential in cell phenotype. In particular, certain mutants such as P52L, A127T, P233L, T255I, D399N, and P487L lead to dramatic differences in focal adhesions, lamellipodia, filopodia, and/or mobility when expressed in a common HEK-293 cell model system. In further analysis of wild-type PXN vs. select mutants, there was alteration of the mitochondrial localization and dynamics. It has been shown previously that there are differential fusion/fission mitochondrial dynamics for cancers.36 The overexpression of wild-type PXN caused alteration of mitochondria, and the PXN mutations produced other alterations.
The present investigations further revealed tight and differential associations of PXN and of mitochondrial morphogenic proteins with BCL-2 and in response to HGF stimulation of the MET receptor tyrosine kinase. The D399N, A127T, and P233L PXN mutations co-immunoprecipitated with BCL-2 and were accompanied by DRP-1 and MNF-2. We have recently shown that DRP-1 and MFN-2 can be abnormal in lung cancers.13 Here, we have demonstrated that the complexing of PXN with mitochondrial morphogens provides a potential mechanism to explain the mitochondrial dynamics observed in the HEK-293 model, a relatively simple system free from effects of other oncoproteins. Our results demonstrate PXN clearly has unanticipated effects on mitochondrial dynamics in addition to well-known effects on cytoskeletal dynamics and function.37 It is quite possible that the abnormality of the mitochondria is also related to the actin cytoskeleton, a more direct target of PXN. Random mutations of PXN have been generated in vitro and have led to effects on focal adhesion.37 Since lung cancer is a very heterogeneous disease, it will now be important to determine how the wild-type and mutant PXN interact with various oncogenes and tumor suppressor genes. It has been determined that there are various mutations of EGFR and MET receptor tyrosine kinases in lung cancer.11,14,38 It would be valuable to determine if PXN synergizes with these receptor tyrosine kinases to enhance lung tumorigenesis. Also, these may be important in mechanisms of metastasis for lung cancer. Not only are oncogenes important, but tumor suppressors such as p53 are an essential component to transformation of lung epithelial cells. Thus, it would be highly beneficial to systematically analyze PXN and/or mutant PXN interactions with these various molecules in the future.
There is a large molecular heterogeneity in lung cancer, especially non-small cell lung cancer. Particularly in adenocarcinoma, mutations/genetic alterations of EGFR, ALK, ROS1, MET, and KRAS as prime examples. In squamous cell carcinoma, there are alterations of FGFR1, PIK3CA, and DDR2 to name a few. As more and more molecular “dissection” is performed on tumor tissues, we will identify that individual patients will have individual genetic/proteomic alterations. Interestingly, the downstream effectors of these oncogenes and/or tumor suppressors are just beginning to be studied. We, and others have previously shown that PXN can be a target of various oncogenes such as BCR/ABL, Crk, Src, and E6 and tumor suppressors such as p53 to lead to multiple signal transduction and cell motility/migration cascades. We had also previously shown that PXN can be amplified and/or mutated in lung cancer tumor tissue specimens. Thus, the functionality of mutations is crucial to study. It is interesting that the A127T paxillin mutant leads to increased angiogenesis phenotype (as previously published by our group). Now, we show that there are a number of cell motility changes as well as mitochondrial changes with not only A127T, but with others such as P52L and D399N. As we go forward, we should investigate further each genotype and link to phenotype.
The mutant PXN appear to have considerable gain of function. In the NSCLC patient sample set, the expression of the phosphorylated form of PXN was increased in the mutants set as compared with the wild-type PXN set. It would be also important to determine the relative levels of these proteins in primary and metastatic tumors. Since PXN was relatively high in primary tumors and increased in expression in micro-emboli, we predict that the primary and metastatic sites would be different in expression and potentially even in localization.
Lung cancer can be treated with surgery in early stage disease, and potentially radiation therapy/chemotherapy for later stage disease. In particular, cisplatin chemotherapy is the mainstay of therapy for lung cancers. We have identified that there is a differential sensitivity to cisplatin with regards to wild-type vs. mutant PXN. This sensitivity correlates with PXN effects on mitochondrial size.39 It would now be useful to determine if chemotherapy can be combined with mitochondrial inhibitors in the context of PXN wild-type or mutant form. Also, due to the histological and molecular heterogeneity of lung cancers, it would also be important to perform this in relation to the various subsets of lung cancers (such as adenocarcinomas or squamous cell carcinomas). Animal modeling of PXN mutants should be established to determine the efficacy in vivo as well as the potential to contribute to metastasis.
In summary, we had previously identified several mutations of PXN in lung cancer. Now, in a systematic fashion we show that some of these mutations have considerable effects on cytoskeletal function and mitochondrial function. It would be important to determine the relationship of PXN and its mutants with cytoskeletal function and mitochondrial dynamics in lung cancer cells as well as bronchial epithelial cells. Also, this could eventually lead to a novel therapeutic opportunity.
Materials and Methods
Cell line
Cells were maintained at 37 °C and 5% CO2 in a humidified incubator except where noted. The human embryonic kidney epithelial cells, HEK-293, (American Type Culture Collection) were grown in Dulbecco's Modified Eagle Medium supplemented with 10% fetal bovine serum.
PXN cloning and mutagenesis
The coding region of PXN cDNA was subcloned into the mammalian expression vector pAcGFP-N1 (Clontech, Palo Alto, CA). Briefly, primers 5′-CACCATGGAC GACCTCGACG CC-3′ and 5′-TCACTTGTAC AGCTCATCCA TGC-3′ were used to amplify the coding region. The PCR product was TOPO cloned into pENTR/SD/D-TOPO vector (Gateway; Invitrogen). The insert was transferred into pAcGFP-N1 after converting it into a destination expression vector using Gateway Conversion System (Invitrogen) by a recombination reaction using LR clonase II. Various mutations were introduced into the wild-type PXN insert in the pAcGFP-N1vector using the Quick Change Site-Directed Mutagenesis XL kit (Stratagene). Standard bidirectional sequencing of the inserts confirmed the incorporation of various point mutations.
Transient transfection and live-cell confocal microscopy
Wild-type PXN and 17 PXN mutants generated by site-directed mutagenesis were used. For semi-quantitative analysis, the empty vector and all PXN constructs were transiently transfected into HEK-293 cells using the Mirus Bio TransIT®-293 Transfection Reagent (Mirus). Forty-eight hours later, cells were transferred to glass-bottomed Fluorodish (World Precision Instruments), and allowed to grow for 24 h. Live-cell confocal microscopy was performed 72 h post transfection on heated platform (37 °C) with 5% CO2. Transfectants were analyzed for effects of PXN mutations and to test if the mutations were sufficient for transformation into cancer-like cells. Since all transfected cells were GFP tagged, they were visualized through the green filter at 525 nm. Imaging was done for 90–120 min for each dish containing a specific PXN construct. Five data points with 5‒10 unique cells were imaged in each dish. Transfection was repeated 2‒4 times with each construct so that at least 15–30 unique cells were available for data analysis. Cells were imaged at different z-planes with an Olympus DSU spinning disc confocal microscope on a IX81 platform, 100× NA 1.45 objective, Hamamatsu C9100–12 back-thinned EM-CCD, and SlideBook acquisition software (Intelligent Imaging Innovations, Inc.). Visual observation was used to score cellular phenotype. ImageJ software (National Institute of Mental Health) was used for quantitative analyses. All microscopy described were conducted at the Integrated Microscopy Core Facility of the University of Chicago.
Semi-quantitative analysis of effect of PXN mutation
The images of transiently transfected HEK-293 cells tracked for 90–120 min as described above were rendered into time-lapse videos. Each video was visually scored for six cellular features: cell size, focal adhesion, filopodia formation, and lamellipodia formation, cell mobility, which is here defined as the ability of the cell to make moving motions without actually moving away from where it is, and cell displacement, which is here defined as the ability of the cell make moving motions as well as to move across a spatial plane. Seven to 14 cells were tracked for each mutant and the acquired data compiled in a semi-quantitative fashion.
Semi-quantitative analysis of effect of PXN mutation on mitochondrial dynamics
Transiently transfected HEK-293 cells were grown on glass bottom Fluorodishes. Immediately prior to confocal imaging, cells were stained with 50 nM MitoTracker Red (Invitrogen, M22426) in phenol red free DMEM for 15 min in an incubator and gently washed with same media. Thereafter, images were captured and analyzed in a semi-quantitative fashion to determine distribution and localization of mitochondria within the cellular compartments in each mutant. In order to further analyze the phenomenology observed for the mitochondria, the acquired images were further analyzed with a macro developed for measurement of different cellular and mitochondrial parameters. The calculations used to quantitate these features were by a novel method developed in the laboratory, which are briefly described here as Number of Mitochondria (approximate total number of individual mitochondria in each cell), Mean Mitochondrial Intensity (the mean brightness or intensity of red mitochondrial staining in the entire cell indicating mitochondrial energy levels) and Mean Volume (mitochondrial volume in an image divided by the number of nuclei, indicating how much mitochondria is present in the cell).
Stable transfection
HEK-293 cells were stably transfected with pAcGFP-N1 vector containing wild-type paxillin and each of six mutants of paxillin (P52L, A127T, P233L, T255I, D399N, and P487L) using the Mirus Bio TransIT®-293 Transfection Reagent. Twenty-four hours after transfection, cells were treated with Geneticin (1000 μg/mL) to select for transfected cells. The media was replaced with fresh media every 72 h. After two weeks, the cells were harvested and sorted for GFP using FACSAria at the University of Chicago Flow Cytometry Core Facility. The collected GFP-positive cells were maintained in 200 μg/mL Geneticin and tested for GFP positivity by fluorescent microscopy as well as FACS analysis.
Confocal microscopy with HGF stimulation
Mitochondria volumes were analyzed in control and HGF-stimulated cells to investigate dynamics of PXN interaction with mitochondria. HEK-293 cells were grown on glass bottom 35 mm dishes, stained with 50 nM MitoTracker Red and 1 μg/mL Hoechst (Invitrogen, 33342) for 15 min in phenol red free DMEM, gently washed with same media followed by treatment with HGF (50 ng/mL, 15 min). Live imaging was performed on a Marianas (Yokogawa CSU-X1) mSAC spinning disk confocal system (Intelligent Imaging Innovation Co.) on an Axio Observer Z1 platform using a 100X NA 1.45 oil objective (Carl Zeiss, Jena, Germany), AOTF-switched 405, 488 and 561 excitation and 482/38, 525/50, and 607/43 nm emission filters for Hoechst, GFP, and MitoTracker Red, respectively. Images were captured on an Evolve back-thinned EM-CCD camera (Roper) under control of SlideBook software. Temperature was controlled by a full-enclosure microscope incubator (OKO Labs, Ottaviano, Italy). Images were deconvolved using Huygens Pro software (SVI, Netherlands), and 3-D surface reconstructions were fit to the MitoTracker channel using Imaris software (Bitplane, South Windsor, CT). In addition to the above described Number of Mitochondria and Mean Mitochondrial volume, the Mean intensity of GFP fluorescence within the fit MitoTracker surfaces representing either GFP inside mitochondria or GFP at the surface of mitochondria was also analyzed. All treatments were captured, fit, and analyzed using the same parameters.
Immunoprecipitation
Cells were serum starved for six hours and then treated with HGF (100 ng/mL) for 15 min. Cells were lysed in ice cold 1% NP-40 in 25 mM Tris-HCL with 150 mM NaCl, 1 mM EDTA, 0.4 mM Na3VO4, 40 mM NaF, 50 mM okadaic acid, 0.2 mM phenylmethylsulfonyl fluoride, 1:250 dilution of Calbiochem protease inhibitor mixture III. Immunoprecipitation was performed using the Pierce Crosslink Immunoprecipitation kit (Thermo Scientific). Briefly, 10 μg of antibody (Santa Cruz Biotechnology, anti-BCL-2, SC-783) was covalently attached to 10 μl of settled Protein A/G resin using a DSS (disuccinimidyl suberate) cross linker. Equal quantities of the lysates were precleared using control agarose resin and then incubated at 4 °C on a rotator overnight with the antibody-bead complexes. Samples were then washed three times with ice-cold NP-40 buffer to remove non-bound proteins. Finally, the antibody-bound protein was eluted using the supplied elution buffer. The eluted proteins were then mixed with 5X sample buffer supplied in kit and heated to 95 °C for five minutes. Samples were stored at -20 °C prior to immunoblotting.
Immunoblotting
Protein samples were run on 4–15% Mini-protean TGX gels (Bio-Rad) and transferred onto ImmobilonTM membranes (Millipore). Blots were blocked using 5% BSA in TBST for one hour and probed with primary antibody overnight at 4 °C. After washing in TBST, blots were incubated with HRP-conjugated secondary antibodies for one hour at room temperature. The blots were then washed again and visualization of immunoreactive bands was achieved using LumiGLO® enhanced chemiluminescence (Cell Signaling Technology) and imaged. Antibodies and other reagents used were PXN (Invitrogen, AHO-0492), Phospho-PXN (Abcam, 32115), BCL-2 (Santa Cruz Biotechnology, SC-783), Phospho-BCL-2 (Cell Signaling, 2875), Phospho-DRP-1 (Cell Signaling, 3455, 4867), DRP-1 (Novus Biologicals, NB110–55288), MFN-2 (Epitomics, 3272–1), Actin (Sigma-Aldrich), and HRP-secondary antibodies (Cell Signaling). All other reagents were obtained from Sigma-Aldrich. Densitometry, or quantification of protein levels, was performed on the tiff-formatted images with the Image J software. The intensities of bands were compared according to their gray scale to determine relative density. Adjusted density values were determined by dividing the relative density value for each sample lane by the relative density of the loading control for the same lane.
Cisplatin sensitivity assay
Cells trypsinized from subconfluent cultures were resuspended in culture medium and seeded into triplicate wells of a 96-well plate (100 µl/well) at concentrations of 2.5 × 105 cells/ml at standard culture conditions of 5% CO2 in air at 37 °C. After an initial 24 h period to allow for cell attachment, the media was replaced with fresh media containing Cisplatin (Sigma Aldrich) at a final concentration of 2.5 μM. The cells were cultured for 72 h with fresh drug introduced every 48 h. The viability of cells was assessed with colorimetric MTA assay under 570 AB using a BioTek plate reader. DMSO was used as a negative control and drug vehicle.
Tissue sample acquisition
The NSCLC samples used here were previously reported by our lab to have PXN mutations at various unique domains.5 Six NSCLC patient samples bearing the mutations P46S, P47L, A127T, E355K, K506R and P487L and seven NSCLC samples bearing wild-type PXN were selected. The formalin-fixed, paraffin-embedded tissues samples were acquired from the archives of the University of Chicago Human Tissue Resource Center (HTRC) available to us through IRB-approved protocols. An experienced pathologist (ANH) analyzed all samples using conventional light microscopy and determined the histological subtype and tumor grade. These data were entered into our HIPAA regulated database, which included the demographic and clinical data and treatment outcomes.
Immunohistochemistry
Samples for this study were obtained from formalin-fixed, paraffin-embedded tissues from University of Chicago HTRC through IRB-approved protocols. Tissue samples were sectioned at 5-μm thickness for immunohistochemistry (IHC). The samples were probed using specific antibodies for PXN (Invitrogen, AHO0492), Phospho-PXN (Abcam, 4832), FAK (Millipore, 05–537), Phospho-FAK (Invitrogen, 44624G), Bcl-2 (Epitomics, 1017–1) COX-IV (Cell Signaling, 4844), DRP-1 (Novus Biologicals, NB110–55288), and MFN-2 (Epitomics, 3272–1). Following an antigen retrieval protocol, tissues were deparaffinised, blocked with BSA, probed with primary and secondary antibodies, and visualized using light microscopy. Tumour sections were analyzed by two experienced pathologists (ANH, QA), and staining intensity scored by conventional light microscopy using a 0/1+/2+/3+ scale. The histological subtype and tumor grade were confirmed as well. These data were entered into our database, which included the demographic, clinical information and PXN mutational status; the correlation analyzed manually.
Supplementary Material
Acknowledgments
The project described was supported by the Clinical and Translational Science Award (CTSA) program through the NIH National Center for Advancing Translational Sciences (NCATS), grant UL1TR000427. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH (DW). D.L.W., V.N., E.E.V., P.A.S. and R.S. conceived the strategies and supervised the project. R.H, I.K, F.E.L, and P.U. designed and performed experiments, analyzed data. Y.C.T. and S.K. built initial constructs and validated them. V. P. B. performed live cell confocal microscopy and developed image analysis tools, analyzed data. M.K.F. and R.S. treated patients and provided archival tissue samples. Q.A., M.T. and A.N.H analyzed immunohistochemistry data. G. C. and M.R. maintained Thoracic Oncology Database. R.D.H. gave technical support. R.H., P.A.S. and R.S. wrote the manuscript. V.P.B., S.K., T.M.B., M.I., D.L.W. and V. N. edited the manuscript. Supported in part by NIHR01 CA29501-05; Respiratory Health Association of Metroplitan Chicago; Geelard Memorial Foundation (R.S.)
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/25091
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Yuan Y, Liao YM, Hsueh CT, Mirshahidi HR. Novel targeted therapeutics: inhibitors of MDM2, ALK and PARP. J Hematol Oncol. 2011;4:16. doi: 10.1186/1756-8722-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen QY, Xu LQ, Jiao DM, Yao QH, Wang YY, Hu HZ, et al. Silencing of Rac1 modifies lung cancer cell migration, invasion and actin cytoskeleton rearrangements and enhances chemosensitivity to antitumor drugs. Int J Mol Med. 2011;28:769–76. doi: 10.3892/ijmm.2011.775. [DOI] [PubMed] [Google Scholar]
- 4.Mackinnon AC, Tretiakova M, Henderson L, Mehta RG, Yan BC, Joseph L, et al. Paxillin expression and amplification in early lung lesions of high-risk patients, lung adenocarcinoma and metastatic disease. J Clin Pathol. 2011;64:16–24. doi: 10.1136/jcp.2010.075853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jagadeeswaran R, Surawska H, Krishnaswamy S, Janamanchi V, Mackinnon AC, Seiwert TY, et al. Paxillin is a target for somatic mutations in lung cancer: implications for cell growth and invasion. Cancer Res. 2008;68:132–42. doi: 10.1158/0008-5472.CAN-07-1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sattler M, Pisick E, Morrison PT, Salgia R. Role of the cytoskeletal protein paxillin in oncogenesis. Crit Rev Oncog. 2000;11:63–76. doi: 10.1615/CritRevOncog.v11.i1.30. [DOI] [PubMed] [Google Scholar]
- 7.Turner CE. Paxillin interactions. J Cell Sci. 2000;113:4139–40. doi: 10.1242/jcs.113.23.4139. [DOI] [PubMed] [Google Scholar]
- 8.Vande Pol SB, Brown MC, Turner CE. Association of Bovine Papillomavirus Type 1 E6 oncoprotein with the focal adhesion protein paxillin through a conserved protein interaction motif. Oncogene. 1998;16:43–52. doi: 10.1038/sj.onc.1201504. [DOI] [PubMed] [Google Scholar]
- 9.Turner CE, Glenney JR, Jr., Burridge K. Paxillin: a new vinculin-binding protein present in focal adhesions. J Cell Biol. 1990;111:1059–68. doi: 10.1083/jcb.111.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deakin NO, Turner CE. Paxillin comes of age. J Cell Sci. 2008;121:2435–44. doi: 10.1242/jcs.018044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lawson C, Schlaepfer DD. Integrin adhesions: who’s on first? What’s on second? Connections between FAK and talin. Cell Adh Migr. 2012;6:302–6. doi: 10.4161/cam.20488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–9. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 13.Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, et al. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012;26:2175–86. doi: 10.1096/fj.11-196543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maulik G, Shrikhande A, Kijima T, Ma PC, Morrison PT, Salgia R. Role of the hepatocyte growth factor receptor, c-Met, in oncogenesis and potential for therapeutic inhibition. Cytokine Growth Factor Rev. 2002;13:41–59. doi: 10.1016/S1359-6101(01)00029-6. [DOI] [PubMed] [Google Scholar]
- 15.Ma PC, Jagadeeswaran R, Jagadeesh S, Tretiakova MS, Nallasura V, Fox EA, et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005;65:1479–88. doi: 10.1158/0008-5472.CAN-04-2650. [DOI] [PubMed] [Google Scholar]
- 16.Knott AB, Bossy-Wetzel E. Impairing the mitochondrial fission and fusion balance: a new mechanism of neurodegeneration. Ann N Y Acad Sci. 2008;1147:283–92. doi: 10.1196/annals.1427.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu J, Yang Y, Wu J. Bcl-2 cleavages at two adjacent sites by different caspases promote cisplatin-induced apoptosis. Cell Res. 2007;17:441–8. doi: 10.1038/cr.2007.36. [DOI] [PubMed] [Google Scholar]
- 18.Stathopoulos GP, Boulikas T. Lipoplatin formulation review article. J Drug Deliv. 2012;2012:581363. doi: 10.1155/2012/581363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Warburg O. On Metabolism of tumors (Constable, London; 1930). [Google Scholar]
- 20.Plathow C, Weber WA. Tumor cell metabolism imaging. J Nucl Med. 2008;49(Suppl 2):43S–63S. doi: 10.2967/jnumed.107.045930. [DOI] [PubMed] [Google Scholar]
- 21.Plas DR, Thompson CB. Cell metabolism in the regulation of programmed cell death. Trends Endocrinol Metab. 2002;13:75–8. doi: 10.1016/S1043-2760(01)00528-8. [DOI] [PubMed] [Google Scholar]
- 22.Maechler P, Wollheim CB. Mitochondrial function in normal and diabetic beta-cells. Nature. 2001;414:807–12. doi: 10.1038/414807a. [DOI] [PubMed] [Google Scholar]
- 23.Cannino G, Di Liegro CM, Rinaldi AM. Nuclear-mitochondrial interaction. Mitochondrion. 2007;7:359–66. doi: 10.1016/j.mito.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 24.Brini M. Ca(2+) signalling in mitochondria: mechanism and role in physiology and pathology. Cell Calcium. 2003;34:399–405. doi: 10.1016/S0143-4160(03)00145-3. [DOI] [PubMed] [Google Scholar]
- 25.Gunter TE, Buntinas L, Sparagna G, Eliseev R, Gunter K. Mitochondrial calcium transport: mechanisms and functions. Cell Calcium. 2000;28:285–96. doi: 10.1054/ceca.2000.0168. [DOI] [PubMed] [Google Scholar]
- 26.Gunter TE, Yule DI, Gunter KK, Eliseev RA, Salter JD. Calcium and mitochondria. FEBS Lett. 2004;567:96–102. doi: 10.1016/j.febslet.2004.03.071. [DOI] [PubMed] [Google Scholar]
- 27.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009;28:1589–600. doi: 10.1038/emboj.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–62. doi: 10.1038/nature05111. [DOI] [PubMed] [Google Scholar]
- 30.Karbowski M, Arnoult D, Chen H, Chan DC, Smith CL, Youle RJ. Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J Cell Biol. 2004;164:493–9. doi: 10.1083/jcb.200309082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skulachev VP, Bakeeva LE, Chernyak BV, Domnina LV, Minin AA, Pletjushkina OY, et al. Thread-grain transition of mitochondrial reticulum as a step of mitoptosis and apoptosis. Mol Cell Biochem. 2004;256-257:341–58. doi: 10.1023/B:MCBI.0000009880.94044.49. [DOI] [PubMed] [Google Scholar]
- 32.Wallace DC, Brown MD, Lott MT. Mitochondrial DNA variation in human evolution and disease. Gene. 1999;238:211–30. doi: 10.1016/S0378-1119(99)00295-4. [DOI] [PubMed] [Google Scholar]
- 33.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delivani P, Adrain C, Taylor RC, Duriez PJ, Martin SJ. Role for CED-9 and Egl-1 as regulators of mitochondrial fission and fusion dynamics. Mol Cell. 2006;21:761–73. doi: 10.1016/j.molcel.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 35.Salgia R, Li JL, Lo SH, Brunkhorst B, Kansas GS, Sobhany ES, et al. Molecular cloning of human paxillin, a focal adhesion protein phosphorylated by P210BCR/ABL. J Biol Chem. 1995;270:5039–47. doi: 10.1074/jbc.270.10.5039. [DOI] [PubMed] [Google Scholar]
- 36.Grandemange S, Herzig S, Martinou JC. Mitochondrial dynamics and cancer. Semin Cancer Biol. 2009;19:50–6. doi: 10.1016/j.semcancer.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 37.Turner CE. Paxillin and focal adhesion signalling. Nat Cell Biol. 2000;2:E231–6. doi: 10.1038/35046659. [DOI] [PubMed] [Google Scholar]
- 38.Veale D, Ashcroft T, Marsh C, Gibson GJ, Harris AL. Epidermal growth factor receptors in non-small cell lung cancer. Br J Cancer. 1987;55:513–6. doi: 10.1038/bjc.1987.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santin G, Piccolini VM, Barni S, Veneroni P, Giansanti V, Dal Bo V, et al. Mitochondrial fusion: a mechanism of cisplatin-induced resistance in neuroblastoma cells? Neurotoxicology. 2013;34:51–60. doi: 10.1016/j.neuro.2012.10.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.