

Abstract
Based on the observation that wild-type Kaposi's sarcoma-associated herpesvirus (KSHV) DNA can be detected in the oral cavity of healthy, immunocompetent individuals, we hypothesized that epithelial cells could be infected in vitro by wild-type (WT) KSHV isolated from immunocompetent individuals. Primary oral epithelial (P-EPI) cells and telomerase-immortalized oral epithelial cells were generated from human gingival tissue and were then infected in vitro with WT KSHV isolated from throat wash samples. Markers of lytic and latent KSHV infection were detected in cultures by 24 h postinfection by immunofluorescence confocal microscopic assays. The infectivity of the WT and BCBL virus was blocked by neutralizing antibodies against KSHV gB. The presence of KSHV DNA in these cells was confirmed by real-time PCR amplification of different regions of the viral genome. The significant in vitro viral replication that had occurred was inhibited by ganciclovir and by neutralizing antibodies against gB. When infected cultures were examined by scanning electron microscopy, thousands of KSHV particles were clearly visible across the surfaces of P-EPI cells. The detection of enveloped particles indicated that the infectious cycle had proceeded through assembly and egress. We thus demonstrated that oral WT KSHV isolated from immunocompetent individuals was able to infect and replicate in vitro in a relevant primary cell type. Furthermore, our results provide compelling evidence for KSHV transmission within infected oral epithelial cells derived from healthy, immunocompetent populations.
Kaposi's sarcoma-associated herpesvirus (KSHV; also called human herpesvirus 8 [HHV-8]) is the etiologic agent of Kaposi's sarcoma (KS) (12) and peripheral effusion lymphoma (PEL) (10). The human herpesviruses are generally ubiquitous among human populations, are generally shed in the oral cavity, and are primarily transmitted via the salivary route. However, current serological tests for KSHV have determined that the seroprevalence within the general U.S. population is low, suggesting that, unlike the other human herpesviruses (with the exception of herpes simplex virus type 2 [HSV-2]), KSHV is not ubiquitous among healthy human populations.
The primary mode of KSHV transmission remains unresolved, as extensive evidence exists for both sexual (8, 25, 30, 31) and oral (5, 29, 34) transmission routes in immunosuppressed individuals. However, recent epidemiology studies suggested that oral transmission of KSHV does occur among healthy populations (14, 15). Complicating the transmission issue, the identities of the cell types harboring KSHV in vivo in the oral cavity and producing the virus detected in saliva are not known. Endothelial cells are the primary infected cells in KS lesions (7). Recently, KSHV antigens were detected in oral epithelial cells of an early KS lesion (44). In addition, the ability of the virus to infect primary human keratinocytes has also been demonstrated (3, 9). Based on the above evidence, we hypothesized that the wild-type (WT) isolates present in the oral cavity of healthy, HIV-negative individuals might differ in tropism from PEL-associated laboratory KSHV strains. We therefore employed an infection protocol initially described by Vieira and colleagues (42) to characterize in vitro WT KSHV infections of cultured epithelial cells.
We also hypothesized that KSHV infections are likely spread between healthy individuals by virus shed into saliva. To test this hypothesis, we isolated WT KSHV from throat wash (TW) samples of healthy, immunocompetent donors and used it as an inoculum to infect primary oral epithelial (P-EPI) cells and telomerase-immortalized oral epithelial (T-EPI) cells derived from human gingival tissue. Our results provide compelling evidence that KSHV is an opportunistic pathogen that replicates in oral epithelial cells.
MATERIALS AND METHODS
Cells and media.
Human melanoma (MeWo) cells were cultured in Eagle's minimal essential medium (GIBCO-BRL) containing 10% fetal bovine serum (FBS) (Sigma) and supplemented with nonessential amino acids (GIBCO-BRL), penicillin, streptomycin, and l-glutamine (GIBCO-BRL) as previously described (23). A293T cells, a simian virus 40 (SV40) T-cell-transformed clone of the HEK293 line (22), were a gift from Lishan Su (University of North Carolina, Chapel Hill) and were cultured and transfected as previously described (18). BJAB and BCBL1 cell lines were cultured in RPMI 1640 medium supplemented with 10% FBS and containing penicillin, streptomycin, sodium pyruvate, and l-glutamine (GIBCO-BRL). P-EPI cells were generated by a modified protocol initially described by Oda and Watson (32). Fragments of gingival tissue removed after dental extractions from healthy, HIV-negative individuals and obtained with informed consent were washed four times in keratinocyte-SFM medium (KGM) containing bovine pituitary extract and epidermal growth factor supplements furnished by the manufacturer (Invitrogen) as well as penicillin, streptomycin, sodium pyruvate, and l-glutamine (GIBCO-BRL). For prevention of the growth of bacterial and fungal contaminants, the KGM was further supplemented with Normocin and Fungin at the concentrations recommended by the manufacturer (Invivogen, Inc.). The fragments were floated in a 1:1 solution of KGM and 0.25% trypsin (GIBCO-BRL) in a small culture dish for 10 to 14 h at 4°C. The epithelial and fibroblast layers were then separated, and the epithelial layers were placed in a 15-ml conical tube with 5 ml of phosphate-buffered saline (PBS) containing 2% FBS (Sigma) and shaken vigorously for several minutes. The remains of the epithelial fragments were removed from the tube, and the shaking was repeated with fresh PBS-2% FBS in another tube. The remains of the tissue were removed, and the two cell suspensions were pooled and pelleted by centrifugation at 400 × g for 5 min at 4°C. The cell pellet was washed once in KGM, resuspended in 2 ml of fresh KGM, plated in a 60-mm-diameter culture dish, and incubated at 36°C in 5% CO2. One volume of fresh, warm medium was added every 48 h, and the medium was changed completely after 5 to 6 days of culture. The cells began to adhere to the dish after 4 to 5 days in culture, and they became 50% confluent after an additional 10 to 15 days. P-EPI monolayers were split 1:3 when they were 40 to 60% confluent. All cells stained positive for epidermal growth factor receptor, indicating their epithelial origin.
Transduction.
P-EPI cells from passages 1 and 2 were immortalized by transduction with a retrovirus vector containing the human telomerase gene (h-tert) (6). P-EPI cells (5 × 105) were seeded onto a 60-mm-diameter culture dish and allowed to adhere overnight. The KGM was removed, and 2 ml of fresh, cell-free h-tert-containing retrovirus supernatant from A293T cells at 48 h posttransfection was added to the P-EPI monolayer. After 3 h of incubation at 37°C, 2 ml of fresh KGM was added to the culture dish. After overnight incubation, the medium was changed, with fresh KGM added to the monolayers. The medium was changed every 24 to 48 h, and these T-EPI cells were split 1:3 every 4 to 7 days.
Virus isolation.
TW samples in 10 ml of sterile PBS were obtained with informed consent from nine different healthy, HIV-negative, KS-negative individuals for use in infections. Wild-type KSHV was isolated from these samples as previously described (42). Nine different donors were employed for at least three independent infection experiments with each cell type. Briefly, after incubation with 1 mM dithiothreitol at ambient temperature, the oral cells were removed from the samples by centrifugation. The cell pellets were frozen for DNA isolation and/or spotted onto slides for confocal immunofluorescence assays (IFAs), described below. The TW fluid supernatant was filtered and underwent high-speed ultracentrifugation at 4°C in an SWTi 41 rotor (Beckman Instruments, Inc., Palo Alto, Calif.). The supernatant was discarded, and the virus pellets were resuspended in 50 to 100 μl of PBS. Similarly, the replication of latently infected KSHV in BCBL1 cells was induced by a 2-h incubation with 25 ng of tetradecanoyl phorbol acetate (TPA)/ml, after which the medium was replaced with fresh RPMI. Cells were cultured for an additional 48 to 72 h, and KSHV virus was isolated by high-speed centrifugation of the filtered culture supernatant as described above. The supernatant from uninfected BJAB cells served as a negative control.
Conditions of infection.
Cell monolayers were trypsinized, washed, resuspended in fresh culture medium, mixed with the resuspended WT virus, BCBL virus, or BJAB culture medium (mock/endogenous infection), and plated in eight-well chamber slides (Falcon) or 35-mm-diameter well culture plates at a concentration of 2.5 × 105 cells/ml. At various hours postinfection (hpi), the monolayers were further processed for IFA or DNA isolation, as described below. The replication of KSHV in T-EPI and A293T cells was blocked by the addition of 2.5 mg of ganciclovir (Roche Laboratories, Inc.)/ml or 10 mg of acyclovir (Sigma)/ml to the culture medium at the time of infection. The infection of T-EPI and A293T cells was blocked by preincubation of the resuspended virus pellet with 60 μg of neutralizing anti-gB antibody or the UK-218 control antibody (a gift from B. Chandran, University of Kansas, Kansas City)/ml as described previously (1).
IFAs.
The detection of KSHV antigens in infected cell monolayers was performed as previously described (44). Briefly, monolayers were fixed in 1:1 methanol-acetone, washed in PBS, and blocked overnight at 4°C with 20% normal goat serum (NGS) in PBS. Cells were incubated overnight at 4°C with the following primary antibodies diluted in 20% NGS: a mouse monoclonal antibody against the lytic infection-associated viral glycoprotein K8.1 (11), purified rabbit polyclonal antibodies against the viral glycoprotein gB (a gift from B. Chandran, University of Kansas) (1), rabbit polyclonal antibodies against KSHV latency-associated nuclear antigen (LANA) (a gift from D. Ganem, University of California, San Francisco), or a murine isotype control antibody or rat monoclonal antibody against KSHV LANA (27) (Advanced Biotechnologies, Inc., Columbia, Md.). A murine immunoglobulin G1 isotype control antibody (CalTag, Inc.) was used to optimize KSHV K8.1 primary antibody concentrations. Lastly, cells were washed, stained with goat anti-mouse antibody (K8.1), goat anti-rat secondary antibody (LANA) conjugated to AlexaFluor 488, or goat anti-rabbit secondary antibody (gB) conjugated to AlexaFluor 547 and ToPro-3 DNA stain (Molecular Probes, Inc.) for 1 h at ambient temperature, and analyzed by confocal microscopy on an Olympus FV500 microscope. Oropharyngeal cells isolated from TW samples were smeared onto slides and stained as described above, except that they were blocked in 20% NGS for 2 to 5 days at 4°C prior to staining to minimize background signals from oral flora.
Imaging by SEM.
P-EPI-3 passage 3 monolayers were infected with BCBL1-derived or WT virus as described above and seeded onto eight-well chambered borosilicate coverslips (Nalge Nunc International). At 48 hpi, the monolayers were washed with PBS, fixed in PBS containing 1% glutaraldehyde, and shipped overnight on ice to the University of Iowa Central Microscopy Research Facility. High-resolution scanning electron microscopy (SEM) was performed by methods specifically optimized for the visualization of herpesvirus particles on the surfaces of infected cells (24, 33, 39).
DNA isolation and PCR amplification.
Infected cell monolayers were removed from the plastic culture surface by incubation with trypsin-EDTA (Gibco) for 5 min, which ensured that viral particles associated with the outer cell surfaces were removed. DNAs were isolated from TW cells and from infected cell monolayers by using a DNEasy kit protocol recommended by the manufacturer (Qiagen, Inc., Valencia, Calif.). Cellular DNAs were employed for nested PCR amplifications to detect KSHV ORF 26 DNA. Briefly, 300 ng of cellular DNA was used as a template for the first-round PCR with an outer primer pair in a 50-μl reaction volume, and 5 μl of the first-round reaction was used as template DNA for the second round with an inner primer pair. DNAs from uninfected BJAB and latently infected BCBL1 cells were used as negative and positive controls, respectively. Three hundred nanograms of cellular DNA was also employed for real-time PCR detection of KSHV ORF 73, using primer and probe sequences (Qiagen Operon Technologies, Inc.) that were previously described by Dittmer et al. (17). Real-time PCR detection of lytic K8.1 from 150 ng of cDNA was also employed, using primer sequences (Lineberger Cancer Center Nucleic Acid Core Facility, University of North Carolina) that were previously described by Fakhari and Dittmer (19). Real-Time PCRs were performed by using an ABI Prism 7000 sequence detection system, with Taqman Universal PCR master mix and No AmpEraseUNG (Applied Biosystems, Foster City, Calif.). DNA from latently infected BCBL1 cells served as a positive control. DNA from uninfected BJAB cells and reactions run in the absence of added DNA served as negative controls. The cellular genes β-actin and glyceraldehyde-6-phosphate dehydrogenase were amplified to ensure the quality of the cDNAs and to serve as loading controls (data not shown). Analysis of the data was performed with the associated Prism 7000 software (Applied Biosystems), and relative KSHV DNA copy numbers were calculated by employing a standard curve of induced BCBL1 DNA serially diluted 10-fold in DG75 cell DNA. The undiluted copy number was arbitrarily set at 107 copies/300 ng of DNA based on quantitative results with other primer-probe sets. The PCR products generated by real-time PCR from cDNAs were electrophoresed in a 2% agarose gel and subsequently visualized by Southern blot hybridization with a 32P-labeled probe internal to the primer pair, as previously described (19).
RNA isolation and reverse transcription-PCR amplification.
RNAs from infected and uninfected cells were isolated with an RNeasy kit as directed by the manufacturer (Qiagen, Inc.). Contaminating DNAs were removed by use of a DNAfree kit (Ambion). cDNA was generated by using 0.1 μM RNA, a 0.5 mM concentration of each deoxynucleoside triphosphate, and 25 nM random-6 oligonucleotides (Qiagen, Inc.) in a 20-μl reaction volume. The reaction was heated to 65°C for 5 min and cooled to 4°C for 2 min. One microliter of Super Script II (Gibco), 4 μl of 5× SSII buffer, and 10 mM dithiothreitol were then added to the reaction, which was heated for 1 h at 42°C and 70°C for 15 min and then quantified. Duplicate reactions were performed without the addition of enzyme to generate reverse transcription-negative controls. The cDNA was subsequently employed for PCR amplification of lytic and latent KSHV gene sequences as described above.
RESULTS
Endogenous WT KSHV isolated from primary oral epithelial cells.
For characterization of the state of the WT virus infections in the donor oral cavities, oropharyngeal cells isolated from TW samples employed as sources of WT KSHV were analyzed for the expression of latent and lytic viral proteins (Fig. 1A, panel 2). Bacterial DNA was often detected on the oropharyngeal cells isolated from TW samples (Fig. 1A, panel 2, lower right). In addition, relative amounts of viral DNA isolated from oropharyngeal cells of several donors were compared to that detected in BCBL1 cells (Fig. 1C). Both LANA and the K8.1 viral glycoprotein associated with lytic infections were detected in 3 to 15% of the cells isolated from different TW donors, and the numbers of antigen-containing cells remained similar in consecutive samples taken at intervals of 10 to 30 min and 6 days to several weeks from the same donor (data not shown). The KSHV ORF 26 gene was amplified from DNA isolated from the oropharyngeal TW cells (Fig. 1B, lanes 1 to 7), with one exception (lane 7). KSHV ORF 26 was also detected in consecutive TW samples taken from the same donor at 30-min intervals on two separate occasions (Fig. 1B, lanes 1 and 2 and lanes 3 and 4), demonstrating that the presence of WT KSHV in cells of the oral cavity was not unusual for this individual. Donor 5 was seropositive, donor 6 is known to be seronegative, and the status of the remaining donors is unknown. None of the donors were men who have sex with men. In addition, real-time PCR amplification of the ORF 73 gene from these DNA samples was detected above the threshold level at cycle numbers 31 to 39 and with relative copy numbers that ranged from 3 to 1,500 per 300 ng of DNA, while the BJAB negative control remained below the threshold (Fig. 1C). Interestingly, the signal from the known seropositive donor sample crossed the threshold after 31 cycles, which was several logs higher than the other donors (Fig. 1C).
FIG. 1.

Detection of endogenous KSHV present in oropharyngeal cells of WT KSHV donors and early-passage primary epithelial cell cultures. (A) Immunofluorescent confocal microscopic detection of KSHV antigens. Monolayers were fixed and incubated with isotype antibody or an antibody against KSHV LANA or glycoprotein K8.1. Secondary antibodies conjugated to AlexaFluor 488 were used to detect the primary antibody bound to the KSHV protein, and ToPro-3 dye was used to detect nuclear DNA. Panel 1, antigen-positive controls (TPA-induced BCBL1 cells). Upper photos show the Alexa 488 fluorescence, and lower photos show the nuclei stained with ToPro-3. Panel 2, donor oropharyngeal cells. Upper photos show the Alexa 488 fluorescence, and lower photos show the nuclei stained with ToPro-3. Panel 3, P-EPI-2 passage 4. Upper photos show the Alexa 488 fluorescence, and lower photos show the nuclei stained with ToPro-3 merged with the Alexa 488 fluorescence and the bright-field images. Panel 4, T-EPI 1 passage 4. Upper photos show the Alexa 488 fluorescence, and lower photos show the nuclei stained with ToPro-3 merged with the Alexa 488 fluorescence and the bright-field images. (B) Detection of KSHV ORF 26 by conventional PCR. DNAs from uninfected BJAB and latently infected BCBL1 cells were used as negative and positive controls, respectively. Lanes 1 to 7, oropharyngeal DNA isolated from donor TW samples; lane 8, P-EPI-2 passage 7 cell DNA; lane 9, BCBL1 cell DNA; lane 10, BJAB cell DNA; lane 11, no template DNA; lane 12, T-EPI 1 passage 6 DNA (this sample was run on another gel, but the PCRs were performed in the same experiment as the rest of the lanes). (C) Real-time PCR amplification plot of KSHV ORF 73 in donor oropharyngeal cell DNA. DNAs isolated from TW donor oropharyngeal cells from one KSHV-seropositive (sero+) donor and six other donors (TW donors) were used as templates for real-time PCR detection of KSHV ORF 73 DNA. DNA isolated from uninfected BJAB and latently infected BCBL1 cells served as negative and positive controls, respectively. KSHV DNA copy numbers relative to a standard curve of 10-fold serially diluted BCBL1 DNA are shown in parentheses. n. det., not detectable.
The detection of WT KSHV antigens in all donor samples suggested that the P-EPI cells and the T-EPI cells might also have been infected with endogenous WT KSHV. An analysis of P-EPI-2 and T-EPI-1 cultures at different passages confirmed the presence of endogenous WT KSHV in both cultures, which were generated from tissues of two different HIV-negative individuals (Fig. 1A, panels 2 and 3, and Fig. 1B, lanes 8 and 12). The P-EPI-2 cells remained positive for KSHV LANA and K8.1 antigens through passage 7 (Fig. 1B), at which time the culture senesced, although the percentage of positive cells decreased with each passage (data not shown). By passage 10, the immortalized T-EPI-1 cells were negative for KSHV DNA and KSHV antigen expression by real-time PCR amplification and IFA, respectively (see Fig. 3C; also data not shown), indicating that the endogenous virus did not persist for long in primary oral epithelial cultures. Nevertheless, our results demonstrated the presence of endogenous WT KSHV in primary epithelial cell cultures and the ability of this virus to express both latent and lytic proteins in cells isolated from the oral cavities of immunocompetent, HIV-negative individuals.
FIG. 3.

Detection of WT KSHV infection of primary oral epithelial cells. P-EPI-2 passage 5 (A) and T-EPI 1 passage 10 (B) monolayers were fixed and incubated with isotype antibody, or antibodies against KSHV LANA or K8.1 glycoprotein at 24, 48, 72, and 98 hpi with virus isolated from throat wash samples from 4 different donors; one donor used for infections shown in Fig. 3A, and virus isolated from three other donors was used for panels B to E. Virus isolated from TPA-induced-BCBL1 and uninfected BJAB supernatant (Endog.) were used as positive and negative controls, respectively. (A) Expression kinetics of latent (LANA) and lytic (K8.1) KSHV antigens in P-EPI cells. Secondary antibodies conjugated to AlexaFluor 488 were used to detect the primary antibody bound to KSHV antigens, and ToPro-3 dye was used to detect the cell nuclei. Upper photos in each row show the AlexaFluor 488 channel, and lower photos show the AlexaFluor 488 and ToPro-3 and bright field. Row 1, 48 hpi K8.1 expression (left panels, isotype control); row 2, 48 hpi LANA expression; row 3, 72 hpi K8.1 expression; row 4, 72 hpi LANA expression. (B) Expression kinetics of latent (LANA) and lytic (K8.1) KSHV antigens in T-EPI cells. Secondary antibodies conjugated to AlexaFluor 488 were used to detect the primary antibody bound to KSHV antigens, and ToPro-3 dye was used to detect the cell nuclei. Upper photos in each row show the AlexaFluor 488 channel, and lower photos show the AlexaFluor 488, ToPro-3 and bright field, or AlexaFluor 547 overlay. Row 1, 24 hpi K8.1 expression; row 2, 24 hpi LANA expression; row 3, 96 hpi K8.1 expression; row 4, 96 hpi LANA expression. Secondary antibodies conjugated to AlexaFluor 488 were used to detect the primary antibody bound to KSHV antigens, and ToPro-3 dye was used to detect the cell nuclei. Upper photos in each row show the AlexaFluor 488 channel, and lower photos show the AlexaFluor 488, ToPro-3 and bright field, or AlexaFluor 547 overlay. (C) More lytic than latent antigen expression in T-EPI cells. Secondary antibodies conjugated to goat anti-rabbit AlexaFluor 547 (red) or goat anti-rat or mouse AlexaFluor 488 (green) were used to detect the primary antibody bound to KSHV antigens, and ToPro-3 dye (blue) was used to detect the cell nuclei. Upper panels show the AlexaFluor 488 and AlexaFluor 547 channels; and lower panels show the AlexaFluor 488, AlexaFluor 547, and ToPro-3 overlay. Left photos, negative and positive controls in BCBL1 cells. Center photos, faint LANA expression (red),cytoplasmic K8.1 (red) expression, and DNA (blue) in T-EPI cells 38 hpi with WT KSHV. Right photos, cytoplasmic gB (red) and perinuclear LANA expression (green). (D) Detection of K8.1 cDNA in P-EPI cells. cDNA generated from RNA isolated from 105 PEPI cells at 96 hpi was used as template for real-time PCR amplification of a 161-bp region of the K8.1 gene containing a 94-bp splice (19). The PCR was electrophoresed on a 2% agarose gel. A 32P-labeled oligonucleotide probe internal to the primer pair was hybridized to the amplification products after Southern blot transfer. Induced BCBL1 cells served as positive controls. Duplicate cDNA samples generated with (RT+ lanes; 161-bp product from spliced template) and without added reverse transcriptase (RT− lanes; 255-bp product from unspliced template) served as controls to detect genomic DNA. (E) Real-time PCR detection of KSHV DNA in passage 7 T-EPI 1-infected cells. DNA was isolated from cells infected with BCBL virus or WT TW virus (WT) or mock infected. Cells harvested at 2 days postinfection (top panel) or after 1 passage at 8 days postinfection (bottom panel) were used as templates for real-time PCR detection of KSHV ORF 73 DNA. DNA from TPA-activated BCBL1 cells served as a positive control (upper panel). Endog, DNA from an untreated low passage T-EPI cell monolayer containing endogenous virus. KSHV DNA copy numbers relative to a standard curve of 10-fold serially diluted BCBL1 DNA are in parentheses. n. det., not detectable.
Differences in replication characteristics in epithelial cell lines.
The replication of KSHV isolated from latently infected PEL cell lines such as BCBL1 induced to replicate by activation with phorbol esters has been well characterized in many different cell types (3, 4, 37). For determination of the extent to which WT KSHV could infect transformed epithelial cell lines, a melanoma cell line (MeWo) and the SV40 T-antigen-transformed A293T cell line were infected with WT virus isolated from TW samples. A recent report demonstrating that KSHV efficiently utilizes the α3β1 integrin for entry (2) suggested that in vitro infection might be more efficient if the cells were in the process of adhering, rather than already adherent, when exposed to the virus. Therefore, the cells were trypsinized just prior to infection with WT KSHV and BCBL1-derived virus. Both WT and BCBL viruses were able to establish a latent infection in MeWo cells by 24 hpi, as demonstrated by IFA detection of the LANA protein in these cells (Fig. 2A, left panel). Lytic infection, defined by IFA detection of the K8.1 glycoprotein, appeared to be rare, as it was only detected twice in the BCBL virus-infected cells (Fig. 2A, right panel). Viral DNA was readily detectable by PCR amplification of the ORF 26 gene at 48 hpi (Fig. 2B). Latent infection was still detectable by IFA at 48 hpi, but not at 72 hpi (Table 1; data not shown). Real-time PCR analyses of cellular DNA at various time points postinfection suggested that the KSHV infection was transient in MeWo cells; the amount of viral DNA decreased steadily during the first 96 hpi (Fig. 2C). The BCBL1-derived virus was more prevalent at the early times postinfection (perhaps due to an occasional lytic event, as illustrated in Fig. 2A), and remained detectable longer by both real-time and conventional PCR (Fig. 2C; data not shown).
FIG.2.

Detection of KSHV in epithelial cell lines infected with WT KSHV. (A) Detection of WT KSHV antigens in MeWo cells. Monolayers were fixed and incubated with antibodies against KSHV LANA or K8.1 glycoproteins at 24 h postinfection. Secondary antibodies conjugated to Alexa 488 were used to detect the primary antibody bound to the KSHV protein, and ToPro-3 dye was used to detect the cell nuclei. Upper photos show the Alexa 488 channel, and lower photos show the Cy5 channel and the ToPro-3-stained nuclei. (B) PCR detection of KSHV ORF 26 DNA in MeWo cells. Three hundred nanograms of cellular DNA was used as template for the first round PCR, and 5 μl of the first round reaction was use as template DNA for the second round. Lane 1, mock-infected cells; lane 2, 48 hpi BCBL1 virus-infected cells; lane 3, 48 hpi WT virus-infected cells; lanes 4 and 5, DNA isolated from BCBL1 or BJAB cells that was used as positive and negative controls, respectively; lane 6, no DNA. (C) Real-time PCR detection of WT KSHV ORF 73 DNA in MeWo cells. Three hundred nanograms of DNA isolated from infected with either BCBL1 virus or WT TW virus (WT virus) or mock-infected cells and harvested at 16 and 96 h postinfection was used as template for real-time PCR detection of KSHV ORF 73 DNA. DNA from TPA-activated BCBL1 cells served as a positive control. KSHV DNA copy numbers relative to a standard curve of 10-fold serially diluted BCBL1 DNA are in parentheses. n. det., not detectable. (D) Detection of WT KSHV antigens in A293T cells at 24 to 96 hpi. Infected monolayers were fixed and incubated with antibody against KSHV K8.1 glycoprotein. Secondary antibodies conjugated to AlexaFluor 488 were used to detect the primary antibody bound to the KSHV protein, and ToPro-3 dye was used to detect the cell nuclei.
TABLE 1.
IFA detection of lytic and latent WT KSHV infection of epithelial cells
| Cell type | Virus | Detection of lytic virus/detection of latent virus at indicated hpia
|
||||
|---|---|---|---|---|---|---|
| 12 | 24 | 48 | 72 | 96 | ||
| MeWo | BCBL | −/+ | ±/++ | −/+ | −/− | −/− |
| WT | −/+ | −/++ | −/± | −/− | −/− | |
| A293T | BCBL | ND | +/± | +/± | ±/− | ±/− |
| WT | ND | +/± | ++/± | +++/− | ++++/− | |
| P-EPI | BCBL | ND | ++/++ | +/++ | +/+ | −/± |
| WT | ND | ++/++ | +/++ | +++/+ | ++++/+ | |
| T-EPI | BCBL | ND | ++/++ | +/++ | +/+ | −/± |
| WT | ND | ++/++ | +/++ | ++/+ | +++/+ | |
Lytic virus was measured by counting the K8.1-positive cells in the monolayer. −, not detectable; ±, <1%; +, 1 to 2%; ++, 3 to 5%; +++, 6 to 10%; ++++, >10%. Latent virus was measured by counting the LANA-positive cells in the monolayer. −, not detectable; ±, <1%; +, 1 to 5%; ++, 6 to 10%. ND, not done.
Surprisingly, the infection of A293T cells was primarily lytic in nature; LANA was not expressed at detectable levels with either the anti-rat monoclonal antibody or the anti-rabbit polyclonal antibody beyond 12 hpi (Table 1). Interestingly, the WT virus replicated more efficiently in these cells than the BCBL virus, as demonstrated by the consistent observation of increasing foci of K8.1 glycoprotein expression observed in WT-infected monolayers over time (Fig. 2D; Table 1). Indeed, the WT virus appeared to mediate cytopathic effects in these cells, specifically cell membrane fusion formation at 48 to 96 hpi, which created large K8.1 glycoprotein-positive foci containing multiple nuclei (Fig. 2D). Collectively, these results suggested that interesting phenotypic differences existed between WT KSHV isolated from the oral cavity and the PEL-associated BCBL virus. Interestingly, tonsillar B lymphocytes were also successfully infected with KSHV (data not shown).
Replication differences between TW KSHV and BCBL1-derived KSHV in primary oral epithelial cells.
To test the ability of WT KSHV and the PEL-associated BCBL1 virus to replicate in a primary oral epithelial cell model, we inoculated P-EPI and T-EPI cells with WT and BCBL1-derived viruses (Fig. 3). Infections of each cell type were repeated at least three times with WT KSHV virus isolated from TW samples of different donors, with similar results. In both P-EPI and T-EPI cell cultures, the WT KSHV isolates replicated more efficiently than the BCBL1-derived virus over a 96-h interval, and two late lytic antigens, gpK8.1 and gB, could be visualized in these cells, although more lytic than latent antigen was consistently observed (Fig. 3A to C). Furthermore, this WT virus could be induced to replicate with the addition of phorbol esters, such as TPA and sodium butyrate (data not shown). The results of these experiments are summarized in Table 1. Relative differences in replication rates between the viruses were also detected by real-time PCR amplification of lytic and latent genes from cDNA or DNA isolated from cells at different times postinfection (Fig. 3D and E). Cell monolayers were trypsinized and washed prior to DNA isolation to remove any remaining input virus from the cell membranes. At early time points postinfection, the relative amount of BCBL1-derived viral DNA present in infected cell lysates was larger than that of the WT, TW-derived virus (Fig. 3D, upper panel). However, by 8 days postinfection, relative levels of WT viral DNA in infected cell lysates were exponentially higher than levels of BCBL1-derived viral DNA (Fig. 3E, lower panel). LANA expression decreased more rapidly in BCBL virus-infected cells than in WT virus-infected cultures over the 96-h period (Table 1).
Visualization of WT KSHV particles on primary epithelial cell membranes.
Enveloped virions formed in cultured cells infected with the alpha- and betaherpesviruses have been visualized by high-resolution SEM technology (24, 33, 39). To determine whether KSHV virions were detected on the membranes of primary oral epithelial cells, we performed SEM on P-EPI-3 passage 3 monolayers that were endogenously infected or infected with either BCBL1-derived or TW-derived WT virus. Because of the reported low titers of KSHV in healthy people, we postulated that this herpesvirus would be more difficult to detect than other herpesviruses. Surprisingly, KSHV particles were visualized as easily as other herpesviruses by SEM (Fig. 4). The virion size was variable, with the majority of particles having a diameter between 100 and 150 nm, although a few larger particles were also seen (Fig. 4). Considerably more virions were present on the WT and BCBL1 virus-infected monolayers than on the endogenously infected monolayers (Fig. 5). The virion surfaces were pleiomorphic in character, a common finding among herpesviruses, particularly those considered to be more cell associated. These results documented the presence of KSHV virions on the surfaces of endogenously infected primary oral epithelial cells. More importantly, the markedly increased numbers of virions present on both BCBL1- and WT virus-infected P-EPI membranes confirmed the fact that viral replication and virion assembly occurred in these cells.
FIG. 4.
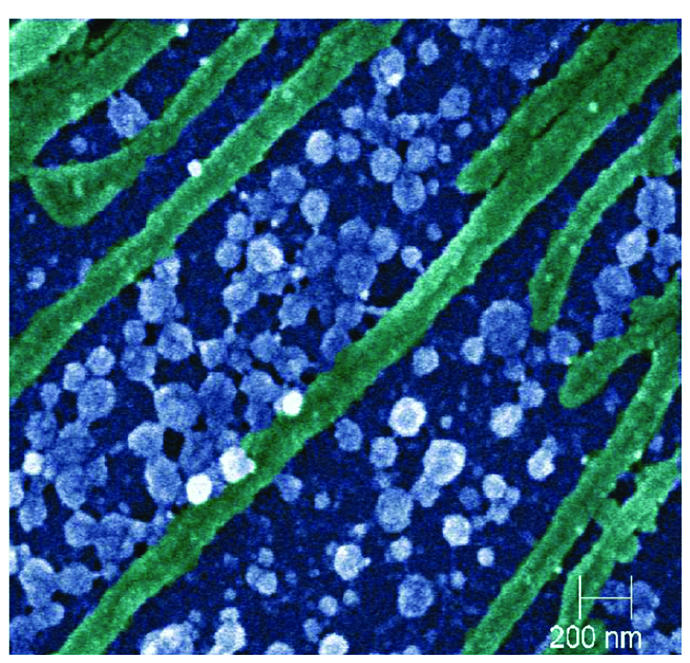
Representative scanning electron micrograph of KSHV virions (blue or white) at ×40,000 magnification on infected P-EPI cells (epithelial cell plasma membrane extensions shown in green). P-EPI cells infected with either WT or BCBL1 KSHV were cultured for 48 h prior to processing.
FIG. 5.

SEM detection of KSHV virions on P-EPI cell membranes. Endogenously infected P-EPI-3 passage 3 cells (E and F) were infected with WT KSHV (C and D) or control BCBL1 (A and B) virus and cultured for 48 h prior to processing. Magnification: ×15,000 for panels A, C, and E; ×5,000 for panels B, D, and F.
Inhibition of WT KSHV replication by antiviral compounds and inhibition of infection by neutralizing antibodies.
To further confirm WT KSHV replication in epithelial cells and to rule out detection of contaminating viruses bound to cell surfaces, we used the antiviral compounds acyclovir and ganciclovir to block virus replication. At the time of infection, the cells were cultured in the presence or absence of the antiviral compound. In the presence of 2.5 mg of ganciclovir/ml, K8.1 glycoprotein expression was abolished in P-EPI-2 cells by 24 hpi (Fig. 6A). As expected, expression of the LANA antigen was not affected by the presence of ganciclovir (data not shown). Furthermore, the addition of ganciclovir to the P-EPI-2 cells blocked the replication of the endogenous KSHV in these cells (Fig. 6A). Acyclovir has also been shown to decrease KSHV replication, although it does not completely inhibit the virus (26). In the presence of 10 mg of acyclovir/ml, WT KSHV K8.1 glycoprotein expression detected at 48 hpi in A293T cells was reduced by 50%, from >200 positive foci/well in the absence of the drug to 100 positive foci/well in the presence of the drug (data not shown). In addition, the K8.1-positive foci were smaller (compare foci in Fig. 2D to those in Fig. 6B). In the presence of acyclovir, the BCBL1-derived virus was reduced from three foci/well to one barely detectable focus of K8.1 expression (Fig. 6B).
FIG. 6.

Inhibition of WT KSHV replication or infection in epithelial cells. (A) Inhibition of lytic replication in primary epithelial cells by ganciclovir. P-EPI-2 passage 5 cells were infected with WT KSHV or control BCBL1 virus and cultured with or without 2.5 mg of ganciclovir/ml for 24 h prior to IFA detection of glycoprotein K8.1. Left panel, no ganciclovir; right panel, ganciclovir added. Upper photos show the Alexa 488 filter, and lower photos show the Alexa 488 and ToPro-3 and bright-field overlay. (B) Inhibition of WT KSHV lytic replication in A293T cells by acyclovir. A293T cells were infected in the presence of 10 mg of acyclovir/ml, and examined at 48 hpi. Upper photos show the Alexa 488 channel, and lower photos show the Alexa 488, ToPro-3 and bright-field overlay. (C) Inhibition of WT KSHV infectivity by neutralizing antibody against gB. Virus suspension was incubated with 60μg of anti-gB or negative control UK218 antibody/ml for 1 h prior to infection of T-EPI or A293T cells. At 96 hpi, cells were processed for IFA. Photos show the Alexa 488 and ToPro-3 overlay. (D) Ganciclovir-mediated inhibition of lytic gene expression in A293T cells. A293T cells were infected in the presence or absence of 2.5-mg/ml ganciclovir (± GAC) for 24 h (upper panel) or 72 h (lower panel). cDNA with or without reverse transcriptase (RT±) was generated from isolated RNA and used as template for real-time PCR amplification of a region of the K8.1 gene containing a 94-bp splice (19). The PCR was electrophoresed on a 2% agarose gel. A 32P-labeled oligonucleotide probe internal to the primer pair was hybridized to the amplification products after Southern blot transfer. Induced BCBL1 cells served as positive controls. Duplicate cDNA samples generated with (RT+ lanes; 161-bp product from spliced template) and without added reverse transcriptase (RT− lanes; 255-bp product from unspliced template) served as controls to detect genomic DNA.
Akula et al. previously demonstrated the efficacy of neutralizing antibodies against gB for blocking in vitro KSHV infections (1). To confirm that the epithelial cells were being infected by the resuspended WT and BCBL virus pellets, we incubated the virus pellets with either neutralizing antibodies to KSHV gB or a control antibody prior to infection. Neutralizing antibodies dramatically decreased the ability of WT and BCBL viruses to infect T-EPI cells (Fig. 6C). In addition, the infection of A293T cells in the presence of ganciclovir inhibited WT and BCBL lytic replication, as demonstrated by the absence of spliced K8.1 cDNA (Fig. 6D). The results of these inhibition experiments further substantiated that the WT virus entered and replicated within the in vitro oral epithelial cell model system.
DISCUSSION
We have shown that WT KSHV is present in the oral cavities of healthy individuals and that this virus is capable of permissive infections of oral epithelial cells. The present data demonstrate for the first time that normal human oral epithelial cells can be productively infected by KSHV in vivo and are capable of infection transfer in vitro. The infection of P-EPI and T-EPI cells with WT KSHV isolated from healthy individuals resulted in productive viral expression. Additional experiments detected the infection of naïve epithelial cells with endogenous WT KSHV virions, suggesting that oral epithelial cells may constitute an infectious reservoir for KSHV, as has previously been documented for Epstein-Barr virus and murine homologues (20, 35, 40, 41). In addition, we detected KHSV virions in buccal cells from healthy TW donors by transmission electron microscopy and detected both lytic and latent viral proteins by immunogold labeling (data not shown). While the early B cell is the principally infected cell type in the peripheral blood of KSHV-seropositive patients with KS, our data suggested that the expression of the integrin receptor might render oral epithelium a cellular portal for KSHV infection via oral mucosal exposure (2, 38).
The data are consistent with previous studies showing infection of epithelial cells derived from foreskin keratinocytes (3, 9). Oral epithelial cell types ranging from primary to transformed were susceptible substrates. The evidence of infection included the detection of viral gene products, increases in viral DNA in infected cell lysates over time, and drug inhibition studies, all of which were indicative of productive infections in vitro.
Previous studies have shown that it is difficult to sustain rounds of serial KSHV passaging. Increasing amounts of lytic protein expression and the increased quantities of viral DNA over time subsequent to infection of oral epithelial cells and A293T cells with WT virus demonstrated productive infections in vitro rather than lingering input virus (Fig. 2 and 3). In addition, the virus from endogenously infected lower passage P-EPI cells could be isolated from the culture medium and used to infect higher passage T-EPI cells from which the virus had been lost (data not shown).
Similar to the case with other gammaherpesviruses, the expression of viral gene products in oral epithelial cells suggests that the oral mucosa may represent the predominant reservoir of infectious virus for KSHV transmission. Mucosal epithelial sites are frequently the site of lytic gammaherpesviral infections, as determined by infection of murine lung epithelial cells and gastric epithelial cells with murine herpesvirus 68 (35, 41). The AIDS-associated oral hairy leukoplakia lesion, the only pathological manifestation of permissive Epstein-Barr virus infection, exhibits abundant viral replication within oral epithelial cells (45). The oral cavity is also a reservoir for Epstein-Barr virus infection in healthy individuals (40, 43). Other herpesviruses, such as HHV-6 and HHV-7, are also present in the oral cavity. However, the presence of these viruses was not examined in this study. The present data documented low but detectable levels of KSHV in primary oral epithelial cell cultures (Fig. 1 and 4). However, the endogenous virus present in these primary cells could be induced to replicate. We have shown that P-EPI and T-EPI cells were readily infectable, with apparent replication differences depending on whether the source of the virus was the WT from immunocompetent individuals or laboratory-adapted BCBL1-derived virus (Fig. 2 and 3). Oral epithelial cells more readily sustained infection by WT isolates than by BCBL1-derived virus, revealing potential differences in pathogenicity.
The scanning electron micrographs confirmed that numerous viral particles are present on the surfaces of P-EPI cells (Fig. 4 and 5). When viewed at a lower magnification, HSV-1 emerges uniformly over the infected cell surface (33, 39), whereas varicella-zoster virus (VZV) often emerges in a pattern called viral highways (24, 33). KSHV emerged in clusters, with larger numbers of particles at the leading edge of an infectious focus. Although differences in the egress patterns may appear subtle after a cursory examination, the inspection of hundreds of micrographs has defined distinguishing features for each of the human herpesviruses. When the surfaces of the different herpesvirus particles were compared at higher magnifications (>40,000×), HSV-1 was the most prototypic whereas VZV was the most pleiomorphic, with only a small fraction of surface virions having a prototypic appearance. HSV-2 was more pleiomorphic than HSV-1 but less than VZV. In turn, KSHV fell between HSV-2 and VZV. Based on the above SEM observations, increased pleiomophism correlates with the property of increased cell-associated nature of growth in cell culture. To the extent that P-EPIs can sustain low levels of productive infection, these cells may provide a foothold for KSHV in the body and subsequently transfer infection to more productive cell types, such as B lymphocytes trafficking through the oral mucosa, in which latency may be established, and endothelial cells during KS development. Consistent with this hypothesis, we recently observed the in vitro transfer of infection from KSHV-infected T-EPI to tonsillar lymphocytes (Fig. 7). Such results are analogous to infection patterns for the related gammaherpesvirus Epstein-Barr virus and lead us to propose a model in which virus cycles from epithelial cells to the lymphoid compartment (Fig. 7).
FIG. 7.

Model of KSHV infection and reactivation in the oral cavity of immunocompetent individuals. Lytic replication in the oral epithelium may serve as a reservoir of infectious virus. (A) K8.1-positive oral epithelial cell. Virus from this reservoir may then infect trafficking lymphocytes and lymphoid tissue in the oropharynx, replicate and then establish latency. (B) K8.1-positive tonsil cell 24 hpi with TW-derived virus. This may in turn provide a latent virus reservoir. Purple circles in nuclei represent latently infected cells.
While evidence of in vitro lytic KSHV infection in the literature is sparse, it has been described in both endothelial cells and in the transformed epithelial cell line HEK293 (13, 16, 21, 28). Recently, Bechtel and colleagues (3) demonstrated that multiple cell lines latently infected with BCBL-derived KSHV were readily induced to replicate after transduction with an adenovirus expressing the immediate-early transactivator Rta, suggesting that lytic KSHV replication is cell type dependent (4). Abundant KSHV lytic replication was detected at early time points in P-EPI and T-EPI cells and occurred in the absence of a detectable latent phase in A293T cells, an SV40-transformed clone of HEK 293 (Fig. 2). Megakaryon formation due to infected cell membrane fusion is a phenomenon commonly associated with lytic herpesvirus infections (20). KSHV-mediated membrane fusion has been observed in 293 and B cells transiently transfected with gB, gH, and gL glycoproteins (36). Megakaryon formation in the absence of detectable latency was observed with the WT KSHV infection of the A293T cell line, but was not detected in cells infected with BCBL1-derived virus (Fig. 2D), again suggesting a pathogenic difference between isolates. Similarly, cell-cell fusion was also observed at later time points in P-EPI and T-EPI monolayers infected with WT but not BCBL1-derived KSHV (Fig. 3A, K8.1 - 72 hpi), although the areas of fusion were not as extensive as those observed in the transformed cell lines. These results were consistent with those of Pertel (36) and suggested that WT KSHV is able to mediate cell fusion in vitro. Taken together with the observed replication kinetics, these data point to differences between WT KSHV and laboratory-adapted, PEL-derived viruses that have been employed in the majority of previous studies.
In conclusion, we have shown that KSHV is harbored in the oral cavity of healthy individuals and that this virus is capable of permissive infection in oral epithelial cells. To our knowledge, these studies provide the first direct comparison of in vitro infection of WT KSHV from the oropharynx of healthy individuals to PEL-derived, lab-adapted virus. In the absence of severe immunosuppression, the infection may persist at a low level for the lifetime of the host, being controlled by the intact immune response. Further studies will help us to identify factors that influence the vulnerability of oral epithelial cells to KSHV infection and to more fully define their role in conveying infection to lymphocytes.
Acknowledgments
We thank Robert Holzknecht and Deborah Granger for technical assistance, Dirk Dittmer for helpful suggestions, Blossom Damania for constructs, Bala Chandran for the gift of the gB neutralizing rabbit antibody, Don Ganem for the gift of rabbit polyclonal antibody against LANA, and Lishan Su for the A293T cells.
This investigation was supported in part by USPHS grants K23 DE 00460-01 and R03 DE14444-01 from the National Institute of Dental and Craniofacial Research, National Institutes of Health, and by UNC grant CFAR 9P30AI50410. C.G. is supported by USPHS grants AI 22795 and AI 56223.
REFERENCES
- 1.Akula, S. M., N. P. Pramod, F. Z. Wang, and B. Chandran. 2001. Human herpesvirus 8 envelope-associated glycoprotein B interacts with heparan sulfate-like moieties. Virology 284:235-249. [DOI] [PubMed] [Google Scholar]
- 2.Akula, S. M., N. P. Pramod, F. Z. Wang, and B. Chandran. 2002. Integrin alpha3beta1 (CD 49c/29) is a cellular receptor for Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 108:407-419. [DOI] [PubMed] [Google Scholar]
- 3.Bechtel, J. T., Y. Liang, J. Hvidding, and D. Ganem. 2003. Host range of Kaposi's sarcoma-associated herpesvirus in cultured cells. J. Virol. 77:6474-6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blackbourn, D. J., E. Lennette, B. Klencke, A. Moses, B. Chandran, M. Weinstein, R. G. Glogau, M. H. Witte, D. L. Way, T. Kutzkey, B. Herndier, and J. A. Levy. 2000. The restricted cellular host range of human herpesvirus 8. AIDS 14:1123-1133. [DOI] [PubMed] [Google Scholar]
- 5.Blackbourn, D. J., E. T. Lennette, J. Ambroziak, D. V. Mourich, and J. A. Levy. 1998. Human herpesvirus 8 detection in nasal secretions and saliva. J. Infect. Dis. 177:213-216. [DOI] [PubMed] [Google Scholar]
- 6.Bodnar, A. G., M. Ouellette, M. Frolkis, S. E. Holt, C. P. Chiu, G. B. Morin, C. B. Harley, J. W. Shay, S. Lichtsteiner, and W. E. Wright. 1998. Extension of life-span by introduction of telomerase into normal human cells. Science 279:349-352. [DOI] [PubMed] [Google Scholar]
- 7.Boshoff, C., T. F. Schulz, M. M. Kennedy, A. K. Graham, C. Fisher, A. Thomas, J. O. McGee, R. A. Weiss, and J. J. O'Leary. 1995. Kaposi's sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat. Med. 1:1274-1278. [DOI] [PubMed] [Google Scholar]
- 8.Casper, C., A. Wald, J. Pauk, S. R. Tabet, L. Corey, and C. L. Celum. 2002. Correlates of prevalent and incident Kaposi's sarcoma-associated herpesvirus infection in men who have sex with men. J. Infect. Dis. 185:990-993. [DOI] [PubMed] [Google Scholar]
- 9.Cerimele, F., F. Curreli, S. Ely, A. E. Friedman-Kien, E. Cesarman, and O. Flore. 2001. Kaposi's sarcoma-associated herpesvirus can productively infect primary human keratinocytes and alter their growth properties. J. Virol. 75:2435-2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cesarman, E., Y. Chang, P. S. Moore, J. W. Said, and D. M. Knowles. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186-1191. [DOI] [PubMed] [Google Scholar]
- 11.Chandran, B., C. Bloomer, S. R. Chan, L. Zhu, E. Goldstein, and R. Horvat. 1998. Human herpesvirus-8 ORF K8.1 gene encodes immunogenic glycoproteins generated by spliced transcripts. Virology 249:140-149. [DOI] [PubMed] [Google Scholar]
- 12.Chang, Y., E. Cesarman, M. S. Pessin, F. Lee, J. Culpepper, D. M. Knowles, and P. S. Moore. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865-1869. [DOI] [PubMed] [Google Scholar]
- 13.Ciufo, D. M., J. S. Cannon, L. J. Poole, F. Y. Wu, P. Murray, R. F. Ambinder, and G. S. Hayward. 2001. Spindle cell conversion by Kaposi's sarcoma-associated herpesvirus: formation of colonies and plaques with mixed lytic and latent gene expression in infected primary dermal microvascular endothelial cell cultures. J. Virol. 75:5614-5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cook, R. D., T. A. Hodgson, E. M. Molyneux, E. Borgstein, S. R. Porter, and C. G. Teo. 2002. Tracking familial transmission of Kaposi's sarcoma-associated herpesvirus using restriction fragment length polymorphism analysis of latent nuclear antigen. J. Virol. Methods 105:297-303. [DOI] [PubMed] [Google Scholar]
- 15.Cook, R. D., T. A. Hodgson, A. C. Waugh, E. M. Molyneux, E. Borgstein, A. Sherry, C. G. Teo, and S. R. Porter. 2002. Mixed patterns of transmission of human herpesvirus-8 (Kaposi's sarcoma-associated herpesvirus) in Malawian families. J. Gen. Virol. 83:1613-1619. [DOI] [PubMed] [Google Scholar]
- 16.Delecluse, H. J., M. Kost, R. Feederle, L. Wilson, and W. Hammerschmidt. 2001. Spontaneous activation of the lytic cycle in cells infected with a recombinant Kaposi's sarcoma-associated virus. J. Virol. 75:2921-2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dittmer, D., C. Stoddart, R. Renne, V. Linquist-Stepps, M. E. Moreno, C. Bare, J. M. McCune, and D. Ganem. 1999. Experimental transmission of Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) to SCID-hu Thy/Liv mice. J. Exp. Med. 190:1857-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duus, K. M., E. D. Miller, J. A. Smith, G. I. Kovalev, and L. Su. 2001. Separation of human immunodeficiency virus type 1 replication from Nef-mediated pathogenesis in the human thymus. J. Virol. 75:3916-3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fakhari, F. D., and D. P. Dittmer. 2002. Charting latency transcripts in Kaposi's sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J. Virol. 76:6213-6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fields, B. N., D. M. Knipe, and P. M. Howley (ed.). 1996. Fields virology, 3rd ed. Lippincott-Raven Publishers, Philadelphia, Pa.
- 21.Foreman, K. E., J. Friborg, Jr., W. P. Kong, C. Woffendin, P. J. Polverini, B. J. Nickoloff, and G. J. Nabel. 1997. Propagation of a human herpesvirus from AIDS-associated Kaposi's sarcoma. N. Engl. J. Med. 336:163-171. [DOI] [PubMed] [Google Scholar]
- 22.Graham, F. L., J. Smiley, W. C. Russell, and R. Nairn. 1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36:59-74. [DOI] [PubMed] [Google Scholar]
- 23.Grose, C., and P. A. Brunel. 1978. Varicella-zoster virus: isolation and propagation in human melanoma cells at 36 and 32 degrees C. Infect. Immun. 19:199-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harson, R., and C. Grose. 1995. Egress of varicella-zoster virus from the melanoma cell: a tropism for the melanocyte. J. Virol. 69:4994-5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howard, M. R., D. Whitby, G. Bahadur, F. Suggett, C. Boshoff, M. Tenant-Flowers, T. F. Schulz, S. Kirk, S. Matthews, I. V. Weller, R. S. Tedder, and R. A. Weiss. 1997. Detection of human herpesvirus 8 DNA in semen from HIV-infected individuals but not healthy semen donors. AIDS 11:F15-F19. [DOI] [PubMed] [Google Scholar]
- 26.Kedes, D. H., and D. Ganem. 1997. Sensitivity of Kaposi's sarcoma-associated herpesvirus replication to antiviral drugs. Implications for potential therapy. J. Clin. Investig. 99:2082-2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kellam, P., D. Bourboulia, N. Dupin, C. Shotton, C. Fisher, S. Talbot, C. Boshoff, and R. A. Weiss. 1999. Characterization of monoclonal antibodies raised against the latent nuclear antigen of human herpesvirus 8. J. Virol. 73:5149-5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lagunoff, M., J. Bechtel, E. Venetsanakos, A. M. Roy, N. Abbey, B. Herndier, M. McMahon, and D. Ganem. 2002. De novo infection and serial transmission of Kaposi's sarcoma-associated herpesvirus in cultured endothelial cells. J. Virol. 76:2440-2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lampinen, T. M., S. Kulasingam, J. Min, M. Borok, L. Gwanzura, J. Lamb, K. Mahomed, G. B. Woelk, K. B. Strand, M. L. Bosch, D. C. Edelman, N. T. Constantine, D. Katzenstein, and M. A. Williams. 2000. Detection of Kaposi's sarcoma-associated herpesvirus in oral and genital secretions of Zimbabwean women. J. Infect. Dis. 181:1785-1790. [DOI] [PubMed] [Google Scholar]
- 30.Lin, J. C., S. C. Lin, E. C. Mar, P. E. Pellett, F. R. Stamey, J. A. Stewart, and T. J. Spira. 1995. Is Kaposi's-sarcoma-associated herpesvirus detectable in semen of HIV-infected homosexual men? Lancet 346:1601-1602. [DOI] [PubMed] [Google Scholar]
- 31.Martin, J. N., and D. H. Osmond. 1999. Kaposi's sarcoma-associated herpesvirus and sexual transmission of cancer risk. Curr. Opin. Oncol. 11:508-515. [DOI] [PubMed] [Google Scholar]
- 32.Oda, D., and E. Watson. 1990. Human oral epithelial cell culture. I. Improved conditions for reproducible culture in serum-free medium in vitro. Cell Dev. Biol. 26:589-595. [DOI] [PubMed] [Google Scholar]
- 33.Padilla, J. A., S. Nii, and C. Grose. 2003. Imaging of the varicella zoster virion in the viral highways: comparison with herpes simplex viruses 1 and 2, cytomegalovirus, pseudorabies virus, and human herpes viruses 6 and 7. J. Med. Virol. 70:S103-S110. [DOI] [PubMed] [Google Scholar]
- 34.Pauk, J., M. L. Huang, S. J. Brodie, A. Wald, D. M. Koelle, T. Schacker, C. Celum, S. Selke, and L. Corey. 2000. Mucosal shedding of human herpesvirus 8 in men. N. Engl. J. Med. 343:1369-1377. [DOI] [PubMed] [Google Scholar]
- 35.Peacock, J. W., and K. L. Bost. 2000. Infection of intestinal epithelial cells and development of systemic disease following gastric instillation of murine gammaherpesvirus-68. J. Gen. Virol. 81:421-429. [DOI] [PubMed] [Google Scholar]
- 36.Pertel, P. E. 2002. Human herpesvirus 8 glycoprotein B (gB), gH, and gL can mediate cell fusion. J. Virol. 76:4390-4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Renne, R., D. Blackbourn, D. Whitby, J. Levy, and D. Ganem. 1998. Limited transmission of Kaposi's sarcoma-associated herpesvirus in cultured cells. J. Virol. 72:5182-5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Renne, R., M. Lagunoff, W. Zhong, and D. Ganem. 1996. The size and conformation of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) DNA in infected cells and virions. J. Virol. 70:8151-8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santos, R. A., J. A. Padilla, C. Hatfield, and C. Grose. 1998. Antigenic variation of varicella zoster virus Fc receptor gE: loss of a major B cell epitope in the ectodomain. Virology 249:21-31. [DOI] [PubMed] [Google Scholar]
- 40.Sitki-Green, D., M. Covington, and N. Raab-Traub. 2003. Compartmentalization and transmission of multiple Epstein-Barr virus strains in asymptomatic carriers. J. Virol. 77:1840-1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stewart, J. P., E. J. Usherwood, A. Ross, H. Dyson, and T. Nash. 1998. Lung epithelial cells are a major site of murine gammaherpesvirus persistence. J. Exp. Med. 187:1941-1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vieira, J., M. L. Huang, D. M. Koelle, and L. Corey. 1997. Transmissible Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in saliva of men with a history of Kaposi's sarcoma. J. Virol. 71:7083-7087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walling, D. M., A. L. Brown, W. Etienne, W. A. Keitel, and P. D. Ling. 2003. Multiple Epstein-Barr virus infections in healthy individuals. J. Virol. 77:6546-6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Webster-Cyriaque, J. 2002. Development of Kaposi's sarcoma in a surgical wound. N. Engl. J. Med. 346:1207-1210. [DOI] [PubMed] [Google Scholar]
- 45.Webster-Cyriaque, J., J. Middeldorp, and N. Raab-Traub. 2000. Hairy leukoplakia: an unusual combination of transforming and permissive Epstein-Barr virus infections. J. Virol. 74:7610-7618. [DOI] [PMC free article] [PubMed] [Google Scholar]