

Abstract
Adults with relapsed B-acute lymphoblastic leukemia (ALL) have a dismal prognosis. Only those patients able to achieve a second remission with no minimal residual disease (MRD−) have a hope for long-term survival in the context of a subsequent allogeneic hematopoietic stem cell transplantation (allo-HSCT). We have treated 5 relapsed B-ALL subjects with autologous T cells expressing a CD19-specific CD28/CD3ζ second generation dual-signaling chimeric antigen receptor (CAR) termed 19-28z. All patients with persistent morphological disease or MRD+ disease upon T cell infusion demonstrated rapid tumor eradication and achieved MRD-negative complete remissions as assessed by deep sequencing PCR. Therapy was well tolerated although significant cytokine elevations, specifically observed in those patients with morphologic evidence of disease at the time of treatment, required lymphotoxic steroid therapy to ameliorate cytokine-mediated toxicities. Significantly, cytokine elevations directly correlated to tumor burden at the time of CAR modified T cell infusions. Tumor cells from one patient with relapsed disease after CAR modified T cell therapy, ineligible for additional allo-HSCT therapy, exhibited persistent expression of CD19 and sensitivity to autologous 19-28z T cell mediated cytotoxicity suggesting potential clinical benefit of additional CAR modified T cell infusions. These results demonstrate the marked anti-tumor efficacy of 19-28z CAR modified T cells in patients with relapsed/refractory B-ALL and the reliability of this novel therapy to induce profound molecular remissions, an ideal bridge to potentially curative therapy with subsequent allo-HSCT.
Introduction
Despite available chemotherapy and allogenic hematopotetic stem cell transplantation (allo-HSCT), adult patients with relapsed B cell acute leukemia (B-ALL) have a very poor prognosis. Long-term survival of adult patients with relapsed B-ALL is dependent upon achieving a complete remission (CR) induced through salvage chemotherapy followed by allo-HSCT (1, 2). Unfortunately, many patients never receive a potential life saving allo-HSCT due to a failure in achieving a second CR following salvage chemotherapy (1). Further, in patients undergoing an allo-HSCT, those with minimal residual disease (MRD+) by FACS or PCR have a significantly worse prognosis compared to patients with no evidence of MRD (MRD−) at the time of allo-HSCT (3). For this reason, novel therapeutic regimens for this patient population are needed.
A patient’s own T cells may be genetically modified to express an artificial T cell receptor termed a chimeric antigen receptor (CAR) designed to be specific to a tumor associated antigen. We and others have previously reported on promising pre-clinical outcomes of CAR modified T cells targeted to the B cell CD19 antigen (4-7). CD19 is expressed on normal B cells as well as most B cell malignancies including low-grade chronic lymphocytic leukemias (CLL), B cell non-Hodgkins lymphomas, as well as more aggressive B-ALL. Despite differences in CAR and clinical trial designs, expansion of this technology to treat patients with low-grade B cell malignancies (CLL and follicular lymphoma) at 3 different centers have all demonstrated significant anti-tumor responses following infusion of CD19 targeted autologous T cells (8-12). While promising clinical outcomes have been reported in patients with low-grade B cell tumors, to date there are no reported clinical outcomes utilizing this CD19-targeted adoptive T cell therapy approach in patients with relapsed B-ALL, a far more aggressive disease with a markedly worse prognosis.
We have treated 5 relapsed B-ALL adult patients with autologous second generation CD19 targeted CAR (19-28z) T cells following salvage chemotherapy. We report the dramatic ability of autologous 19-28z CAR modified T cells to induce MRD− CRs in patients with relapsed and/or chemotherapy refractory B-ALL. Further, we demonstrate that post T cell infusion cytokine mediated toxicities, similar to reported toxicities (9-12) in low grade B cell malignancies with CD19 targeted CAR modified T cells, correlate to the degree of tumor burden at the time of CAR modified T cell infusion. Our data demonstrate the life-saving potential of this technology for the treatment of relapsed B-ALL.
Results
Infusion of 19-28z CAR modified T cells induce MRD-remissions
Patients with relapsed B-ALL, not previously treated with allo-HSCT, independent of remission status following salvage chemotherapy, were eligible for therapy with autologous 19-28z+ T cells on this clinical protocol (figs. S1-S2). Patients were treated at a dose of 1.5-3 × 106 autologous 19-28z+ T cells/kg (13) (table S1) following prior conditioning therapy with 1.5-3.0 gm/m2 cyclophosphamide. For ethical reasons, and as per the protocol, following adoptive T cell therapy, eligible patients underwent subsequent allo-HSCT limiting the time for follow-up observation of CAR modified T cells and long-term molecular remissions in 4 of 5 patients treated to date.
Of the 5 treated subjects, patients MSK-ALL04 and MSK-ALL05 exhibited persistent chemotherapy refractory disease with 63 and 70% blast cells in the bone marrow respectively following salvage chemotherapy; two other patients had achieved a morphologic CRs with evidence of MRD+ by deep sequencing PCR and FACS (MSK-ALL01 and MSK-ALL06) (Table 1). Subsequent to adoptive 19-28z+ T cell therapy, all patients were MRD− as assessed by deep sequencing PCR demonstrating the loss of detectable malignant clone IgH rearrangements. Of those patients with morphologic evidence of disease at the time of modified T cell therapy, MSK-ALL04 achieved a morphologic CR by day 11 and MRD− status by day 59; MSK-ALL05 dramatically achieved both a morphologic CR as well as MRD− status within 8 days of T cell infusion (Fig. 1, Table 2). MRD+ patient MSK-ALL01 achieved MRD− status by day 28 while MRD+ patient MSK-ALL06 was MRD− by deep sequencing on day 30 and remained with MRD-disease up to the time of allo-HSCT 122 days after therapy (Table 2). Collectively, all patients achieved an MRD− status as the optimal disease response following 19-28z+ T cell infusion. Assessment of the durability of MRD-responses in this study were limited by patients subsequently undergoing allo-HSCT 1-4 months following therapy in 4 of 5 patients with the longest MRD-status reported in this cohort out to 122 days post CAR modified T cell therapy.
Table 1. Patient Characteristics and Response Summary.
Cy indicates cyclophosphamide; Vinc, vincristine; Pred, prednisone; Etop, Etoposide; Peg, pegylated asparaginase; Mito, mitoxantrone; CR1, first complete remission; MRD, minimal residual disease as assessed by deep sequencing (see Supplemental Data); Allo-SCT, allogeneic stem cell transplant. All patients were treated with cyclophosphamide before T cell infusion, either 1.5 gm/m2 (MSK-ALL04, MSKALL05, MSK-ALL06) or 3.0 gm/m2 (MSK-ALL01, MSK-ALL03).
| Patient ID* |
Age | FISH/Cyto genetics |
Initial therapy | Duration of CR1 |
Salvage therapy |
Disease response to Salvage therapy** |
Disease Response |
Steroids | Outcome |
|---|---|---|---|---|---|---|---|---|---|
| MSK- ALL01 |
66 | Normal karyotype |
Mito/Cy → Vinc/Pred → Cy→ Etop/Cy |
27 weeks | Vinc/Pred/Peg | MRD+ | MRD− | N | Allo-SCT |
| MSK- ALL03 |
56 | Normal karyotype |
HyperCVAD | 45 weeks | Inotuzumab ozogamicin → Vinc/Pred/Peg |
MRD− | MRD− | N | Allo-SCT |
| MSK- ALL04 |
59 | t(9;11), 9p21 deletion |
ECOG2993(24) | 5 weeks | Vinc/Pred | Refractory disease, 63% blasts in BM |
MRD− | Y | Ineligible for Allo-SCT, relapse 90 days |
| MSK- ALL05*** |
58 | 9p21 deletion |
ECOG2993 | 28 weeks | HIDAC/Mito | Refractory disease, 70% blasts in BM |
MRD− | Y | Allo-SCT |
| MSK- ALL06 |
23 | Normal karyotype |
NYII ref (25) |
34 months |
Modified NYII Consolidation I ref (25) |
MRD+ | MRD− | N | Allo-SCT |
MSK-ALL02 patient was removed from the study prior to the planned T cell infusion because they deferred T cell infusion for an allo-SCT.
Disease status within 1 week of infusion with CD19-targeted T cells
This patient’s T cells were harvested while in remission. All other patients listed had their T cells harvested while they had relapsed disease.
Figure 1. Rapid anti-tumor effects mediated by 19-28z T cells.

A. BM aspirates pre- and post-treatment with 19-28z T cells in 2 patients with morphologic chemotherapy-refractory B-ALL. Cyclophosphamide was given at Day −1 and CD19 CAR-targeted T cells were infused on Days 1 and 2. Left panels. BM prior to CAR modified T cell therapy demonstrated predominant blast cells with an absence of normal BM precursors. For MSK-ALL04 the left panel includes an inset with 100× magnification. Middle panels are BM aspirates done shortly after 19-28z T cell infusion and is hypocellular with normal stromal elements, histiocytes, and no evidence of blasts. Right panels. By 1 to 2 months after CAR modified T cell therapy there is BM recovery with normal hematopoiesis and no evidence of abnormal blasts. B. Flow cytometry for CD19 and CD10 expression in BM pre- and post-treatment. Cells were gated on CD45+7AAD− cells.
Table 2. Deep Sequencing for IgH rearrangements before and after CD19 CAR-targeted T cell therapy.
Adaptive Biotechnologies performed multiplex PCR and Deep Sequencing on genomic DNA prepared from BM aspirated on the noted day (see Supplemental Data for further detail). Malignant IgH rearrangement refers to IgH rearrangements associated with the B-ALL tumor cells. Total # of IgH rearrangements are derived from both malignant and nonmalignant B cells.
| Patient ID | Day of Treatment | Total # IgH rearrangements |
Total # malignant IgH rearrangements |
|---|---|---|---|
| MSK- ALL01* |
−1: 55: |
57,480 15,925 |
58 0 |
| MSK- ALL03* |
−5: 30: |
1,084 0 |
0 0 |
| MSK- ALL04 |
-4: 11: 24: 59: |
2,430,058 2,407 1,144 995,563 |
2,426,898 1,316 637 0 |
| MSK- ALL05* |
−1: 8: 29: |
3,307,494 1,880 8,270 |
3,300,732 0 0 |
| MSK- ALL06 |
−34: 18: 39: |
255,301 5,429 1,866,851 |
174,698 4 0 |
Patient has gone to allo-SCT and is off-study.
One patient, MSK-ALL04, ineligible for either allo-HSCT or additional CAR T cell therapy relapsed 90 days after treatment in the setting of prior high dose steroid therapy for cytokine mediated toxicities which may in turn have abrogated CAR modified T cell persistence, enhancing the ability of the B-ALL CD19+ tumor cells to escape immune surveillance mediated by the infused CAR modified T cells. Despite this isolated case of relapsed disease, the presented clinical outcomes on this cohort of patients collectively, for the time, demonstrate the profound clinical benefit of CD19-targeted CAR technology in the setting of relapsed adult B-ALL, a highly aggressive and predominantly fatal condition (1, 2).
Cytokine mediated toxicities correlate to tumor bulk at the time of CAR T cell infusion
Therapy with CD19 targeted CAR modified T cells have reported toxicities of high fevers, hypotension, and elevated pro-inflammatory serum cytokines in patients with chronic lymphocytic leukemia (CLL) and B cell non-Hodgkin lymphoma (NHL) (9, 10, 12). For this reason, serum cytokine levels were serially monitored in all treated patients in order to correlate cytokine levels to subsequent treatment related toxicities and patient clinical outcomes (table S2). Patients with evidence of persistent disease at the time of CAR modified T cell infusions generally showed early pro-inflammatory cytokine responses starting 3-5 days after modified T cell infusion, albeit at markedly different levels. The highest cytokine elevations, including sIL2Rα, IFNγ, IL6, and IP10, were seen in MSK-ALL04 and MSK-ALL05, the two patients with the highest tumor burden at the time of T cell infusion (Fig. 2A, fig. S3); these two patients also had the largest number of CD19 CAR-targeted T cells (Fig. 2B, and table S3). Cytokine elevations were far more modest or undetectable in the other MRD+ and MRD− patients (Fig. 2A). The degree of cytokine elevation thus correlated to the bulk of residual disease at the time of adoptive T cell infusion (Fig. 2C), and was coincident with post infusion fevers and episodes of relative hypotension (Figure 3). Three of 5 patients, all with the highest levels of measurable disease, experienced transient fevers following adoptive T cell therapy (Figure 3, and table S2). Increased fever severity and persistence accompanied by relative hypotension and transient mental status changes were observed in MSK-ALL04 and MSK-ALL05 (Figure 3, and table S2). Both MSK-ALL04 and MSK-ALL05 received high-dose lymphotoxic steroid therapy starting day 6 followed by a slow taper which rapidly ameliorated constitutional symptoms (Figure 3) and concomitantly normalized serum cytokine levels (Fig. 2A). Our data, for the first time, presents a positive correlation between cytokine surges with associated clinical toxicities in patients treated with CD19 targeted CAR T cells, to the degree of tumor burden at the time of CD19 targeted T cell infusions (Fig. 2C).
Figure 2. Cytokine release and T cell persistence are increased in patients with high tumor burdens.

A) Pre- and post-treatment serum from patients were evaluated for listed cytokines (pg/mL). * marks initiation of steroids for MSK-ALL04 and MSK-ALL05. MSK-ALL04 started dexamethasone 20 mg every 8 hours on Day 6, then held for one day, and restarted on Day 8. It was tapered off over 2 weeks. MSK-ALL05 was started on dexamethasone 20 mg every 12 hours and it was tapered off over 12 days. B) 19-28z T cells in the blood were detected by qPCR as described (8). From these results absolute 19-28z T cell counts were calculated. MSK-ALL04 is missing a time-point immediately before administration of steroids. C) Correlation between maximum IFNγ (pg/mL) and tumor burden for each patient (left-panel) and between the maximum 19-28z T cell count and tumor burden for each patient were calculated as the Spearman rank correlation coefficient (r), which is listed on both panels. Tumor burden is the number of malignant IgH clonotypes identified in the pre-treatment BM. Tumor burden, cytokines, and 19-28z T cell counts were rank-ordered to calculate the correlation coefficient. The Spearman rank correlation coefficients for tumor burden and maximum IP10 (r=0.91), IL2 (r=0.88), and IL6 (r=0.72) were also calculated.
Figure 3. Persistent fevers in patients with high tumor burden after infusion with 19-28z CAR+ T cells.
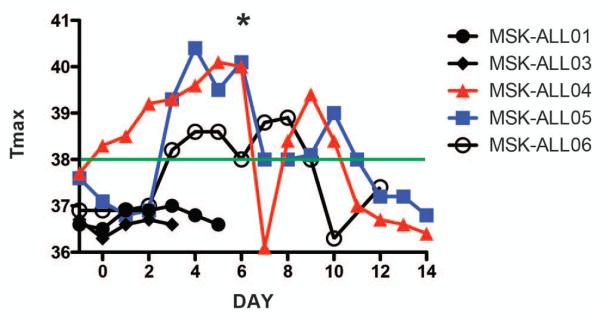
The maximum temperature (°C) in a 24-hour period is noted for all patients. Days listed range from Day −1 (cyclophosphamide) to 14 days after CD19 CAR-targeted T cell infusion. The * marks Day 6 when MSK-ALL04 and MSK-ALL05 were both started on high-dose steroids. The green line marks the minimum temperature for a fever (38°C).
CD19 targeted 19-28z+ T cells exhibit significant in vivo expansion and persistence
In vivo expansion and persistence of 19-28z+ T cells over time is likely to be a critical variable with respect to clinical outcomes. To this end we assessed both expansion and persistence of CAR modified T cells by FACS and PCR in this cohort of patients. Overall, CAR modified T cells were detectable by FACS or RT-PCR in the blood and bone marrow with a range of 3-8 weeks post 19-28z+ T cell infusion (Fig. 2B and 4). In vivo expansion, as measured by peak levels of detectable CAR modified T cells, additionally positively correlated to tumor burden at the time of modified T cell infusion (Fig. 2C). Analysis of T cell persistence in this study is compromised in part by the fact that eligible patients undergo allo-HSCT relatively soon after infusion with CAR modified T cells (1-4 months). Analyses of long-term CAR-modified T cell persistence in MSK-ALL04 and MSK-ALL05 were additionally compromised by prolonged high dose steroid therapies to treat cytokine mediated toxicities which coincided with a rapid drop in CAR-modified T cell counts and clinical evidence of cytokine meditated toxicities (Fig 2B). However, while loss of malignant clones by deep sequencing PCR was uniformly noted in all patients with persistent disease at the time of 19-28z+ T cell therapy, we concomitantly observed recovery of normal B cell clones in all patients consistent with waning persistence of CAR modified T cells and importantly, recovery of normal B cell lymphopoiesis (Table 2). Of note, MSK-ALL05, who rapidly became MRD− (by day 8), exhibited a remarkable rapid neutrophil recovery in the context of chronic daily GCSF infusions, despite prolonged neutropenia prior to T cell therapy, demonstrating the preservation and rapid restoration of normal hematopoiesis in the context of CD19 targeted19-28z+ T cell therapy (fig. S4).
Figure 4. 19-28z T cells can be detected in the blood of treated patients.
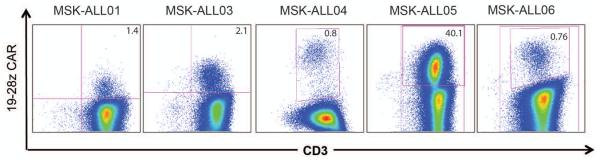
Within 1 week of 19-28z T cell infusion, peripheral T cells are isolated from the blood and activated ex vivo as described in Supplementary Methods and as reported elsewhere (8). Re-activated T cells were evaluated by flow cytometry for CD3 and 19-28z CAR expression. Cells displayed within the FACs plots have been gated as CD45+7AAD-.
Durable remissions occur after treatment with CD19 targeted 19-28z+ T cells and allo-SCT
Of the 5 patients treated on this protocol, 4 have undergone subsequent allo-HSCT (Table 1). MSK-ALL01 died of a suspected pulmonary embolus 2 months after allo-SCT, while in CR without any evidence of disease. The other 3 patients have had no significant complications post allo-SCT and all remain in CR. MSK-ALL03 remains in a CR 18 months after allo-SCT. MSK-ALL05 is in a CR 3 months after allo-SCT. MSK-ALL06 was in a MRD− remission for 122 days post-T cell infusion before being treated with an allo-SCT. To date, this patient remains MRD− 6 weeks after allo-SCT.
Relapsed B-ALL disease after 19-28z+ T cell therapy retains CD19 expression as well as sensitivity to CAR T cell mediated lysis
MSK-ALL04, who was ineligible for allo-HSCT due to multiple, pre-existing co-morbidities, was observed expectantly following CAR modified T cell therapy. This patient relapsed 13 weeks after T cell therapy and subsequently expired (Table 1). The relapsed tumor cells expressed the same malignant IgH rearrangement identified in the initial malignant clone and expressed the target CD19 antigen at pre-treatment levels (Fig. 5A). Further, the recurrent tumor cells retained sensitivity to lysis by autologous 19-28z+ T cells as assessed by a 51Chromium release assay (Fig. 5B). The relapse observed in this patient, despite previously having achieved MRD− status, is therefore not due to antigen escape but may be imputed, at least in part, to the abrogated persistence of the infused 19-28z+ T cells caused by the requisite need for high-dose steroid therapy to treat cytokine mediated toxicities (Fig. 2B).
Figure 5. MSK-ALL04 relapsed B-ALL tumor cells retain CD19 expression and sensitivity to 19-28z T cell mediated killing.

A. CD19 and CD10 expression of B-ALL tumor cells from the initial diagnostic whole blood sample (left panel) and the post 19-28z relapsed sample. Displayed cells have been gated on CD45+7AAD− cells. B. Untransduced (UNT) T cells from the leukapheresis product or 19-28z T cells from the End-of-Product formulated cells were incubated with the post 19-28z relapsed B-ALL tumor cells. Effectors were incubated with tumor cells (radiolabeled with 51Cr) at a 36:1 E:T ratio for 4 hours. 51Cr release was measured and calculated as a killing efficiency as described (8).
Discussion
Adult patients with relapsed B-ALL have an overall poor prognosis. Those patients who relapse following multi-agent chemotherapy protocols have an even more dismal prognosis. The standard of care for these patients is salvage chemotherapy followed by allo-HSCT if in remission and clinically eligible to undergo allo-HSCT. However, many such patients fail to achieve a second CR and/or are ineligible for additional transplantation therapy (1).
Treatment of patients with relapsed indolent B cell malignancies, including CLL and B cell follicular lymphomas, using autolgous T cells targeted to the CD19 antigen through the retroviral or lentiviral introduction of a CD19 specific CAR have demonstrated very promising clinical responses (8-12). Despite a growing body of clinical evidence for efficacy using CAR T cell technology in these indolent B cell tumors, it remains unknown whether this same technology may similarly exhibit significant anti-tumor efficacy in adults with far more aggressive and often fatal relapsed B-ALL.
The limitations of our study include the short-follow up post T cell infusion as well as the relatively small number of patients treated to date with the 19-28z T cells. The treatment of these patients with allo-SCT, the standard of care for relapsed B-ALL patients, after 19-28z T cell infusion, makes it indeed difficult to discern the curative or long-term remission potential of CD19 CAR-targeted T cells. However, it is unequivocal that patients who were ineligible for all-SCT and had no further therapeutic option in the face of rapidly progressing refractory disease, were promptly brought into profound molecular remission by a single infusion of 19-28z T cells.Despite these limitations, our work thus strongly supports the efficacy of 19-28z T cells in adults with acute leukemia.
In this report we summarize the outcomes of 5 adults with relapsed B-ALL, 4 which demonstrated persistent disease following salvage chemotherapy at the time of CAR T cell infusion. Overall, we report that in all patients, ranging from overt morphologic disease to MRD, treatment with autologous T cells uniformly resulted in MRD-CRs independent of tumor burden at the time of T cell therapy. These results demonstrate a potent anti-tumor efficacy of autologous T cells genetically modified to target the CD19 antigen expressed on adult B-ALL tumor cells. These consistent clinical outcomes are meaningful for several reasons: First, these data demonstrate the potential and rapid kinetics of 19-28z CD19-targeted T cells in eradicating chemotherapy resistant adult B-ALL; and second, the ability to rapidly and reliably achieve MRD− status dramatically improves the prognosis of these patients by providing a bridge to allo-HSCT under optimal conditions, the standard of care for these patients. Specifically, in the absence of CAR modified T cell therapy, two patients treated on this cohort would not have been eligible for allo-HSCT, while an additional two patients would have undergone this therapy in the MRD+ setting with predicted poor clinical outcomes (3).
Additionally, our studies demonstrate that cytokine-mediated toxicities, which are a significant previously published impediment to the application of this therapy, correlates to tumor bulk at the time of modified T cell infusion. Specifically, we demonstrate an improved side-effect profile with CAR modified T cells if infused in the context of a lower disease burden. These findings suggest that optimal safety of modified T cell infusions is either in the MRD+ setting or perhaps soon after initial salvage chemotherapy in the context of a largely aplastic bone marrow. Importantly, we find that a T cell-mediated cytokine release syndrome is not requisite to achieving optimal CAR T cell mediated anti-tumor efficacy resulting in MDR− complete remissions of disease.
Sufficient persistence of CD19-targeted CAR-modified cells to achieve tumor eradication (MRD− status) is, in theory, essential to optimally cancer anti-tumor efficacy. However, optimal anti-CD19 targeted T cell therapy as presented by collaborators at UPenn and other institutions present a balanced analysis with respect to the risks of potentially life threatening cytokine storms and lifelong B cell aplasias (9, 10, 12). In the present study, we observed a rapid onset of cytokine elevations in parallel with in vivo T cell expansion correlating to T cell clearance 3-8 weeks after infusion as assessed by FACS, RT-PCR, and in vitro expansion assays. Steroid therapy to ameliorate cytokine driven toxicity in 2 patients markedly reduced modified T cell populations at a time of maximum detectable CAR modified T cells (Fig. 2C) similar to a previously reported CLL patient at UPenn treated with high dose steroid therapy to control T cell mediated cytokine toxicities following infusion with CD19 targeted T cells (9). Nonetheless, in our studies, despite the steroid limited duration of 19-28z CAR modified T cells, CD19 targeted modified T cell persistence was sufficient to convert all patients with detectable disease to an MRD− status (Table 2). Further studies of modified T cell persistence in patients moving on to allo-HSCT were precluded by this additional standard of care therapy. Patient MSK-ALL04, of note, was managed expectantly due to co-morbidities prohibiting allo-HSCT and infusion of additional 19-28z CAR modified T cells. The patient, followed by modified CAR T cell infusion required additional lymphotoxic steroid therapy soon after modified T cell infusion (day 6) curtailing 19-28z+ T cell persistence and ultimately resulting in the relapse of the patient disease. Significantly, in vitro studies demonstrate that the relapsed clone retained CD19 expression and sensitivity to 19-28z+ T cells ex vivo. These results are consistent with the potential of lengthening molecular remissions, converting relapsed patients back to an MRD-status, or ultimately fully eradicating CD19+ malignant tumor clones in those patients otherwise ineligible for allo-HSCT through additional infusions of CAR modified T cells. Considering the decreased tumor burden at the time of second line therapy with CAR modified T cell infusion, the latter is likely to induce more modest cytokine elevations and therefore to be better tolerated.
The required dose of CAR-modified T cells was successfully generated in all patients within 2 weeks. To date, we have enrolled a total of 14 patients to this Phase I protocol. Of these 14 patients, 2 are in a CR and are not eligible for T cell infusion at this time, 3 deferred T cell infusion to go directly to allo-SCT, 3 died during salvage chemotherapy, 5 patients were successfully infused. One patient was lost to follow-up after enrollment. On an intent-to-treat basis we have generated 8 T cell products for enrolled patients including the 5 described in this study and 1 who went straight to allo-SCT and 2 who died during salvage chemotherapy.
The gene-transfer efficiency for 19-28z ranged from 5 to 14.2% (Table S1). While there is no clear etiology for this modest gene-transfer efficiency, it may be in part due to the nature of acute leukemia or the numerous cycles of high-dose lymphotoxic chemotherapeutic agents received by the patients before T cell harvest. Regardless, it does not appear that the lower CAR-transduction efficiency inhibited the anti-leukemia effect of the 19-28z T cells.
While we and others have previously reported clinical trial outcomes of CAR T cells targeted to either CD19 or CD20 in patients with low grade lymphomas and CLL (8-11, 14, 15) and summarized in (16), herein we are the first to present highly effective clinical outcomes with CD19-targeted CAR T cells in adults in the setting of far more aggressive B-ALL.
Recently published reports from the University of Pennsylvania (UPenn) demonstrate promising clinical outcomes in CLL patients treated with a second generation CAR derived from initial studies published elsewhere (17) containing the 4-1BB co-stimulatory signaling domain. In these studies, investigators demonstrate marked anti-tumor efficacy, profound cytokine mediated toxicity, and long-term persistence of both B cell aplasias and autologous T cells expressing this 4-1BB containing CAR (9, 12). Although results in adult ALL have not yet been reported with 4-1BB/CD3ζ CARs, some differences between the kinetics and magnitude of the cytokine response between the two CARs are noteworthy. In CLL patients, the 4-1BB/CD3ζ CAR yielded delayed peak cytokine responses seen greater than 10 days out from infusion, in contrast to a more immediate cytokine release (day 3-5) seen in our CD28/CD3ζ-containing 19-28z CARs. Additionally, chronic B cell aplasias were associated with long-term persistence of the 4-1BB/CD3ζ T cells, in contrast to the recovery of normal B lymphopoeisis we report here (Table 2). The more limited expansion and persistence of 19-28z+ T cells enables normal B cell recovery over time in the context of a more modest cytokine mediated toxicity profile. These findings are consistent with murine studies reported so far, which suggest a slower kinetic but ultimately higher accumulation and persistence of 4-1BBz T cells that occurs without antigen. In xenogeneic NSG chimeras, Milone et al (7) reported similar therapeutic efficacy of CD28/CD3z and 4-1BB/CD3z T cells in treating CD19+ leukemia, with the latter accumulating to higher levels in delayed fashion. In our own preclinical studies (18), 4-1BB/CD3z T cells were not as effective as CD28z T cells in treating B-ALL in scid/beige xenochimeras (19). Collectively, our studies treating patients with the CD28 containing 19-28z+ CAR modified T cells provides an alternate approach to using CAR technology, one in which multiple (perhaps two or three) infusions of modified T cells induce CRs in the context of lower tumor burdens, which in turn carry a diminished risk for cytokine-mediated toxicities and long-term B cell aplasias.
Adults with relapsed chemotherapy refractory B-ALL have a dismal prognosis outside the context of an allo-HSCT. We report, for the first time, the successful induction of MRD− remissions in adults with chemotherapy refractory B-ALL by treatment with CD19-targeted CAR-modified T cells. We acknowledge limited post treatment follow-up due to the fact that 4 of 5 patients subsequently received standard of care therapy with allo-HSCT as stipulated in the protocol and were therefore removed from further follow-up on this protocol. The 5th patient treated was ineligible for allo-HSCT and relapsed 3 months after therapy although this relapse may in part have been precipitated by high dose lymphotoxic steroid therapy to treat T cell mediated cytoline storms soon after modified T cell infusion limiting which in turn limited persistence of 19-28z CAR modified T cells. Further, these results are consistent with the potential of lengthening molecular remissions or alternatively reconverting relapsed patients to MRD-status through additional infusions of 19-28z CAR modified T cells. Whether additional infusions with CAR modified T cells in this particular patient may have changed the clinical outcome remains to be and will be investigated in future relapsed refractory B-ALL patients similarly ineligible for additional therapy with allo-HSCT. The rapid kinetics of 19-28z T cells may prove to be an essential asset in patients with lower tumor burdens but at high risk for an overt relapse. This approach provides a bridge for patients otherwise either ineligible or eligible under very suboptimal conditions (MRD+) to receive potentially life saving therapy with an allo-HSCT. Furthermore, this therapy has the potential to decrease the relapse rate post allo-HSCT, which can occur in up to one-third of patients (20, 21). In addition, repeated infusions of 19-28z+ T cells in patients unable to undergo allo-HSCT may prolong remission. Based on the presented data, treatment of relapsed chemotherapy refractory B-ALL in adults with 19-28z+ autologous T cells is a very promising and novel approach worthy of continued study. Finally, given these clinical outcomes, the addition of 19-28z CAR modified T cells for inclusion into currently up-front adult B-ALL treatment protocols warrants serious consideration.
Materials and Methods
Clinical Protocol design
This is a clinical trial designed to assess the safety of infusing autologous T cells modified to express the CD19 specific CAR 19-28z into patients with relapsed B cell ALL (ClinicalTrials.gov #NCT01044069). Patients presenting in CR1 or with relapsed disease are eligible for enrollment (Figures S1 and S2). However, patients enrolled during CR1 are only treated if they develop relapsed disease (Figure S2). Patients with relapsed disease receive a chemotherapy regimen chosen by their treating physician and are treated upon either count recovery or evidence of persistent disease. Patients receive high dose cyclophosphamide chemotherapy (1.5-3.0 gm/m2) (day −1) followed by a split dose infusion of CAR modified T cells on days 1 and 2. The primary endpoint of this study is safety of CAR modified T cell infusion. Secondary endpoints include studies to detect 19-28z+ T cell persistence, as well as assays to assess morphologic, cytogenetic, and molecular anti-tumor responses.
Patient Clinical Background
Five patients with relapsed B cell ALL were treated to complete the first cohort of patients on this clinical trial infused with the lowest planned dose of 19-28z modified T cells (1.5-3.0 × 106 CAR+ T cells/kg) (Table 1 and Table S1).
MSK-ALL01, a 66-year old male with normal cytogenetics received induction chemotherapy with high dose cytarabine and mitoxantrone achieving CR1. Following 3 cycles of consolidation chemotherapy, the patient was enrolled on trial with subsequent collection of T cells by leukapheresis. Following peripheral blood mononuclear cell (PBMC) collection, the patient was found to have relapsed disease with 50% blasts in the BM. The patient subsequently achieved a second morphological remission with minimal residual disease (MRD+) following re-induction chemotherapy with prednisone, vincristine, and PEG-Asparaginase. Upon bone marrow recovery, the patient received high dose cyclophosphamide (3 gm/m2) followed by split dose infusion of 19-28z+ T cells. He was referred for an allogeneic SCT while MRD- and was removed from study.
MSK-ALL03, a 56-year old male diagnosed with B-ALL with normal cytogenetics, achieved a CR1 that persisted throughout HyperCVAD chemotherapy (22) but relapsed during maintenance therapy with 6-mercaptopurine and methotrexate. The patient was subsequently treated on protocol with inotuzumab ozogamicin (23), achieving a very brief remission of disease. Upon relapse, the patient was enrolled on the CAR modified T cell trial, leukapheresed in the setting of relapsed disease, and achieved a third CR following salvage therapy with prednisone, vincristine, and PEG-Asparaginase. Upon BM recovery, the patient was treated with high dose cyclophosphamide (3 gm/m2) followed by split dose 19-28z+ T cells. He was referred for an allo-SCT while MRD- and removed from study.
MSK-ALL04, a 59 year old male diagnosed with high risk B cell ALL (WBC count 147.8 K/μL at diagnosis) with a 9p21 deletion (9p21−) received initial induction chemotherapy on protocol ECOG 2993 (24) achieving a CR after induction phase I but presented with relapsed disease following induction phase II. The patient was enrolled on study, leukapheresed in relapse (WBC=54 K/μL), and was subsequently re-induced with a combination of vincristine and prednisone. Upon completion, the patient exhibited persistent disease with 63% blasts present in a hypocellular bone marrow (BM). The patient subsequently received high dose cyclophosphamide (1.5 gm/m2) (day −1) followed by split dose infusion of 19-28z modified T cells on days 1 and 2. Long-standing co-morbidities (poorly controlled diabetes, renal insufficiency, and coronary artery disease) precluded an allo-SCT so he was monitored expectantly.
MSK-ALL05 is a 58 year-old male diagnosed with B-ALL and was induced into CR1 on the ECOG2993 regimen (24). He was treated with both phases of induction and an intensification phase but relapsed with 48% blasts in the BM. FISH and cytogenetic analysis of his relapsed B-ALL tumor cells demonstrated they were hyperdiploid, including an extra copy of the MLL gene, and had homozygotic deletion of the p16 gene. He was enrolled, leukapheresed, and treated with high-dose cytarabine (3 gm/m2) and mitoxantrone (40 mg/m2) but his disease persisted with 70% blasts in a hypocellular BM. He was treated with cyclophosphamide (1.5 gm/m2) at day −1 followed by split dose infusion of 19-28z T cells on days 1 and 2. He was referred for an allo-SCT while MRD- and subsequently removed from study after transplant.
MSK-ALL06 is a 23 year-old female with relapsed B-ALL (normal cytogenetics). After diagnosis she was induced into CR1 with a NYII induction regimen (25). She completed all her consolidation and maintenance treatments but relapsed about 9 months after her last maintenance treatment. She was treated with a modified NYII Consolidation I regimen (25) with the goal of inducing a CR2 and referring her to an allo-SCT. However, post-treatment analyses confirmed MRD (0.14% blasts in BM) and she developed a small bowel obstruction and underwent a bowel resection, which demonstrated mucor colitis. She was then enrolled in our trial, leukapheresed, and infused with 19-28z T cells following cyclophosphamide (1.5 gm/m2). After her mucor infection resolved she underwent an allo-HSCT in an MRD-CR at day 122 following CAR modified T cell therapy.
Generation of 19-28z CAR modified T cells
Peripheral blood mononuclear cells (PBMCs) were obtained from enrolled patients by leukapheresis and CAR-modified T cells were produced as described (8, 13). Briefly, leukapheresis product was washed and cryopreserved. T cells from thawed leukapheresis product were isolated and activated using Dynabeads® Human T-Activator CD3/CD28 magnetic beads (Invitrogen) and transduced with gammaretroviral 19-28z supernatant. Transduced T cells were then further expanded using the Wave bioreactor to achieve the desired modified T cell dose.
Assessment of 19-28z CAR modified T cell persistence
Persistence of 19-28z CAR modified T cells in patient peripheral blood and BM was assessed by FACS using biotinylated goat anti-mouse IgG F(ab)2 (Jackson ImmunoResearch) as described (8). In the setting of low lymphocyte numbers in samples, persistence of CAR modified T cells was assessed by FACS following non-specific expansion of T cells ex vivo using Dynabeads® ClinExVivo™ CD3/CD28 as previously described (8).
Assessment of anti-CD19 responses mediated by 19-28z CAR modified T cells
Serial BM and peripheral blood samples were assessed for both detectable tumor (CD19+ CD10+) as well as normal B cells (CD19+ CD10−) by flow cytometry. Persistence of normal B cells and the B-ALL clone were further monitored by deep sequencing (Adaptive Biotechnologies) (9). Briefly, 7.5 μg of genomic DNA (or its cell equivalent) from BM or blood was submitted to Adaptive Biotechnologies for multiplex PCR and re-sequencing. Sequences were then interrogated for copies of the malignant IgH clonotype, which was identified by multiplex PCR and re-sequencing of BM samples obtained during relapse. Additionally, MSK-ALL04 and MSK-ALL05 blood and BM were analyzed by FISH for the 9p21 deletion as an assessment of disease response to modified T cell therapy.
Analysis of cytokine profiles following 19-28z CAR modified T cell infusion
Serial serum samples obtained prior to and following modified T cell infusion were analyzed for cytokine profiles as a surrogate marker for infused T cell activation using the Luminex IS100 system and commercially available 39-plex cytokine detection assays as described (8).
Chromium Release Assay
19-28z T cells were evaluated for CD19-targeted killing by a chromium release assay as described (13).
Clinical Investigation Statement
The IRB at MSKCC reviewed and approved this trial. All patients enrolled and treated on this trial gave written informed consent before participation. All clinical investigation was conducted according to the Declaration of Helsinki principles.
Supplementary Material
Figure S1. Trial schema for adult patients with relapsed B-ALL.
Figure S2. Trial schema for adult patients with B-ALL in CR.
Figure S3. Serum cytokine detection and correlation with tumor burden.
Figure S4. Rapid hematopoietic recovery in MSK-ALL05 after infusion with 19-28z T cells.
Table S1: Characteristics of infused 19-28z transduced T cells; tumor burden in apheresis and bone marrow.
Table S2: Adverse events
Table S3: Percentage of 19-28z CAR+ T cells detected in the CD3+ T cells of the peripheral blood and of the bone marrow (in parenthesis) of patients up to 57 days post-CAR modified T cell therapy.
ACKNOWLEDGEMENTS
We thank the following for financial support: the National Cancer Institute (RB, MLD, IR, MS), the Terry Fox Foundation (RB), the American Society of Hematology-Amos Medical Faculty Development Program (MLD), the Alliance for Cancer Gene Therapy (MS), the Mallah Foundation, the Majors Foundation and Mr Lew Sanders (MS, RB and IR), The Annual Terry Fox Run for Cancer Research (New York, NY) organized by the Canada Club of New York (RB), Kate’s Team, and Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Cancer Foundation for Research and the Experimental Therapeutics Center of MSKCC (MS, RB, and IR).
Footnotes
Competing interests: MS and RB are co-holders of patent #US 7,446,190, which covers the 19-28z receptor.
REFERENCES and NOTES
- 1.Fielding AK, Richards SM, Chopra R, Lazarus HM, Litzow MR, Buck G, Durrant IJ, Luger SM, Marks DI, Franklin IM, McMillan AK, Tallman MS, Rowe JM, Goldstone AH. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood. 2007;109:944. doi: 10.1182/blood-2006-05-018192. [DOI] [PubMed] [Google Scholar]
- 2.Gokbuget N, Stanze D, Beck J, Diedrich H, Horst HA, Huttmann A, Kobbe G, Kreuzer KA, Leimer L, Reichle A, Schaich M, Schwartz S, Serve H, Starck M, Stelljes M, Stuhlmann R, Viardot A, Wendelin K, Freund M, Hoelzer D. Outcome of relapsed adult lymphoblastic leukemia depends on response to salvage chemotherapy, prognostic factors, and performance of stem cell transplantation. Blood. 2012;120:2032. doi: 10.1182/blood-2011-12-399287. [DOI] [PubMed] [Google Scholar]
- 3.Bassan R, Spinelli O, Oldani E, Intermesoli T, Tosi M, Peruta B, Rossi G, Borlenghi E, Pogliani EM, Terruzzi E, Fabris P, Cassibba V, Lambertenghi-Deliliers G, Cortelezzi A, Bosi A, Gianfaldoni G, Ciceri F, Bernardi M, Gallamini A, Mattei D, Di Bona E, Romani C, Scattolin AM, Barbui T, Rambaldi A. Improved risk classification for risk-specific therapy based on the molecular study of minimal residual disease (MRD) in adult acute lymphoblastic leukemia (ALL) Blood. 2009;113:4153. doi: 10.1182/blood-2008-11-185132. [DOI] [PubMed] [Google Scholar]
- 4.Brentjens RJ, Latouche JB, Santos E, Marti F, Gong MC, Lyddane C, King PD, Larson S, Weiss M, Riviere I, Sadelain M. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med. 2003;9:279. doi: 10.1038/nm827. [DOI] [PubMed] [Google Scholar]
- 5.Cooper LJ, Topp MS, Serrano LM, Gonzalez S, Chang WC, Naranjo A, Wright C, Popplewell L, Raubitschek A, Forman SJ, Jensen MC. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood. 2003;101:1637. doi: 10.1182/blood-2002-07-1989. [DOI] [PubMed] [Google Scholar]
- 6.Kochenderfer JN, Yu Z, Frasheri D, Restifo NP, Rosenberg SA. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood. 2010;116:3875. doi: 10.1182/blood-2010-01-265041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, Campana D, Riley JL, Grupp SA, June CH. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Molecular therapy : the journal of the American Society of Gene Therapy. 2009;17:1453. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, Olszewska M, Bernal Y, Pegram H, Przybylowski M, Hollyman D, Usachenko Y, Pirraglia D, Hosey J, Santos E, Halton E, Maslak P, Scheinberg D, Jurcic J, Heaney M, Heller G, Frattini M, Sadelain M. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, Yang JC, Kammula US, Devillier L, Carpenter R, Nathan DA, Morgan RA, Laurencot C, Rosenberg SA. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2011 doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, Morgan RA, Rosenberg SA. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hollyman D, Stefanski J, Przybylowski M, Bartido S, Borquez-Ojeda O, Taylor C, Yeh R, Capacio V, Olszewska M, Hosey J, Sadelain M, Brentjens RJ, Riviere I. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J Immunother. 2009;32:169. doi: 10.1097/CJI.0b013e318194a6e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, Forman SJ. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, Kamble RT, Bollard CM, Gee AP, Mei Z, Liu H, Grilley B, Rooney CM, Heslop HE, Brenner MK, Dotti G. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davila M, Brentjens R, Wang X, Riviere I, Sadelain M. How do CARs work?: Early insights from recent clinical studies targeting CD19. OncoImmunology. 2012;1:1577. doi: 10.4161/onci.22524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, Campana D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18:676. doi: 10.1038/sj.leu.2403302. [DOI] [PubMed] [Google Scholar]
- 18.Brentjens RJ, Santos E, Nikhamin Y, Yeh R, Matsushita M, La Perle K, Quintas-Cardama A, Larson SM, Sadelain M. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2007;13:5426. doi: 10.1158/1078-0432.CCR-07-0674. [DOI] [PubMed] [Google Scholar]
- 19.Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Molecular therapy : the journal of the American Society of Gene Therapy. 2010;18:413. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldberg JD, Linker A, Kuk D, Ratan R, Jurcic J, Barker JN, Castro-Malaspina H, Giralt S, Hsu K, Jakubowski AA, Jenq R, Koehne G, Papadopoulos EB, van den Brink MR, Young JW, Boulad F, Kernan NA, O’Reilly RJ, Prockop SE, Yahalom J, Heller G, Perales MA. T Cell-Depleted Stem Cell Transplantation for Adults with High-Risk Acute Lymphoblastic Leukemia: Long-Term Survival for Patients in First Complete Remission with a Decreased Risk of Graft- versus-Host Disease. Biol Blood Marrow Transplant. 2013;19:208. doi: 10.1016/j.bbmt.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fielding AK, Rowe JM, Richards SM, Buck G, Moorman AV, Durrant IJ, Marks DI, McMillan AK, Litzow MR, Lazarus HM, Foroni L, Dewald G, Franklin IM, Luger SM, Paietta E, Wiernik PH, Tallman MS, Goldstone AH. Prospective outcome data on 267 unselected adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia confirms superiority of allogeneic transplantation over chemotherapy in the pre-imatinib era: results from the International ALL Trial MRC UKALLXII/ECOG2993. Blood. 2009;113:4489. doi: 10.1182/blood-2009-01-199380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kantarjian HM, O’Brien S, Smith TL, Cortes J, Giles FJ, Beran M, Pierce S, Huh Y, Andreeff M, Koller C, Ha CS, Keating MJ, Murphy S, Freireich EJ. Results of treatment with hyper-CVAD, a dose-intensive regimen, in adult acute lymphocytic leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2000;18:547. doi: 10.1200/JCO.2000.18.3.547. [DOI] [PubMed] [Google Scholar]
- 23.Kantarjian H, Thomas D, Jorgensen J, Jabbour E, Kebriaei P, Rytting M, York S, Ravandi F, Kwari M, Faderl S, Rios MB, Cortes J, Fayad L, Tarnai R, Wang SA, Champlin R, Advani A, O’Brien S. Inotuzumab ozogamicin, an anti-CD22-calecheamicin conjugate, for refractory and relapsed acute lymphocytic leukaemia: a phase 2 study. Lancet Oncol. 2012;13:403. doi: 10.1016/S1470-2045(11)70386-2. [DOI] [PubMed] [Google Scholar]
- 24.Rowe JM, Buck G, Burnett AK, Chopra R, Wiernik PH, Richards SM, Lazarus HM, Franklin IM, Litzow MR, Ciobanu N, Prentice HG, Durrant J, Tallman MS, Goldstone AH. Induction therapy for adults with acute lymphoblastic leukemia: results of more than 1500 patients from the international ALL trial: MRC UKALL XII/ECOG E2993. Blood. 2005;106:3760. doi: 10.1182/blood-2005-04-1623. [DOI] [PubMed] [Google Scholar]
- 25.Steinherz PG, Redner A, Steinherz L, Meyers P, Tan C, Heller G. Development of a new intensive therapy for acute lymphoblastic leukemia in children at increased risk of early relapse. The Memorial Sloan-Kettering-New York-II protocol. Cancer. 1993;72:3120. doi: 10.1002/1097-0142(19931115)72:10<3120::aid-cncr2820721038>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Trial schema for adult patients with relapsed B-ALL.
Figure S2. Trial schema for adult patients with B-ALL in CR.
Figure S3. Serum cytokine detection and correlation with tumor burden.
Figure S4. Rapid hematopoietic recovery in MSK-ALL05 after infusion with 19-28z T cells.
Table S1: Characteristics of infused 19-28z transduced T cells; tumor burden in apheresis and bone marrow.
Table S2: Adverse events
Table S3: Percentage of 19-28z CAR+ T cells detected in the CD3+ T cells of the peripheral blood and of the bone marrow (in parenthesis) of patients up to 57 days post-CAR modified T cell therapy.