

Abstract
Two new fernene triterpenoids, fern-9(11)-en-3,19-dione (1) and 3β-acetoxyfern-9(11)-en-19-one (2), together with the known 3β-acetoxyfern-9(11)-en-19β-ol (3) and lichexanthone (4), have been isolated from the acetone extract of the lichen Pyxine berteriana. The structures of the new compounds were established on the basis of IR, extensive 1D and 2D NMR, and MS analyses. Although several fern-9(11)-enes have been isolated from lichens, compounds 1 and 2 are the first examples of naturally occurring fernene triterpenoids with a carbonyl function at C-19.
Lichens are symbiotic associations composed of at least a fungal partner, the mycobiont, and a photosynthetic partner, the photobiont.1 These associations frequently produce characteristic secondary metabolites that are of fungal origin. Most are unique to lichens, and only a small number occur in non-lichenized fungi or higher plants.2 Many of these lichen secondary compounds exhibit antibiotic, antitumor, antimutagenic, allergenic, antifungal, antiviral, enzyme inhibitory, and plant growth inhibitory properties.2,3 Triterpenoids are widely distributed in lichens, being commonly present in genera such as Nephroma and Pseudocyphellaria as well as in different genera of the Physciaceae (e.g., Dirinaria, Physcia, and Pyxine) and the Parmeliaceae (e.g., Parmelia and Evernia).3,4 A previous report on the secondary metabolites of Pyxine berteriana (Physciaceae) from Brazil indicated that it contained atranorin, lichexanthone, methyl pyxinate, and pyxinol, according to TLC analysis.4 In the course of the search for new metabolites from the lichen P. berteriana (Fée) Imshaug we have isolated two new fernene triterpenoids, fern-9(11)-en-3,19-dione (1) and 3β-acetoxyfern-9(11)-en-19-one (2), together with the known 3β-acetoxyfern-9(11)-en-19β-ol (3) and lichexanthone (4), which is a chemical marker of a group of species in the genus Pyxine.5,6 The structure elucidation of compounds 1 and 2 is described herein.
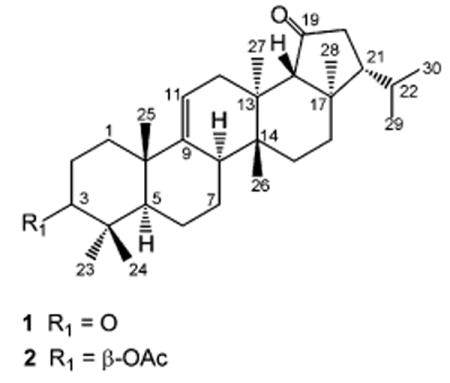
Compound 1 was obtained as a white, amorphous powder and showed a molecular ion at m/z 438.3485 in the HREIMS, indicative of a molecular formula of C30H46O2. Its IR absorption bands at 1731 and 1708 cm−1 suggested the presence of two ketone groups contained in a cyclopentanone7 and a cyclohexanone ring,8 respectively. The assignment of 1H and 13C NMR spectroscopic data of 1 (Table 1) was based on DEPT, HSQC, HMBC, NOESY, and 1H–1H COSY spectra. A DEPT NMR experiment permitted differentiation of the 30 13C NMR resonances into eight methyl, eight sp3 methylene, five sp3 methine, and five sp3 quaternary carbons, in addition to two carbonyls (δC 215.4 and 216.6) and a trisubstituted vinyl group resonating at δH 5.39 and at δC 117.1 and 148.9, characteristic of a Δ9,11 double bond.9 Characteristic resonances in the 1H and 13C NMR spectra (Table 1) for six tertiary methyls [δH/C 0.77/15.6 (C-26), 0.91/15.8 (C-28), 0.97/17.0 (C-27), 1.04/24.3 (C-23), 1.12/21.7 (C-24), 1.30/24.2 (C-25)] and two secondary methyls [δH/C 0.88/22.8 (C-30), 0.99/22.2 (C-29)] indicated a fern-9(11)-ene-type pentacyclic triterpenoid skeleton.9 In accordance with the COSY spectrum, the signal at δH 2.76 (H-2β) showed cross-peaks with the signals at δH 2.20 (H-1β), 2.23 (H-2α), and 1.64 (H-1α). On the basis of the HMBC and HSQC spectra, the signals at δH 2.76 and 2.23 (δC 35.1, H-2) showed cross-peaks with the signals at δC 40.4 (C-1), 216.6 (C-3), and 37.6 (C-10), establishing that C-3 corresponded to the carbonyl group at δC 216.6. Further correlations in the HMBC spectrum of the singlet at δH 1.30 (δC 24.2) with the signals at δC 37.6 (C-10), 40.4 (C-1), 46.4 (C-5), and 148.9 (C-9) allowed us to assign this methyl resonance to C-25. The NOESY correlations between H-5/CH3-23 and CH3-25/CH3-24 in conjunction with HSQC data permitted assignment of the 1H and 13C resonances of CH3-23 (δH 1.04, δC 24.3) and CH3-24 (δH 1.12, δC 21.7). HMBC correlations of these methyl protons with the signal at δC 216.6 confirmed the assignment of this carbonyl group to C-3. The broad doublet at δH 2.09 (δC 38.8) was assigned to H-8 on the basis of the cross-peaks with the signals at δH 1.41 and 1.70 (H-7) in the COSY spectrum, while correlations of the signal at δH 2.09 with H-5 (1.72) and CH3-27 (δH 0.97) in the NOESY spectrum confirmed its α-orientation. We assigned the chemical shift of C-18 at δC 64.6 on the basis of the HMBC correlations of CH3-27 (δH 0.97) and CH3-28 (δH 0.91) to C-18. The NOESY cross-peaks between H-18 (δH 2.18) and CH3-26 (δH 0.77) showed that H-18 had a β-orientation and indicated trans-fusion of the D/E ring. On the other hand, H-18 correlated in the HMBC spectrum with the signals at δC 36.3 (C-13), 43.0 (C-17), and the carbonyl group at δC 215.4. This observation together with the presence of a singlet for H-18 in the 1H NMR spectrum suggested that C-19 corresponded to the carbonyl group at δC 215.4. Further HMBC correlations of H-20β (δH 2.38) to C-17, C-19, and C-21 confirmed the presence of a carbonyl group at C-19. On the basis of the NOESY spectrum, the signal at δH 1.45 (δC 55.2, H-21) showed cross-peaks with H-18β, H-20β, and CH3-26, confirming the β-orientation of H-21, and hence the α-orientation of the isopropyl chain. Thus, this natural product corresponds to fern-9(11)-en-3,19-dione.
Table 1. NMR Spectroscopic Data (500 MHz, CDCl3) of Compounds 1 and 2a.
| position | 1 | 2 | ||
|---|---|---|---|---|
|
|
|
|||
| δC, mult. | δH (J in Hz) | δC, mult. | δH (J in Hz) | |
| 1 | 40.4, CH2 | 1.64, m (H-1α) | 39.0, CH2 | 1.40, m (H-1α) |
| 2.20, m (H-1β) | 1.93, dt (13.6, 3.4) (H-1β) | |||
| 2 | 35.1, CH2 | 2.23, m (H-2α) | 24.6, CH2 | 1.66, m |
| 2.76, td (14.6, 5.6) (H-2β) | ||||
| 3 | 216.6, qC | 80.9, CH | 4.48, dd (9.0, 6.7) | |
| 4 | 48.0, qC | 38.1, qC | ||
| 5 | 46.4, CH | 1.72, m | 44.5, CH | 1.38, m |
| 6 | 19.2, CH2 | 1.40 (H-6α), 1.75 (H-6β), m | 18.8, CH2 | 1.61, 1.76, m |
| 7 | 17.8, CH2 | 1.41, 1.70, m | 17.8, CH2 | 1.27, 1.63, m |
| 8 | 38.8, CH | 2.09, bd (13.6) | 39.0, CH | 2.02, bd |
| 9 | 148.9, qC | 150.1, qC | ||
| 10 | 37.6, qC | 37.6, qC | ||
| 11 | 117.1, CH | 5.39, m | 116.4, CH | 5.32, m |
| 12 | 35.0, CH2 | 2.51, ddd (17.9, 5.6, 1.5) (H-12α), 1.68, m (H-12β) | 35.1, CH2 | 2.49, ddd (17.8, 5.4, 1.8) (H-12α), 1.66, m (H-12β) |
| 13 | 36.3, qC | 36.3, qC | ||
| 14 | 37.3, qC | 37.3, qC | ||
| 15 | 28.9, CH2 | 1.40, m | 28.9, CH2 | 1.36, m |
| 16 | 35.8, CH2 | 1.65, m (H-16β), 1.82, dt (13.2, 3.4) (H-16α) | 35.8, CH2 | 1.64, m (H-16β), 1.83, dt (13.2, 3.3) (H-16α) |
| 17 | 43.0, qC | 43.1, qC | ||
| 18 | 64.6, CH | 2.18, s | 64.7, CH | 2.17, s |
| 19 | 215.4, qC | 215.5, qC | ||
| 20 | 42.4, CH2 | 1.77, dd (18.9, 10.3) (H-20α), 2.38, dd (18.9, 8.2) (H-20β) | 42.4, CH2 | 1.77, dd (18.9, 10.0) (H-20α), 2.38, dd (18.9, 8.0) (H-20β) |
| 21 | 55.2, CH | 1.45, m | 55.2, CH | 1.44, m |
| 22 | 30.2, CH | 1.64, m | 30.2, CH | 1.63, m |
| 23 | 24.3, CH3 | 1.04, s | 27.4, CH3 | 0.84, s |
| 24 | 21.7, CH3 | 1.12, s | 16.1, CH3 | 0.94, s |
| 25 | 24.2, CH3 | 1.30, s | 25.2, CH3 | 1.08, s |
| 26 | 15.6, CH3 | 0.77, s | 15.6, CH3 | 0.73, s |
| 27 | 17.0, CH3 | 0.97, s | 16.9, CH3 | 0.98, s |
| 28 | 15.8, CH3 | 0.91, s | 15.8, CH3 | 0.91, s |
| 29 | 22.2, CH3 | 0.99, d (6.6) | 22.3, CH3 | 0.98, d (6.5) |
| 30 | 22.8, CH3 | 0.88, d (6.6) | 22.8, CH3 | 0.88, d (6.5) |
| CH3COO | 170.9, qC | |||
| CH3COO | 21.3, CH3 | 2.05, s | ||
Assigned by a combination of 1H–1H COSY, NOESY, HSQC, and HMBC experiments.
Compound 2 was obtained as a white, amorphous powder. Its HREIMS showed a molecular ion at m/z 482.3782, indicative of a molecular formula of C32H50O3 and eight unsaturation degrees. Three of these were due to the presence of two carbonyl groups (one band at 1732 cm−1 in the IR consistent with both a cyclopentanone ring and an ester group at δC 170.9 and 215.5 in the 13C NMR spectrum) and one trisubstituted double bond [δH 5.32 and δC 116.4 and 150.1]. The assignment of 1H and 13C NMR spectroscopic data of 2 (Table 1) was based on DEPT, HSQC, HMBC, NOESY, and 1H–1H COSY spectra. The 13C NMR spectrum showed 32 resonances, of which 30 were attributed to a fern-9(11)-ene-type pentacyclic triterpene skeleton and two to an acetyl group (δH 2.05; δC 21.3 and 170.9).9 A DEPT NMR experiment showed the presence of nine methyl, eight sp3 methylene, six sp3 methine (one oxygenated), and five sp3 quaternary carbons, in addition to the carbonyl and olefinic carbons. Analysis of the 13C NMR spectroscopic data of 2 (Table 1) revealed structural similarity to those of 3β-acetoxyfern-9(11)-en-19β-ol (3),9 except for the presence of a ketone functionality (δ 215.5 ppm) and the absence of the hydroxy group at C-19 (δC 71.2 ppm, δH 4.23 ppm). This observation, along with the HMBC correlations from H-18 (δH 2.17) to C-13, C-17, C-28, and the carbonyl signal at δC 215.5, indicated the presence of a C-19 ketone functionality. Further HMBC correlations from H-20 to C-17, C-21, C-22, and the signal at δC 215.5 together with comparison of 1H and 13C NMR data of 2 with those of rings C, D, and E of compound 1 confirmed that both compounds shared the same ketone functionality at C-19 and differed in the substituent at C-3. The NMR data of compound 2 showed close resemblance with those of rings A and B and CH3-23, CH3-24, and CH3-25 of 3β-acetoxyfern-9(11)-en-19β-ol (3).9 On the basis of the HMBC and HSQC spectra, the signals at δH 1.40 and 1.93 (δC 39.0, H-1) showed cross-peaks with the signals at δC 24.6 (C-2), 25.2 (CH3-25), 37.6 (C-10), and 44.5 (C-5), whereas the signal at δH 4.48 (δC 80.9, H-3) correlated with the signals at δC 16.1 (CH3-24), 24.6 (C-2), 27.4 (CH3-23), and 38.1 (C-4). These data together with the HMBC correlations of the methyl singlet at δH 2.05 (δC 21.3, CH3COO) with δC 80.9 (C-3) and 170.9 (CH3COO) established the position of the acetoxy group at C-3. NOESY correlations between H-3 and δH 0.84 (CH3-23), 1.38 (H-5α), and 1.40 (H-1α) confirmed the β-orientation of the acetoxy group. The NOESY cross-peaks between H-5/CH3-23 and CH3-25/CH3-24 in conjunction with HSQC data allowed assignment of the 1H and 13C NMR resonances of CH3-23 (δH 0.84, δC 27.4), CH3-24 (δH 0.94, δC 16.1), and CH3-25 (δH 1.08, δC 25.2). As a consequence, compound 2 is identified as 3β-acetoxyfern-9(11)-en-19-one.
Compounds 1 and 2 are the first naturally occurring examples of fernene-type triterpenoids containing an unprecedented ketone function at C-19. Previously, two semisynthetic derivatives with a carbonyl group at C-19, fern-9(11)-en-19-one and fern-7-en-19-one, were obtained by CrO3 oxidation of two fernene triterpenoids isolated from the rhizomes of Davallia solida.7 Compound 1 is the second example of a natural fernene-type triterpenoid with two ketone groups. Previously, fern-9(11)-en-3,12-dione has been isolated from the lichen Xanthoria resendei.8
Compound 3 was isolated as a white, amorphous powder and identified as 3β-acetoxyfern-9(11)-en-19β-ol by comparison of the NMR and EIMS data with those reported previously.9 Lichexanthone (4) was isolated as a yellow, amorphous powder and identified by EIMS and 1H NMR data and comparison with published data.3
Experimental Section
General Experimental Procedures
Optical rotations were measured on a Perkin-Elmer 343 polarimeter. IR spectra were recorded on a Nicolet Magna-550 FT-IR spectrometer. 1H and 13C NMR spectra were recorded in CDCl3 on a Bruker AM 500 spectrometer. EIMS data were recorded on a Shimadzu QP-5000 mass spectrometer. HREIMS were obtained on a VG ZAB T4 mass spectrometer. Analytical HPLC was carried out on a Gilson 506C HPLC chromatograph using a reversed-phase analytical column (Phenomenex Hypersil; 5 μm pore size, 250 × 4.6 mm). The samples were eluted with a two-solvent system at a rate of 1 mL min−1. Solvent A was 1% aqueous orthophosphoric acid–MeOH (7:3), and solvent B was MeOH. The gradient started with 0% B and increased to 58% B within 15 min, then to 100% B within 15 min, followed by 100% B for 10 min. Compounds were detected using a 170 photodiode array detector set at 245 nm, operated in series with Unipoint System software, recording the absorption spectrum in the range 200–400 nm. TLC was performed on precoated Si gel F254 (cyclohexane–EtOAc (9:1)) and detected by spraying with H2SO4 (5% EtOH).
Lichen Material
Thalli of P. berteriana were collected on M. azedarach by one of the authors (M.T.A.) at Glew, Buenos Aires Province, Argentina, on October 7, 2000. A voucher specimen (39315) was identified by M.T.A. and preserved at the Herbarium of the Faculty of Exact and Natural Sciences (BAFC), Buenos Aires, Argentina.
Extraction and Isolation
The lichen (1.05 g wet weight) was cleaned, cut into small pieces, and extracted in acetone (100 mL) at room temperature. The acetone extract was evaporated under reduced pressure to give a residue (103 mg), which was subjected to silica gel column chromatography using cyclohexane and cyclohexane–EtOAc mixtures (99:1 and 98:2) as eluents to give the pure triterpenoids 1 (8.0 mg), 2 (3.6 mg), and 3 (2.9 mg) and lichexanthone (4) (14.6 mg). Compounds 1 and 2 showed one peak in their HPLC chromatograms at tR 41.4 and 37.5 min, respectively.
Fern-9(11)-en-3,19-dione (1)
white, amorphous powder; [α]20D −9.3 (c 0.33, CHCl3); IR (KBr) νmax 2964, 2936, 2669, 1731, 1708, 1469, 1382, 1110 cm−1; 1H and 13C NMR data, see Table 1; EIMS m/z 438 [M]+, 423, 405; HREIMS m/z 438.3485 (calcd for C30H46O2, 438.3498).
3β-Acetoxyfern-9(11)-en-19-one (2)
white, amorphous powder; [α]20D +6.7 (c 0.15, CHCl3); IR (KBr) νmax 2948, 2854, 1732, 1451, 1376, 1246, 1027 cm−1; 1H and 13C NMR data, see Table 1; EIMS m/z 482 [M]+, 467, 407; HREIMS m/z 482.3782 (calcd for C32H50O3, 482.3760).
Supplementary Material
Acknowledgments
This work was supported by the University of Buenos Aires (Grant X124) and CONICET (PIP 5509). Mass spectrometry was provided by the Washington University Mass Spectrometry Resource, an NIH Research Resource (Grant No. P41RR0954). M.A.F. thanks the University of Buenos Aires for a Doctoral fellowship. M.S.M. and M.T.A. are Research Members of the National Research Council of Argentina (CONICET).
Footnotes
Supporting Information Available: Spectroscopic data of new compounds 1 and 2. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Nash TH., III . In: Lichen Biology. Nash TH III, editor. Cambridge University Press; New York: 1996. pp. 1–7. Chapter 1. [Google Scholar]
- 2.Elix JA. In: Lichen Biology. Nash TH III, editor. Cambridge University Press; New York: 1996. pp. 176–178. Chapter 9. [Google Scholar]
- 3.Huneck S, Yoshimura I. Identification of Lichen Substances. Springer Verlag; Berlin: 1996. pp. 208–209. [Google Scholar]
- 4.Huneck S, Morales Mendez A, Kalb K. J Hattori Bot Lab. 1987;62:331–338. [Google Scholar]
- 5.Kathirgamanathar S, Ratnasooriya WD, Baekstrom P, Andersen RJ, Karunaratne V. Pharm Biol. 2006;44:217–220. [Google Scholar]
- 6.Kalb K. Brasilianische Flechten. 1. Die Gattung Pyxine, Bibliotheca Lichenologica. Band 24. J Cramer: Stuttgart; 1987. pp. 20–21. [Google Scholar]
- 7.Tanaka Y, Kitajima J, Ageta H. Nat Med. 1998;52:409–413. [Google Scholar]
- 8.González AG, Martín JD, Pérez C. Phytochemistry. 1974;13:1547–1549. [Google Scholar]
- 9.Wilkins AL, Elix JA. Aust J Chem. 1990;43:623–627. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.